
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Doping and electronic properties of GaAs grown by close-spaced vapor transport from powder sources for scalable III–V photovoltaics†
Andrew J.
Ritenour
a,
Jason W.
Boucher
b,
Robert
DeLancey
b,
Ann L.
Greenaway
a,
Shaul
Aloni
c and
Shannon W.
Boettcher
*a
aDepartment of Chemistry and Biochemistry, University of Oregon, Eugene, 97403, USA. E-mail: swb@uoregon.edu
bDepartment of Physics, University of Oregon, Eugene, 97403, USA
cThe Molecular Foundry, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA
First published on 1st September 2014
Abstract
We report the use of a simple close-spaced vapor transport technique for the growth of high-quality epitaxial GaAs films using potentially inexpensive GaAs powders as precursors. The free carrier type and density (1016 to 1019 cm−3) of the films were adjusted by addition of Te or Zn powder to the GaAs source powder. We show using photoelectrochemical and electron beam-induced current analyses that the minority carrier diffusion lengths of the n- and p-GaAs films reached ∼3 μm and ∼8 μm, respectively. Hall mobilities approach those achieved for GaAs grown by metal–organic chemical vapor deposition, 1000–4200 cm2 V−1 s−1 for n-GaAs and 50–240 cm V−1 s−1 for p-GaAs depending on doping level. We conclude that the electronic quality of GaAs grown by close-spaced vapor transport is similar to that of GaAs made using conventional techniques and is thus sufficient for high-performance photovoltaic applications.
Broader contextThe high balance-of-system costs of photovoltaic (PV) installations indicate that reductions in cell $ per W costs alone are likely insufficient for PV electricity to reach grid parity unless energy conversion efficiency is also increased. Technologies which yield both high-efficiency cells (>25%) and maintain low costs are needed. GaAs and related III–V semiconductors are used in the highest-efficiency single- and multi-junction photovoltaics, but the technology is too expensive for non-concentrated terrestrial applications. This is due in part to the difficulty of scaling the metal–organic chemical vapor deposition (MOCVD) process, which relies on expensive reactors and employs toxic and pyrophoric gas-phase precursors such as arsine and trimethyl gallium, respectively. We describe GaAs films made by an alternative close-spaced vapor transport (CSVT) technique which is carried out at atmospheric pressure and requires only bulk GaAs, water vapor, and a temperature gradient in order to deposit crystalline films with similar electronic properties to that of GaAs deposited by MOCVD. CSVT is similar to the vapor transport process used to deposit CdTe thin films and is thus a potentially scalable low-cost route to GaAs thin films. |
Introduction
GaAs is interesting for photovoltaic (PV) applications due to its high carrier mobilities, large optical absorption coefficient α(λ), and direct band gap of Eg = 1.42 eV. These properties lead to a theoretical maximum one-sun energy conversion efficiency of η = 33.5% (ref. 1–4) and demonstrated world-record of η = 28.8%.5 However, GaAs is not currently cost-competitive with Si or CdTe flat-panel PVs due in part to the high cost and relatively low throughput of conventional epitaxial growth techniques.6 Low-cost, high-throughput routes to GaAs films are needed.Close-spaced vapor transport (CSVT) is a plausibly scalable technique for making epitaxial GaAs films.7–10 In CSVT, H2O vapor (typically ∼2000 ppm in hydrogen or forming gas) is used to etch a solid GaAs source, generating vapor-phase reactants As2 and Ga2O in situ at atmospheric pressure (Fig. 1).11 The liberated As2 and Ga2O diffuse through a thermal gradient (typically ΔT = 10–50 °C) and re-deposit as GaAs on the cooler substrate suspended <1 mm from the source by a spacer. The CSVT reactor can be relatively simple, inexpensive, and compact. CSVT is capable of high growth rate (up to 1 μm min−1 demonstrated) and >95% overall precursor transport/utilization efficiency from source to substrate.8 The supersaturation can be tuned at the substrate through control of the temperature gradient, facilitating selected-area epitaxy and epitaxial layer overgrowth (ELO) on Si substrates.12,13 These features could potentially enable growth of high-quality GaAs films with large grains14 on thermally/lattice-mismatched15,16 or ceramic17 substrates. This is significant because in order to successfully utilize GaAs for terrestrial PV applications, in addition to developing an efficient growth technique, the substrate must either be inexpensive or reusable (e.g. through epitaxial lift-off processes).6
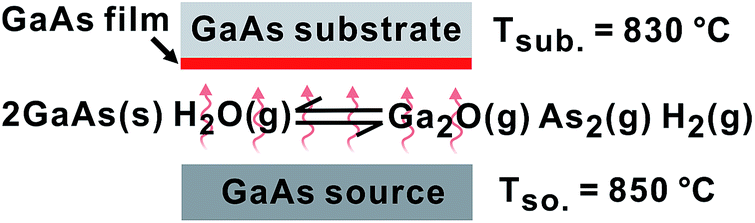 | ||
| Fig. 1 Schematic of CSVT of GaAs with in situ generation of gas-phase precursors. | ||
The use of H2O as a transport agent is an important advantage of CSVT over the conventional metal–organic chemical vapor deposition (MOCVD) process. Although MOCVD produces high-mobility GaAs films, it relies on the use of gas-phase precursors such as arsine (acutely toxic) and trimethylgallium (pyrophoric). The use of these hazardous and expensive precursors contributes to the cost and complexity of MOCVD reactors and makes it less appealing for the growth of GaAs for terrestrial PV applications, which require both low cost and high throughput.
