
Catalytic enantioselective synthesis of α-nitroepoxides via aminolytic kinetic resolution†
Sara
Meninno
,
Loris
Napolitano
and
Alessandra
Lattanzi
*
Dipartimento di Chimica e Biologia, Università di Salerno, Via Giovanni Paolo II, 84084, Fisciano, Italy. E-mail: lattanzi@unisa.it; Fax: 0039 089 969603
First published on 10th October 2014
Abstract
The first enantioselective synthesis of β-aryl-substituted α-nitroepoxides, exploiting an organocatalyzed aminolytic kinetic resolution (AKR), has been developed. Ring-opening reaction of racemic α-nitroepoxides with aniline in the presence of a readily available Cinchona alkaloid-derived thiourea affords unreacted epoxides in up to 95% ee.
The importance of optically active epoxides is widely recognised by their presence as structural motives in several natural products and mostly as highly versatile intermediates frequently involved in total synthesis.1 Indeed, a great variety of functionalised products is achievable via regio- and stereoselective opening of the oxirane ring.2
The asymmetric epoxidation of alkenes is the most appealing and straightforward approach to prepare epoxides.3 Amongst the several methodologies developed so far, a steadily growing number of efficient asymmetric nucleophilic epoxidation reactions have been reported for differently substituted enones4 and enals.5 In contrast, according to the paucity of examples illustrated in the literature,6 the asymmetric epoxidation of nitroalkenes, a class of popular electron-poor alkenes,7 appears to be a challenging process.
Racemic α-nitroepoxides, readily available by epoxidation of the corresponding nitroalkenes with a H2O2/NaOH system, are synthetically useful compounds due to the fact that they undergo complete regioselective ring-opening reactions at the β-position by a variety of nucleophiles to give α-substituted carbonyl compounds,8 functionalised α-aminoacids,6a,b and heterocycles9 (Fig. 1).
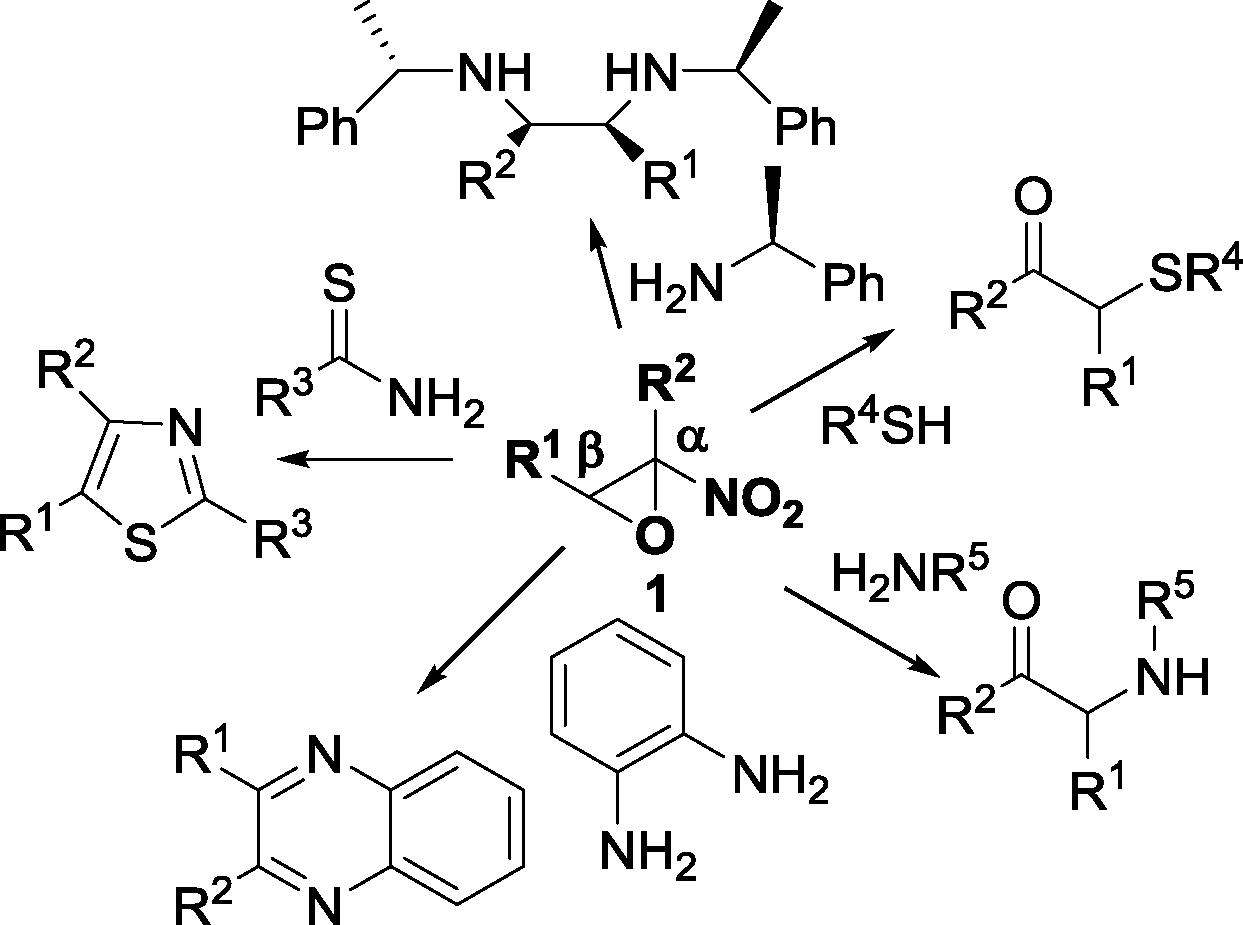 | ||
| Fig. 1 Ring-opening reactions of α-nitroepoxides. | ||