Liquid phase epitaxy (LPE) has also been used for GaAs epitaxy yielding high-mobility (equivalent to MOCVD films) GaAs and also high growth rates (>1 μm min−1), and thus is also appealing for PV applications. LPE is, however, a batch process with likely lower throughput to what is ultimately possible with vapor phase deposition.18 It is also difficult to control/implement heteroepitaxy, especially on lower cost substrates such as Si.
Despite the merits of the CSVT technique, few CSVT GaAs devices have been reported.19–21 We previously demonstrated CSVT n-GaAs films with minority carrier diffusion lengths (LD) > 1 μm (ref. 22) and overall η = 9.3% in a photoelectrochemical (PEC) cell,23 nearly equivalent to MOCVD n-GaAs photoanodes (η = 11%).24 These CSVT n-GaAs films were grown from Si-doped GaAs sources and exhibited free electron concentrations of 0.5–2 × 1017 cm−3. We have since determined that these are doped by S (which outgasses from the graphite heaters upon heating) rather than transport of Si dopants from the Si-doped GaAs source. The S doping is discussed further below (see Results and discussion). Controlling the dopant type and dopant density of CSVT GaAs films is a key step toward fabrication of solid-state PV devices. For PV applications, the films must also possess high carrier mobilities and LD > α(λ)−1.
Here we report n- and p-GaAs films with a range of free electron and free hole concentrations (ND and NA, respectively) grown using CSVT from potentially low-cost powder sources. Mixing powders could also be used to access related ternary III–V materials such as GaAsxP1−x.25ND and NA determined from impedance and Hall-effect measurements agree with dopant concentrations obtained from secondary ion mass spectrometry (SIMS), and demonstrate control over the dopant concentration from ∼1016 cm−3 to ∼1019 cm−3. The LD was up to ∼3 μm for n-GaAs films and up to ∼8 μm for p-GaAs films, determined independently via analysis of the internal quantum efficiency Φint and electron beam induced current (EBIC). These LD are long with respect to α(λ)−1 and consistent with the measured one-sun photocurrents in the PEC configuration (>20 mA cm−2 with no antireflective coating). Hall mobilities of CSVT n- and p-GaAs approach the ionized dopant scattering limit26 and are similar to what has been achieved using MOCVD.27
These results demonstrate that potentially inexpensive powdered GaAs can be used to deposit GaAs films suitable for high performance III–V based PV devices28 at high growth rate, with ∼95% precursor utilization, and at ambient pressures using a simple CSVT reactor.
Experimental
GaAs thin film deposition
The carrier gas for CSVT was H2 (Industrial Source, 99.999%) with water concentration [H2O] = 2000 ppm. [H2O] was controlled by combining a stream of dry H2 with a stream of H2O-saturated (dew point 10 °C) H2 using mass flow controllers (SEC 4400) and monitored with a Panametrics MM4 Hygrometer. PID controllers (Omega CN7800) were used to set the growth temperature to 850 °C and 830 °C for the source and substrate, respectively, as shown in Fig. 1. The temperature was monitored using type K thermocouples embedded in the resistive graphite heaters. A quartz ring 12 mm in diameter and 0.8 mm thick was used as a spacer between the source and substrate. The film thicknesses of all PEC samples were 5–11 μm (measurements obtained from a Zygo 7300 optical profilometer), ensuring all light was absorbed in the CSVT GaAs film rather than the substrate. The growth rate (which depends on [H2O] used during growth23) was ∼0.3 μm min−1. Single-crystal 〈100〉-oriented GaAs wafers grown by the vertical gradient freeze technique29 (AXT, Inc.) were used as substrates. The substrates were epi-ready as received and cleaned by blowing with N2. More details about the CSVT reactor are provided in previous publications.22,23Powder GaAs sources were obtained by grinding undoped GaAs wafers (AXT) in an agate mortar and pestle and pressing at 140 MPa in a 13 mm pellet die. The mortar and pestle were cleaned by submersing in aqua regia and rinsing with 18.2 MΩ cm water. Zn powders were separately weighed and combined with the GaAs powder prior to pressing. Te-doped powders with [Te] < 1019 cm−3 were made by grinding and pressing Te-doped wafers. A source pellet containing [Te] = 1019 cm−3 was made by combining undoped GaAs and Te powder. Single-crystal wafers were also used as sources to provide a comparison to the powders. These were cut into 13 × 13 mm squares and cleaned by blowing with N2.
Photoelectrochemical measurements
Electrodes were fabricated using standard procedures.23 Ohmic contacts were formed on the back of the substrates by thermal evaporation of Au/Zn/Au (20 nm, 30 nm, 50 nm) for p-GaAs or AuGe eutectic (100 nm) for n-GaAs. Contacts were annealed at 450 °C for 2 min in 95% N2/5% H2.30 Ohmic contacts were connected to Sn–Cu wire with conductive Ag adhesive (Pelco 16040–30) and attached to 6 mm diameter glass tubes with non-conductive epoxy (Hysol 1C). A circular electrode active region (∼0.05 cm2) was defined using non-conductive black epoxy (Hysol 9460).Rectifying contacts to n-GaAs for current–voltage (J–E) measurements, impedance measurements, and spectral response measurements were obtained using an electrolyte consisting of 1 M LiClO4 (Alfa-Aesar, 99%, anhydrous), 100 mM ferrocene (Aldrich, 98%, sublimed before use), and 0.5 mM ferrocenium tetrafluoroborate (obtained by oxidizing ferrocene with benzoquinone in the presence of HBF4, recrystallizing in tetrahydrofuran, and drying under vacuum) in dry acetonitrile (Acros, 99.8%, distilled and dried with freshly-activated 3 Å molecular sieves).24,31 For spectral response measurements, the solution was diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 with dry acetonitrile in order to reduce parasitic solution absorbance.
:10 with dry acetonitrile in order to reduce parasitic solution absorbance.
For one-sun J–E measurements of p-GaAs, an aqueous solution of 1 M HI (Aldrich, 99.99%) and 0.125 M I2 (Alfa-Aesar, 99.8+%) was used.32,33 For spectral response and impedance of p-GaAs a non-aqueous electrolyte consisting of 0.1 M NaI (Alfa-Aesar, 99+%, anhydrous), 0.0125 M I2 (Sigma-Aldrich 99.99%, sublimed), and 0.1 M LiClO4 in dry acetonitrile was used (see Results and discussion below).
For all PEC measurements a potentiostat (Bio-Logic SP-200) in three-electrode configuration was used. The GaAs electrode potential (E) was referenced to the potential of a Pt wire poised at the solution potential (Esol) and a Pt mesh was used as the counter electrode. The three electrodes were held in a glass three-neck flask containing the appropriate electrolyte with the GaAs electrode <1 mm from the bottom surface. Mass transport was aided by a magnetic stirrer. Illumination was provided by a solar simulator (Abet Technologies model 10500) for J–E experiments. The light intensity incident on the front face of the glass cell was 100 mW cm−2 as determined using a calibrated photodiode (OSI Optoelectronics UV-005). The photodiode was calibrating using an optical pyrometer (Thor Labs S310C).
Spectral response
Spectral response of the PEC cells was measured using a Bentham PVE300 system. The monochromatic light was chopped at 35 Hz and the nA-range signal was measured using a lock-in amplifier. The chopped signal was free of transients and the amplitude of the signal was independent of chopping frequency from 10–50 Hz.34 A calibrated photodiode (Bentham 11677) was used to determine the incident light intensity so that the nA signal could be converted to external quantum efficiency (Φext). The chopped photocurrent scaled linearly with the incident light intensity over five orders of magnitude (Fig. S6†).Reflectance, R(λ), of the air|glass|acetonitrile|GaAs stack was measured using a spectrometer with an integrating sphere (Perkin Elmer Lambda 1050).34 This data was used to obtain Φint from Φext = Φint [1 − R(λ)]. Although films from wafer sources were not completely specular, all films possessed R(λ) equivalent to a polished single-crystal GaAs wafer when measured in the integrating sphere. Therefore, although some of the films exhibited diffuse reflectance, the total R(λ) was unchanged. All films grown from powder sources were specular.