Chiral non-racemic amines have been recently employed in the ring-opening reaction of racemic α-nitroepoxides to afford, after reduction of the imine intermediate, vicinal diamines in a stereoselective fashion (Fig. 1).10 Hence, the development of processes to access enantioenriched α-nitroepoxides is of great importance, especially in view of the totally unexplored potential of this class of epoxides in asymmetric synthesis.
The kinetic resolution of racemic compounds represents a highly valuable strategy and sometimes the unique tool in the synthesis of a chiral non-racemic compound.11 Specifically, since the seminal work by Jacobsen and co-authors, the hydrolytic kinetic resolution (HKR) of terminal epoxides catalysed by a salen–Co-(III)-based complex12 became widely applied also on the industrial scale11b,13 as a powerful and exclusive approach for the preparation of these challenging compounds in excellent enantioselectivity. In the area of organocatalysis, hydrogen-bonding donors such as hexafluoro-2-propanol,14 achiral thioureas15 and ureas16 have been shown to assist the regioselective ring-opening reaction of epoxides to give differently 1,2-functionalized alcohols. Very recently, a few examples on the desymmetrization of meso-epoxides catalyzed by chiral phosphoric acids17 or sulphonamides18 have been reported.
Driven by our long-standing interest in asymmetric synthesis of epoxides19 and inspired by these reports, we envisaged that an aminolytic kinetic resolution catalyzed by chiral bifunctional organic promoters such as a thiourea amine could be a successful strategy to obtain enantioenriched α-nitroepoxides (Fig. 2).
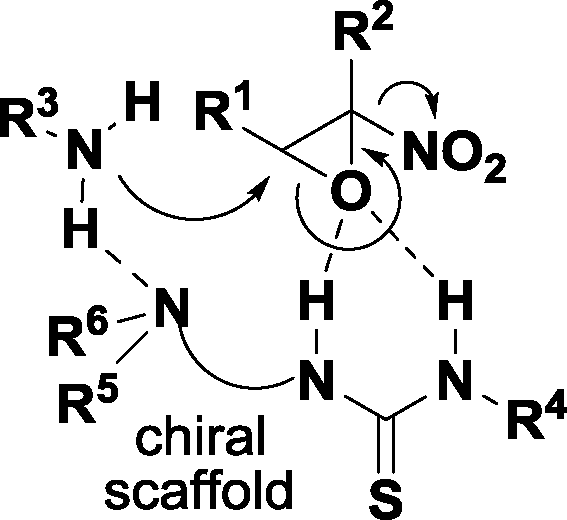 | ||
| Fig. 2 Proposed activation model in the AKR of α-nitroepoxide catalyzed by an amino thiourea. | ||
Activation of the reagents through general acid–base catalysis provided by the bifunctional organocatalyst would have likely led to the preferential opening of one enantiomer of the α-nitroepoxide in a highly regioselective manner.
Herein, we illustrate the first synthesis of enantiomerically enriched aromatic α-nitroepoxides. The kinetic resolution of racemic α-nitroepoxides proceeds via a highly regioselective ring-opening reaction with aniline organocatalysed by a Cinchona alkaloid-derived thiourea.