Impedance spectroscopy
Impedance measurements were conducted with a potentiostat on GaAs electrodes in a dark box. The DC reverse bias was varied between 0–1 V in 10 steps for n-GaAs and 0 to −0.5 V in 10 steps for p-GaAs electrodes with a 10 mV AC bias amplitude. The impedance data were fit to the typical equivalent circuit model to extract the junction capacitance C (Fig. S4†).35 Mott–Schottky analysis was used to determine ND or NA and the barrier height Vbi (Fig. S5†).31,36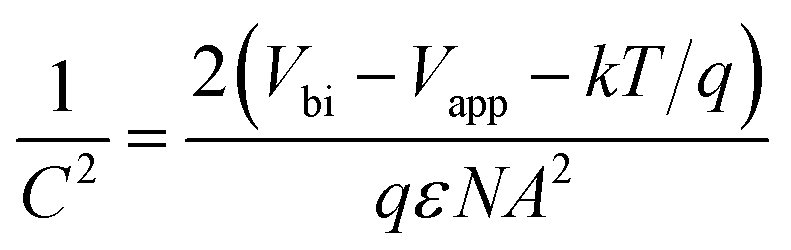 | (1) |
The depletion region thickness W was obtained using:37
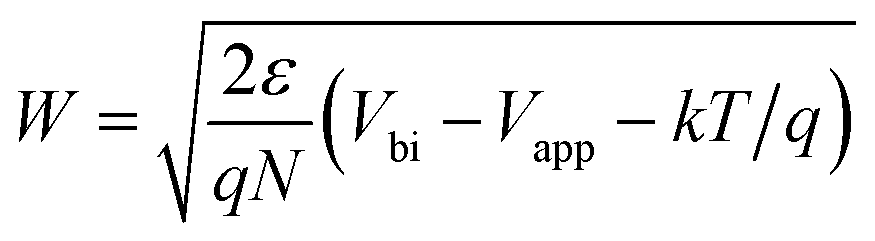 | (2) |
Hall effect measurements
GaAs films deposited on undoped semi-insulating (resistivity ρ > 107 Ω cm) substrates were cut into squares (0.49–0.64 cm2) and ohmic contacts were applied to the corners. The carrier type, ρ, NA or ND, and majority carrier mobility (μe or μh for electron or hole mobility, respectively) were obtained using the Van der Pauw method.38 Hall effect measurements were conducted at 300 K on an Ecopia HMS-5000 Hall effect system.Time-of-flight secondary ion mass spectrometry
Mass spectra were obtained from a Cameca ION-TOF mass spectrometer with Cs+ sputter gun and Bi+ analysis gun. Ion counts were converted to bulk impurity concentrations using relative sensitivity factors (RSF).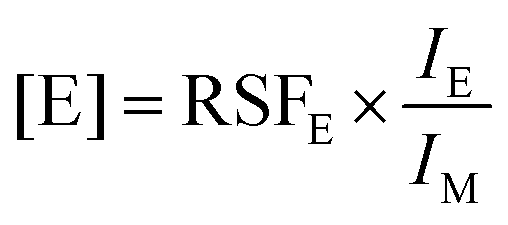 | (3) |
Electron beam-induced current
The EBIC measurements were conducted in a scanning electron microscope (FEI Quanta 200F). The electron beam was scanned toward the Schottky contact and the resulting current transients (Fig. S7†) were recorded from a Matelect ISM-6A induced signal monitor. LD was extracted from the decays according to:| I = qNCe−x/LD | (4) |
Prior to EBIC experiments, surface passivation was needed to lower the surface recombination velocity (SRV). Surface passivation was accomplished by etching with 5 M HCl, rinsing with 18.2 MΩ cm water, immersing in aqueous 1 M Na2S,41 rinsing with water and ethanol, and drying with N2. Samples were immediately pumped into the vacuum chamber and measured within 20 min after passivation. Results obtained without passivation varied as a function of accelerating voltage (Vacc) and did not produce reliable values of LD (see Results and discussion). The signal decays used to extract LD fit eqn (4) over several orders of magnitude of current (Fig. S7†).
Results and discussion
Growth and doping from powder and wafer sources
In order to fabricate high-efficiency GaAs PV devices, it is important to be able to control both NA and ND while maintaining suitable electronic quality of the GaAs layers. Thus to demonstrate the utility of the CSVT technique, it is necessary to determine which dopants are transported by CSVT, how the source dopant concentration is related to the film dopant concentration, and how they affect the electronic quality of the layers.We previously reported the growth of n-GaAs films by CSVT from Si-doped wafer sources and hypothesized that the films were Si-doped since the ND of the films (∼3 × 1017 cm−3) matched the [Si] in the source wafer. However, upon TOF-SIMS analysis we determined that the films were S-doped rather than Si-doped (Fig. S1†). The poor transport efficiency of Si is likely related to the low vapor pressure of SiOx, which forms at high temperatures in the presence of H2O. Unintentional S-doping of GaAs layers grown by CSVT has also been reported in other studies.42,43
The unintentional S impurity is undesirable because it is a compensating defect in p-GaAs films. We used TOF-SIMS to determine that the graphite heaters were a source of S impurity (Fig. S2†). After fabricating purified heaters, the unintentional S-doping of the films decreased to [S] ≤ 7 × 1016 cm−3 as determined by TOF-SIMS analysis of the films. Although the [S] could likely be further reduced by using non-porous, higher-purity heater materials (e.g. more-expensive pyrolysis-derived graphite), for this study the [S] achieved was low enough to permit growth of p-GaAs films with NA ≥ 1017 cm−3, which is appropriate for use as a p-type absorbing layer in a PV device.
Zn is widely used as a p-type dopant in GaAs and has been shown to transport by CSVT.43 GaAs films grown using CSVT from Zn-doped wafers possess ∼1/100th the NA of the source wafer.43 Commercial GaAs wafers were available with [Zn] < 2 × 1019 cm−3, setting an upper limit of NA ≈ 2 × 1017 cm−3 for CSVT films grown from commercially available sources (practically less, due to S compensation). This is problematic since some active PV device components (e.g. emitters and back surface fields) require NA > 2 × 1017 cm−3. In order to grow p-GaAs films with higher NA, we mixed GaAs and Zn powders at the desired ratio and pressed the mixtures into pellets. These pellet sources yielded p-GaAs films with NA up to ∼2 × 1019 cm−3 (Table 1).
| Powder or wafer source | Source dopant species (E) | Source [E]a (cm−3) | Impedance analysis ND − NA (cm−3) (average of three electrodes) | Hall effect ND − NA (cm−3) (one sample) | [E] from SIMS (cm−3) (one sample) |
|---|---|---|---|---|---|
| a For wafer sources the dopant density was provided by the manufacturer; for Zn-doped powder sources the dopant density was calculated from the mass of the GaAs powder and the Zn powder used. b The S dopant was not intentionally added. | |||||
| Wafer | Te | 2–4 × 1018 | 3 × 1018 ± 1 × 1018 | — | 2 × 1018 |
| Powder | Te | 2–4 × 1018 | 4 × 1018 ± 1 × 1018 | 3 × 1018 | — |
| Wafer | Te | 3–6 × 1017 | 4 × 1017 ± 4 × 1016 | 3 × 1017 | 6 × 1017 |
| Powder | Te | 3–6 × 1017 | 7 × 1017 ± 2 × 1017 | 6 × 1017 | — |
| Powder | Zn | 5 × 1021 | −2 × 1019 ± 3 × 1018 | −4 × 1019 | — |
| Powder | Zn | 5 × 1020 | −4 × 1018 ± 7 × 1017 | −4 × 1018 | — |
| Wafer | Zn | 1–2 × 1019 | −2 × 1017 ± 1 × 1016 | −1 × 1017 | 1 × 1017 |
| Powder | Sb | —b | 1 × 1017 ± 4 × 1016 | 2 × 1016 | 3 × 1016 |
| Wafer | Sb | —b | 7 × 1016 ± 3 × 1016 | 8 × 1016 | 7 × 1016 |
In order to control ND, we used Te-doped GaAs sources. It has been shown using impedance profiling that GaAs films grown using CSVT from Te-doped wafers possess ND equivalent to the source wafers.43–45 We reproduced these results by using two n-GaAs:Te sources with different [Te] to grow GaAs films on degenerately-doped GaAs:Si substrates and measuring ND with impedance profiling. In order to confirm that the dopants were transported by CSVT and not diffused from the substrate, we also deposited n-GaAs:Te films on undoped, semi-insulating substrates. Hall effect measurements of these samples confirm the same relationship, ND ≈ source [Te]. We also show using TOF-SIMS that the films possess [Te] similar to the source's [Te] and contain no Si from the GaAs:Si substrate (Fig. 2). All of this data is summarized in Table 1.