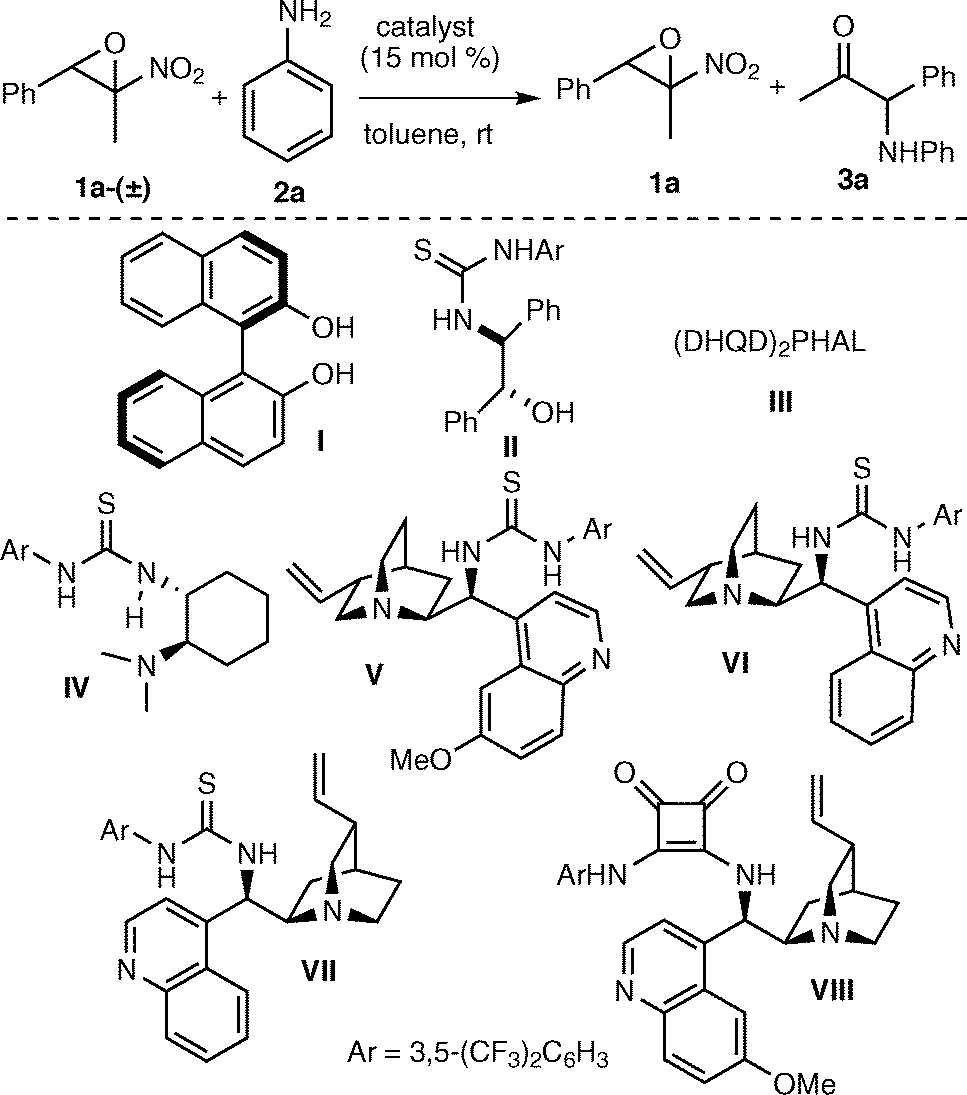
We commenced our study by reacting trans-2-methyl-2-nitro-3-phenyloxirane 1a-(±) and aniline20 in the presence of 15 mol% of chiral H-bonding donors as (R)-BINOL I21 and easily accessible amino alcohol-derived thiourea II22 at room temperature in toluene (Table 1). In the presence of catalyst I, the conversion to the expected ketone 3a was poor after 48 h (entry 1), whereas in the presence of the thiourea alcohol II a slight improvement of the conversion was observed, although the epoxide was isolated almost as a racemic compound (entry 2). A similar outcome was observed when using the Brønsted base catalyst III (entry 3). We were pleased to observe that Takemoto's catalyst IV provided the unreacted epoxide in 50% yield and 23% ee (entry 4). This result showed the requirement of a bifunctional organocatalyst for the reaction to proceed in a more effective way, likely via a cooperative activation of reagents as suggested in Fig. 2. Among the Cinchona alkaloid-derived thioureas, a significant improvement was achieved when using catalyst VI, since the epoxide was isolated in 63% yield and 30% ee (entry 6). Compound VII, the pseudo-enantiomer of VI, catalysed the process less effectively (entry 7). Finally, amine-squaramide VIII afforded a similar result to the one achieved with pseudo-enantiomeric amine thiourea V (entry 8). Catalyst VI was selected for further optimization of the AKR of epoxide 1a-(±) (Table 2).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Time (h) | 3a (%) | Yield 1ac (%) | ee 1ad (%) |
| a All reactions were carried out using 1a-(±) (0.15 mmol), 2a (0.18 mmol), catalyst (15 mol%) in toluene (1.5 ml). b Determined by 1H NMR analysis. c Isolated yield. d Determined by chiral HPLC analysis. Negative ee indicates the formation of the opposite enantiomer. | |||||
| 1 | I | 48 | 12 | nd | nd |
| 2 | II | 48 | 24 | 68 | 2 |
| 3 | III | 48 | 21 | 74 | 6 |
| 4 | IV | 65 | 40 | 50 | −23 |
| 5 | V | 69 | 37 | 51 | −11 |
| 6 | VI | 48 | 33 | 63 | −30 |
| 7 | VII | 48 | 40 | 47 | 28 |
| 8 | VIII | 48 | 35 | 59 | 6 |
| Entry | RNH2 | Solvent | 3 (%) | Yield 1ac (%) | ee 1ad (%) |