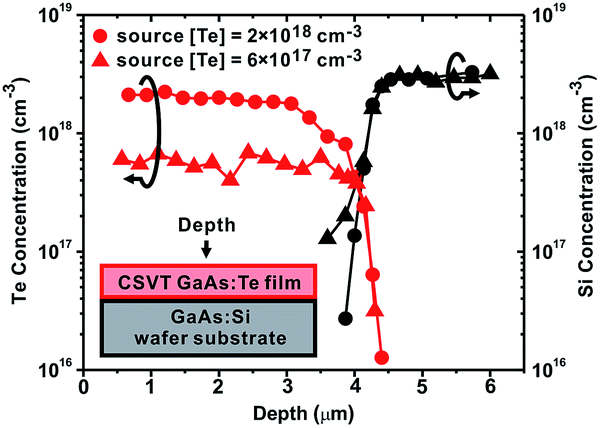 | ||
| Fig. 2 SIMS depth profiles of two n-GaAs films grown from differently-doped GaAs:Te sources on GaAs:Si substrates. The Te depth profiles are shown in red and Si in black. Circles denote the film was grown from a source containing [Te] = 2 × 1018 cm−3, triangles denote the film was grown from a source with [Te] = 6 × 1017 cm−3. | ||
Photoelectrochemical J–E analysis
The PEC GaAs|electrolyte junction is a convenient tool which enables the study of material properties by analyzing photocurrent vs. potential (J–E) response, junction impedance, and Φint without fabrication of solid state devices.23,24,31Electrodes of CSVT n-GaAs films were immersed in a non-aqueous ferrocene/ferrocenium electrolyte (Fc/Fc+) and their J–E response was measured under 100 mW cm−2 of simulated AM1.5G irradiation.24 Commercial 〈100〉-oriented single-crystal wafers were measured as controls. The CSVT samples produced open-circuit voltages (Voc) up to 0.83 V, equivalent to that attained by others using MOCVD n-GaAs.46,47 Short-circuit current density (Jsc) was ∼20 mA cm−2 for moderately-doped samples having ND = 1016 to 1017 cm−3. The performance of all samples exceed the respective bare substrates and similarly-doped GaAs control wafers (Fig. 3A). There were no significant differences between films grown from powder and wafer sources. Lower photocurrent was observed in highly-doped samples, which also exhibit lower μh (and consequently LD) due to carrier scattering by ionized dopant atoms in the lattice (see Hall effect measurements below).48,49
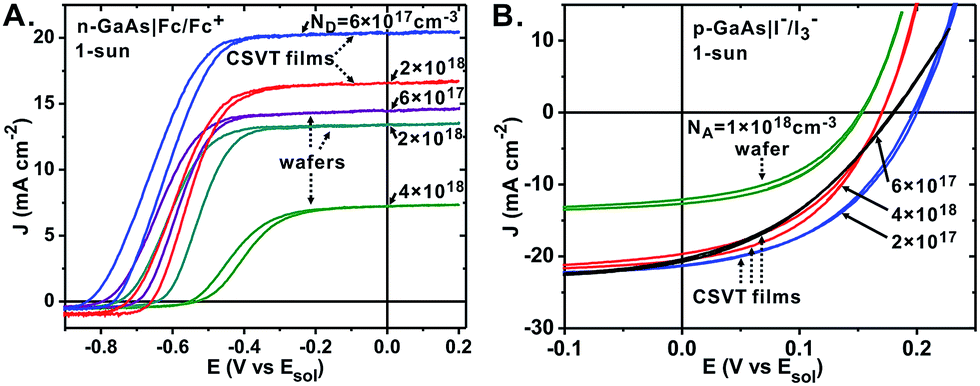 | ||
| Fig. 3 Photoelectrochemical J–E curves of (A) n-GaAs and (B) p-GaAs CSVT films and control wafers. The curves are labeled with the corresponding sample's free carrier concentration in cm−3. The selected curves are representative of other electrodes obtained from the same samples and from other samples with similar free carrier concentrations. | ||
Electrodes of CSVT p-GaAs films and control wafers were immersed in an aq. iodide/triiodide electrolyte (I−/I3−) and their J–E response was measured under 100 mW cm−2 of simulated AM1.5G irradiation (Fig. 3B).32,33 The Voc was 0.15–0.20 V vs. Esol, lower than the n-GaAs samples due to surface pinning of the p-GaAs Fermi level near the valence band edge.32 The best samples exhibited Jsc ∼ 20 mA cm−2, similar to the best n-GaAs samples despite the higher parasitic light absorption of the I−/I3− electrolyte. All CSVT p-GaAs films (including those synthesized with NA > 1018 cm−3) exhibited higher photocurrent than the p-GaAs control wafer (NA = 1 × 1018 cm−3, Jsc = 12 mA cm−2) indicating lower bulk recombination and a longer LD.
Spectral response and LD determination
In order determine the LD for each sample we measured the spectral response using the short-circuit PEC configuration (EWE = 0 V vs. Esol) under low-intensity chopped monochromatic light. For the n-GaAs films such measurements in the Fc/Fc+ electrolyte are well-developed. Spectral response measurements of p-GaAs in aqueous I−/I3−, however, were complicated by the solubility of GaAs in the acidic I−/I3− electrolyte.No etching of p-GaAs was observed in the aq. solution after hours of sustained operation as long as illumination was provided. However, a nA-range anodic current was observed in the aqueous solution when under dark or low-light conditions (<1 μW cm−2). This was problematic for spectral response, (which uses a nA-range chopped light source) and impedance analysis, which is conducted in the dark (Fig. S6†). We suspect the p-GaAs surface, while unstable in H2O especially at low pH,50 is cathodically stabilized by the photo-excited minority carrier electrons, causing it to act as a photo-gated battery.
Therefore we used a non-aqueous solution for the p-GaAs spectral response and impedance measurements. We used NaI to provide I− rather than HI. We also reduced the concentration of redox couple in order to decrease parasitic light absorbance. The low-concentration of redox couple in non-aqueous solution was sufficient to support the nA-range signal and exhibited no photo-gated current (Fig. S6†).
Trends for both n and p-type GaAs in Jsc were mirrored by the spectral response curves (Fig. 4). Due to the wavelength dependence of α(λ), photons with energies near the band-gap Eg are absorbed further from the surface than those with higher energies. Thus Φint decays to zero at Eg. This can be modeled using the Gärtner equation, which assumes no depletion region recombination and that the LD governs bulk recombination:
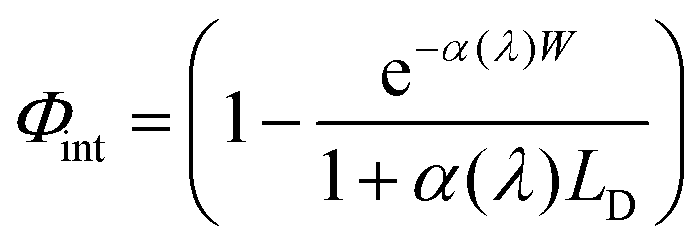 | (5) |
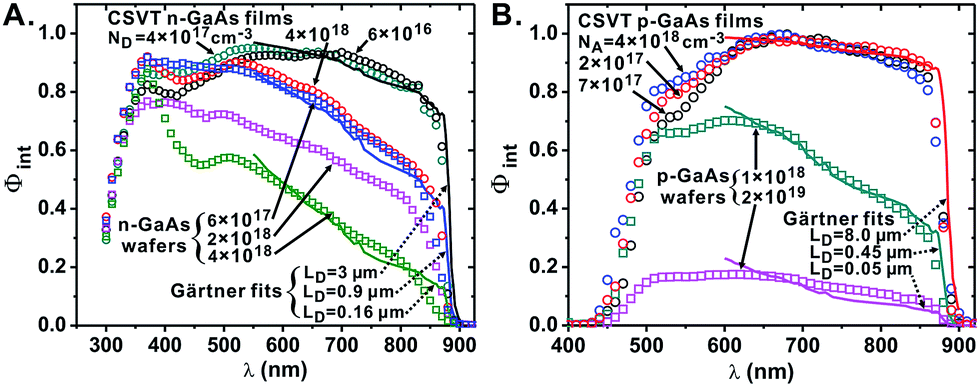 | ||
| Fig. 4 Φ int measurements obtained using PEC on n-GaAs (A) and p-GaAs samples (B). Experimental data is plotted as circles (CSVT GaAs films) or squares (commercial GaAs wafers) and calculated LD fits from eqn (5) are plotted as solid curves. The selected curves are representative of other electrodes obtained from the same samples and from other samples with similar free carrier concentrations. | ||
One parameter fits to Φint from eqn (5) match the experimental data well (Fig. 4). Moderately-doped n-GaAs CSVT films have LD ∼ 2.9 ± 0.2 μm, while CSVT p+-GaAs films possess LD = 5.4 ± 0.1 μm and moderately-doped p-GaAs films possess LD = 7.4 ± 0.4 μm. The LD is higher in p-GaAs because μe is higher than μh, which in turn is due to the curvature of the conduction and valence bands.54 For all CSVT samples, the measured LD was significantly higher than that of the control GaAs wafers (LD = 0.42 μm for n-GaAs:Te, 0.16 μm for n+-GaAs:Si, 0.45 μm for p-GaAs:Zn, and 0.05 μm for p+-GaAs:Zn) and consistent with one-sun Jsc measurements (see above).
Electron beam-induced current
Because GaAs has a direct bandgap and large α(λ),55 when LD exceeds several μm, the fraction of photons absorbed deeper than LD + W is small, making determination of LD by the Gärtner model less precise.In EBIC analysis, the proximity of the excitation source to the charge separating junction is controlled by rastering an electron beam toward a Schottky contact (Fig. 5A), and is thus independent of α(λ). The beam-induced current is measured as a function of the distance between the junction and the excitation source, and LD is determined by fitting the current decay according to eqn (4).