|---|---|---|---|---|---|
| a All reactions were carried out using 1a-(±) (0.15 mmol), 2 (0.18 mmol), VI (15 mol%), solvent (1.5 ml) for 48–113 h. b Determined by 1H NMR analysis. c Isolated yield. d Determined by chiral HPLC analysis. e Reaction carried out with 20 mol% of VI, 1.5 equiv. of aniline at C1a = 0.2 M. f Reaction carried out without a catalyst and with 3.0 equiv of aniline at C1a = 0.1 M for 96 h. g Reaction carried out with 20 mol% of VI, 3.0 equiv. of aniline at C1a = 0.1 M for 96 h. h Reaction carried out without a catalyst and with 3.0 equiv. of aniline at C1a = 0.1 M. | |||||
| 1 | 1-Naphthyl | Toluene | 34 (b) | 64 | 24 |
| 2 | 2-Naphthyl | Toluene | 32 (c) | 61 | 20 |
| 3 | 4-MeOC6H4 | Toluene | 38 (d) | 47 | 40 |
| 4 | Benzyl | Toluene | nd | 34 | 6 |
| 5 | Ph | CHCl3 | 30 (a) | 65 | 18 |
| 6 | Ph | tBuOMe | 52 (a) | 43 | 22 |
| 7 | Ph | p-xylene | 36 (a) | 59 | 30 |
| 8e | Ph | Toluene | 65 (a) | 28 | 59 |
| 9f | Ph | Toluene | 55 (a) | 38 | 50 |
| 10g | Ph | Toluene | 70 (a) | 28 | 72 |
| 11h | Ph | Toluene | 35 (a) | 62 | — |
Sterically demanding 1-, 2-naphthyl amines and 4-methoxyaniline, when reacting with epoxide 1a-(±), proved to be inferior reagents compared to aniline (entries 1–3). More nucleophilic benzyl amine reacted faster and less selectively (entry 4).23 A solvent screening, on model reaction with aniline, confirmed nonpolar aromatic solvents as the most suitable (entries 5–7). The reaction was further optimized in toluene varying the reagent's ratio and concentration. In order to increase the conversion to ketone, the reactions were carried out using 20 mol% catalyst VI, increasing the equivalents of aniline at different concentrations (entries 8–10). The best result was achieved using 3 equivalents of aniline working at C = 0.1 M, since the epoxide was isolated in 28% yield and 72% ee (entry 10). Finally, a control experiment performed without the organocatalyst showed that the background epoxide ring-opening reaction is not a negligible process (entry 11).
Under the optimized conditions a variety of α-nitroepoxides was reacted to study the scope and limitation of the process (Table 3).
|
|
|||||
|---|---|---|---|---|---|
| Entry | R 1 | R 2 | 3 (%) | Yield 1ab (%) | ee 1ac (%) |
| a All reactions were carried out using 1-(±) (0.2 mmol), 2a (0.6 mmol), VI (20 mol%), toluene (2.0 ml) for 84–158 h. b Isolated yield. c Determined by chiral HPLC analysis. | |||||
| 1 | Me | Ph | 65 | 28 (a) | 72 |
| 2 | Me | 4-MeC6H4 | 79 | 17 (e) | 84 |
| 3 | Me | 4-ClC6H4 | 67 | 29 (f) | 86 |
| 4 | Me | 4-CF3C6H4 | 48 | 36 (g) | 77 |
| 5 | Me | 2-Naphthyl | 72 | 26 (h) | 95 |
| 6 | Et | Ph | 59 | 21 (i) | 92 |
| 7 | Et | 3-MeC6H4 | 66 | 31 (j) | 92 |
| 8 | Et | 4-BrC6H4 | 64 | 33 (k) | 90 |
| 9 | Et | 3,4-Cl2C6H3 | 47 | 35 (l) | 61 |
| 10 | Me | Ph(CH2)2 | 28 | 35 (m) | 16 |
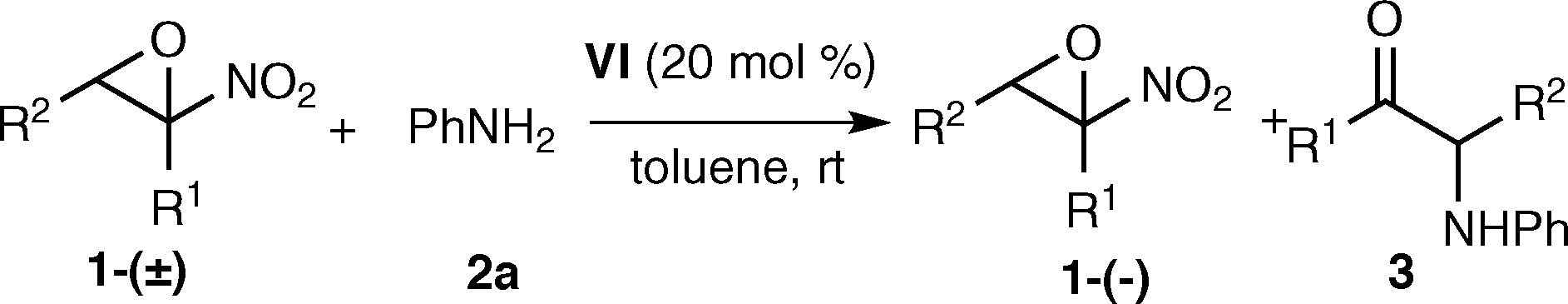
Electron-donating and -withdrawing groups are tolerated in the aromatic ring of compound 1 and unreacted epoxides were recovered in satisfactory yield and good to high enantioselectivity (entries 1–9). The present system appears to be unsuitable for the kinetic resolution of aliphatic α-nitroepoxides. The ring-opening reaction of α-nitroepoxide 1m proceeded at a lower rate and the unreacted epoxide was recovered in 35% yield and 16% ee (entry 10).23 The ee values of α-amino ketone 3 were not determined as it is supposed to be a nearly racemic compound on the basis of the following considerations: i) the estimated stereoselectivity factors of the AKR shown in Table 3 are moderate (3 < S < 7)24 and low ee values25 are expected for the α-amino ketone product 3;11a,b ii) enantiomerically enriched α-amino ketones were reported to be sensitive compounds suffering partial racemization under basic conditions.26
Jackson's6a,b and Aggarwal's27 groups demonstrated that ring-opening reactions of diastereoisomerically pure arylthionitrooxirane or spirocyclic bis-sulfinyl oxiranes with amines proceeded stereospecifically with inversion of the configuration to give enantioenriched α-amino thioesters and amides, respectively.
In order to determine the absolute configuration of optically enriched epoxides and to show the utility of these compounds in asymmetric synthesis, we set up a one-pot two-step diastereoselective route to produce 1,2-amino alcohols (Scheme 1).
 | ||
| Scheme 1 One-pot stereoselective ring-opening/reduction sequence to anti-1,2-amino alcohol 4a. | ||
Enantiomerically enriched 1a-(−), treated with pyrrolidine at 0 °C, afforded the corresponding α-amino ketone, which was in situ reduced in fairly good overall yield and in a highly diastereoselective manner to anti-1,2-amino alcohol 4a.28
Pleasingly, a slight erosion of the enantioselectivity was observed,26,29 as assessed by chiral HPLC analysis on 1-phenyl-1-(pyrrolidin-1-yl)propan-2-ol 4a, whose absolute configuration was determined to be (1R,2S) by comparison of the optical rotation to literature-reported data.30 Consequently, the absolute configuration of 2-methyl-2-nitro-3-phenyloxirane 1a-(−) was assigned to (2R,3S). Finally, other nucleophiles such as thiols and 1,2-diamines are applicable and pharmaceutically important targets such as quinoxalines31 are accessible by exploiting the organocatalysed kinetic resolution of α-nitroepoxides (Scheme 2).
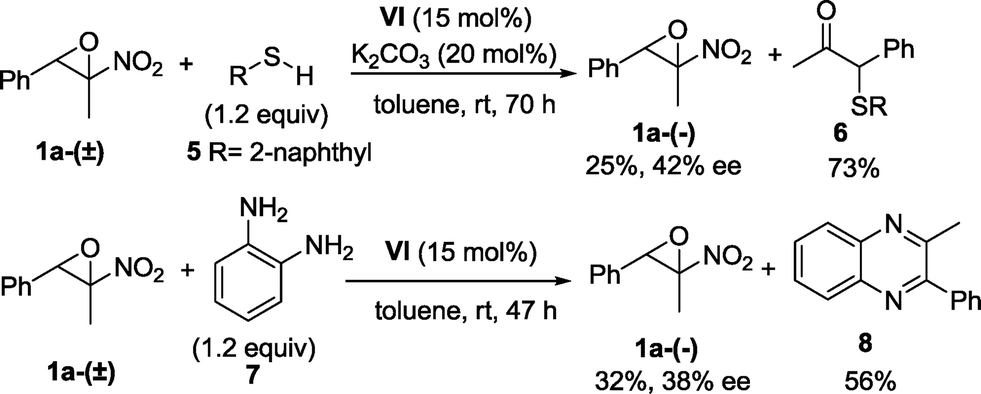 | ||
| Scheme 2 Kinetic resolution of epoxide 1a-(±) with thiol 5 and diamine 7. | ||
Conclusions
In conclusion, we developed the first enantioselective synthesis of β-aryl-substituted α-nitroepoxides, taking advantage of an aminolytic kinetic resolution with aniline catalysed by an easily accessible Cinchona alkaloid-derived thiourea. The epoxides are recovered in acceptable yield and good to high enantioselectivity. Enantioenriched α-nitroepoxides can serve as synthetically useful intermediates, as demonstrated in the one-pot stereoselective approach to highly valuable anti-1,2-amino alcohols.32 Despite the fact that improvements are required for the present AKR to be of practical value, this work highlights the great potential of bifunctional organocatalysts in epoxide ring-opening reactions. Efforts to improve and expand the scope of the organocatalytic kinetic resolution of racemic epoxides are currently under way.Acknowledgements
This work was supported financially by the MIUR. We thank Dr. P. Iannece for the assistance with MS spectral and elemental analyses. A.L. thanks the European COST Action CM0905-Organocatalysis.Notes and references
- For selected reviews, see: (a) J. Marco-Contelles, M. T. Molina and S. Anjum, Chem. Rev., 2004, 104, 2857 CrossRef CAS PubMed; (b) K. Miyashita and T. Imanishi, Chem. Rev., 2005, 105, 4515 CrossRef CAS PubMed; (c) Z.-L. Zhou, Y.-X. Yang, J. Ding, Y.-C. Li and Z.-H. Miao, Nat. Prod. Rep., 2012, 29, 457 RSC.
- For reviews, see: (a) C. Lauret, Tetrahedron: Asymmetry, 2001, 12, 2359 CrossRef CAS; (b) Aziridines and Epoxides in Organic Synthesis, ed. A. K. Yudin, Wiley-VCH, Weinheim, 2006 Search PubMed; (c) G. S. Singh, K. Mollet, M. D'hooghe and N. De Kimpe, Chem. Rev., 2013, 113, 1441 CrossRef CAS PubMed.
- For general reviews, see: (a) T. Katsuki, in Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer, Berlin, 1999, p. 621 Search PubMed; (b) E. M. McGarrigle and D. G. Gilheany, Chem. Rev., 2005, 105, 1563 CrossRef CAS PubMed; (c) Q.-H. Xia, H.-Q. Ge, C.-P. Ye, Z.-M. Liu and K.-X. Su, Chem. Rev., 2005, 105, 1603 CrossRef CAS PubMed; (d) Y. Zhu, Q. Wang, R. G. Cornwall and Y. Shi, Chem. Rev., 2014, 144, 8199 CrossRef PubMed.
- For selected examples, see: (a) D. Enders, J. Zhu and G. Raabe, Angew. Chem., Int. Ed. Engl., 1996, 35, 1725 CrossRef CAS; (b) M. Bougauchi, S. Watanabe, T. Arai, H. Sasai and M. Shibasaki, J. Am. Chem. Soc., 1997, 119, 2329 CrossRef CAS; (c) B. Lygo and P. G. Wainwright, Tetrahedron Lett., 1998, 39, 1599 CrossRef CAS; (d) T. Ooi, D. Ohara, M. Tamura and K. Maruoka, J. Am. Chem. Soc., 2004, 126, 6844 CrossRef CAS PubMed; (e) R. W. Flood, T. P. Geller, S. A. Petty, S. M. Roberts, J. Skidmore and M. Volk, Org. Lett., 2001, 3, 683 CrossRef CAS PubMed; (f) X. Lu, Y. Liu, B. Sun, B. Cindric and L. Deng, J. Am. Chem. Soc., 2008, 130, 8134 CrossRef CAS PubMed; (g) X. Wang, C. M. Reisinger and B. List, J. Am. Chem. Soc., 2008, 130, 6070 CrossRef CAS PubMed.
- For selected examples, see: (a) M. Marigo, G. Franzén, T. B. Poulsen, W. Zhuang and K. A. Jørgensen, J. Am. Chem. Soc., 2005, 127, 6964 CrossRef CAS PubMed; (b) H. Sundén, I. Ibrahem and A. Córdova, Tetrahedron Lett., 2006, 47, 99 CrossRef PubMed; (c) X. Wang and B. List, Angew. Chem., Int. Ed., 2008, 47, 1119 CrossRef CAS PubMed.
- For diastereoselective epoxidations, see: (a) R. F. W. Jackson, J. M. Kirk, N. J. Palmer, D. Waterson and M. J. Wythes, J. Chem. Soc., Chem. Commun., 1993, 889 RSC; (b) R. F. W. Jackson, N. J. Palmer, M. J. Wythes, W. Clegg and M. R. J. Elsegood, J. Org. Chem., 1995, 60, 6431 CrossRef CAS; (c) L. A. Evans, H. Adams, C. G. Barber, L. Caggiano and R. F. W. Jackson, Org. Biomol. Chem., 2007, 3156 RSC; (d) A. Jain, S. Rodríguez, I. López and F. V. González, Tetrahedron, 2009, 65, 8362 CrossRef CAS PubMedFor a stoichiometric enantioselective epoxidation of aliphatic trans-disubstituted nitroalkenes, see: (e) D. Enders, L. Kramps and J. Zhu, Tetrahedron: Asymmetry, 1998, 9, 3959 CrossRef CAS.
- For recent reviews, see: (a) D. Roca-Lopez, D. Sadaba, I. Delso, R. P. Herrera, T. Tejero and P. Merino, Tetrahedron: Asymmetry, 2010, 21, 2561 CrossRef CAS PubMed; (b) T. Ikariya and I. D. Gridnev, Chem. Rec., 2009, 9, 106 CrossRef CAS PubMed; (c) J. L. Vicario, D. Badía and L. Carrillo, Synthesis, 2007, 2065 CrossRef CAS PubMed.
- Y. D. Vankar, K. Shah, A. Bawa and S. P. Singh, Tetrahedron, 1991, 47, 8883 CrossRef CAS.
- For the synthesis of 1,3-thiazoles, see: (a) H. Newman and R. B. Angier, Tetrahedron, 1970, 26, 825 CrossRef CAS; (b) K. M. Weiß, S.-W. Wei and S. B. Tsogoeva, Org. Biomol. Chem., 2011, 9, 3457 RSCFor the synthesis of quinoxalines, see: (c) M. M. Ibrahim, D. Grau, F. Hampel and S. B. Tsogoeva, Eur. J. Org. Chem., 2014, 1401 CrossRef CAS; (d) A. Vidal-Albalat, S. Rodríguez and F. V. González, Org. Lett., 2014, 16, 1752 CrossRef CAS PubMed.
- J. Agut, A. Vidal, S. Rodríguez and F. V. González, J. Org. Chem., 2013, 78, 5717 CrossRef CAS PubMed.
- (a) H. B. Kagan and J. C. Fiaud, Top. Stereochem., 1988, 18, 249 CAS; (b) J. M. Keith, J. F. Larrow and E. N. Jacobsen, Adv. Synth. Catal., 2001, 343, 5 CrossRef CAS.
- M. Tokunaga, J. F. Larrow, F. Kakiuchi and E. N. Jacobsen, Science, 1997, 277, 936 CrossRef CAS.
- (a) J. F. Larrow and P. F. Quigley in Comprehensive Chirality, ed. E. M. Carreira and H. Yamamoto, Elsevier, Oxford, 2012, p. 129. Search PubMedFor other examples on kinetic resolution of racemic epoxides, see: (b) A. Gayet, S. Betilsson and P. G. Andersson, Org. Lett., 2002, 4, 3777 CrossRef CAS; (c) G. Bartoli, M. Bosco, A. Carlone, M. Locatelli, P. Melchiorre and L. Sambri, Org. Lett., 2004, 6, 3973 CrossRef CAS PubMed.
- U. Das, B. Crousse, V. Kesavan, D. Bonnet-Delpon and J.-P. Bégué, J. Org. Chem., 2000, 65, 6749 CrossRef CAS.
- (a) T. Weil, M. Kotke, C. M. Kleiner and P. R. Schreiner, Org. Lett., 2008, 10, 1513 CrossRef CAS PubMed; (b) S. S. Chimni, N. Bala, V. A. Dixit and P. V. Bharatam, Tetrahedron, 2010, 66, 3042 CrossRef CAS PubMed.
- (a) E. M. Fleming, C. Quigley, I. Rozas and S. J. Connon, J. Org. Chem., 2008, 73, 948 CrossRef CAS PubMed; (b) J. Park, K. Lang, K. A. Abboud and S. Hong, Chem. – Eur. J., 2011, 17, 2236 CrossRef CAS PubMed.
- (a) Z. Wang, W. K. Law and J. Sun, Org. Lett., 2013, 15, 5964 CrossRef CAS PubMed; (b) M. R. Monaco, S. Prévost and B. List, Angew. Chem., Int. Ed., 2014, 53, 8142 CrossRef CAS PubMed.
- M. Kumar, R. I. Kureshy, S. Saravanan, S. Verma, A. Jakhar, N. H. Khan, S. H. R. Abdi and H. C. Bajaj, Org. Lett., 2014, 16, 2798 CrossRef CAS PubMed.
- (a) A. Lattanzi, P. Iannece, A. Vicinanza and A. Scettri, Chem. Commun., 2003, 1440 RSC; (b) A. Lattanzi, Org. Lett., 2005, 7, 2579 CrossRef CAS PubMed; (c) C. De Fusco, C. Tedesco and A. Lattanzi, J. Org. Chem., 2011, 76, 676 CrossRef CAS PubMed; (d) A. Russo, G. Galdi, G. Croce and A. Lattanzi, Chem. – Eur. J., 2012, 18, 6152 CrossRef CAS PubMed.
- An excess of aniline (1.2 equiv.) was added to neutralize nitrous acid formed as by-product.
- Y. N. Belokon, V. I. Maleev, M. A. Moskalenko, Yu. V. Samoilichenko, A. S. Peregudov and A. T. Tsaloev, Russ. Chem. Bull., 2013, 62, 1371 CrossRef CAS.
- A. Lattanzi, Synlett, 2007, 2106 CrossRef CAS PubMed.
- Other unidentified products were observed in the crude reaction mixture.
- First order kinetics was assumed for the calculation of the stereoselectivity factors,11a which are underestimated because partially affected by contribution of the background ring-opening reaction.
- As an example, α-amino ketone 3a, reported in entry 1 of Table 3, was recovered with 16% ee.
- (a) B. Tiwari, J. Zhang and Y. R. Chi, Angew. Chem., Int. Ed., 2012, 51, 1911 CrossRef CAS PubMed; (b) C. Kison, N. Meyer and T. Opatz, Angew. Chem., Int. Ed., 2005, 44, 5662 CrossRef CAS PubMed.
- V. K. Aggarwal, J. K. Barrell, J. M. Worrall and R. Alexander, J. Org. Chem., 1998, 63, 7128 CrossRef CAS.
- For diastereoselective reduction of α-amino ketones to 1,2-amino alcohols, see: (a) J.-P. Bégué, D. Bonnet-Delpon and N. Fischer-Durand, Tetrahedron: Asymmetry, 1994, 5, 1099 CrossRef; (b) A. Abouabdellah, J.-P. Bégué, D. Bonnet-Delpon, A. Kornilov, I. Rodrigues and C. Richard, J. Org. Chem., 1998, 63, 6529 CrossRef CAS.
- The ring-opening/reduction sequence was not optimized.
- J.-H. Xie, S. Liu, W.-L. Kong, W.-J. Bai, X.-C. Wang, L.-X. Wang and Q.-L. Zhou, J. Am. Chem. Soc., 2009, 131, 4222 CrossRef CAS PubMed.
- For selected examples, see: (a) S. T. Hazeldine, L. Polin, J. Kushner, J. Paluch, K. White, M. Edelstein, E. Palomino, T. H. Corbett and J. P. Horwitz, J. Med. Chem., 2001, 44, 1758 CrossRef CAS PubMed; (b) D. Kong, E. J. Park, A. G. Stephen, M. Calvani, J. H. Cardellina, A. Monks, R. J. Fisher, R. H. Shoemaker and G. Melillo, Cancer Res., 2005, 65, 9047 CrossRef CAS PubMed.
- For reviews on enantiomerically enriched 1,2-amino alcohols, see: (a) F. D. Klingler, Acc. Chem. Res., 2007, 40, 1367 CrossRef CAS PubMed; (b) J. C. Moore, D. J. Pollard, B. Kosjek and P. N. Devin, Acc. Chem. Res., 2007, 40, 1412 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra and HPLC traces of all of the new compounds. See DOI: 10.1039/c4cy01157k |
| This journal is © The Royal Society of Chemistry 2015 |