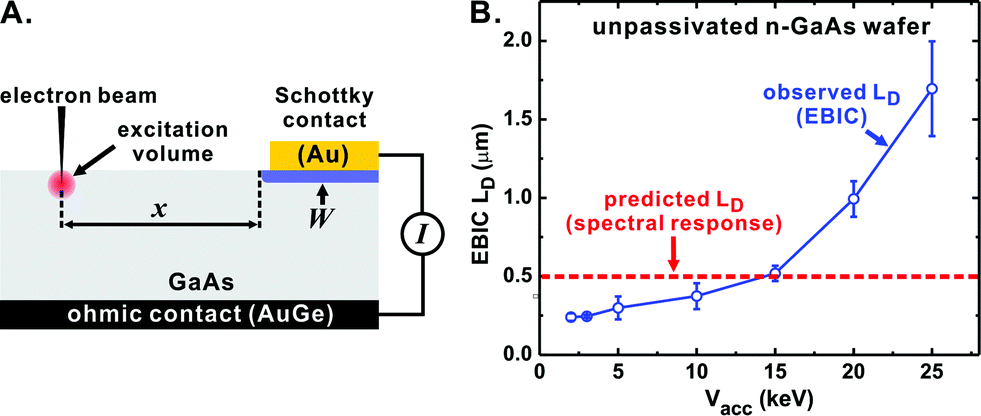 | ||
| Fig. 5 (A) Schematic of the EBIC experiment. X is the separation between the excitation source and the rectifying contact, I is the induced current, and W the depletion region. (B) The observed EBIC decay constant as a function of Vacc. The red dashed line indicates LD predicted by spectral response using the Gärtner method. The sample was an unpassivated n-GaAs:Te wafer with ND = 6 × 1016 cm−3. | ||
After fabricating rectifying Au|n-GaAs junctions and measuring the EBIC response of the junctions, we observed that the response was a function of Vacc (Fig. 5B), complicating accurate extraction of LD.
In an ideal EBIC experiment, the excitation volume (which is proportional to Vacc) is small with respect to LD and the EBIC decay is dominated by bulk recombination with surface recombination playing a negligible role. These assumptions are invalid for unpassivated GaAs, which has a high SRV and a short LD relative to indirect absorbers like Si. Thus at low Vacc the EBIC decay is dominated by surface recombination yielding erroneously low LD, while at high Vacc the excitation volume overlaps with the depletion region, yielding erroneously high LD. We note that in the experiments with unpassivated GaAs, the LD obtained from EBIC analysis coincidentally agrees with spectral response when Vacc = 10–15 keV is used, which matches the Vacc used in other studies.52
In order to obtain EBIC data which is accurately modeled by eqn (4), we used Na2S to passivate the GaAs surface thereby lowering the SRV.56 We also used a low Vacc ≤ 5 keV in order to maintain a small interaction volume. After passivation, both spectral response and EBIC yielded similar values of LD for Vacc ≤ 5 keV (Fig. 6A). Comparing the two techniques we observe more dispersion in the EBIC results (Fig. 6B), but similar overall trends. We suspect the EBIC and spectral response results differ because spectral response averages the current over a relatively large region (generally 0.05 cm2), while EBIC measures the current decay of a line-scan and is therefore more sensitive to local recombination-inducing surface/bulk defects. Nonetheless, the direct measurement of LD by EBIC using Na2S passivation confirms the long LD obtained from fitting PEC spectral response curves.
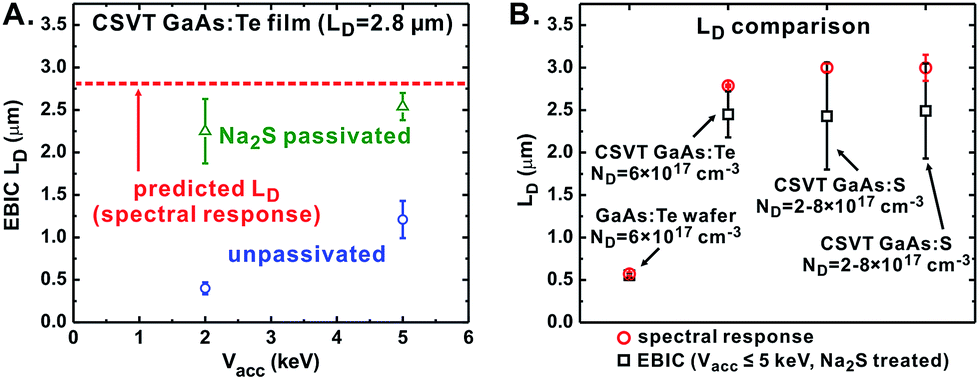 | ||
| Fig. 6 (A) LD measured by EBIC on the same CSVT GaAs film before and after passivation by Na2S. The dashed line indicates the LD predicted by spectral response. (B) Comparison of LD obtained by spectral response and EBIC techniques. | ||
Hall mobility and dopant density
Films grown from a variety of p- and n-GaAs sources were deposited on undoped semi-insulating GaAs for Hall-effect measurements. The data indicates that n-GaAs and p-GaAs films deposited using CSVT (from both powder and wafer sources) have similar μe and μh to films deposited using MOCVD (Fig. 7).27,57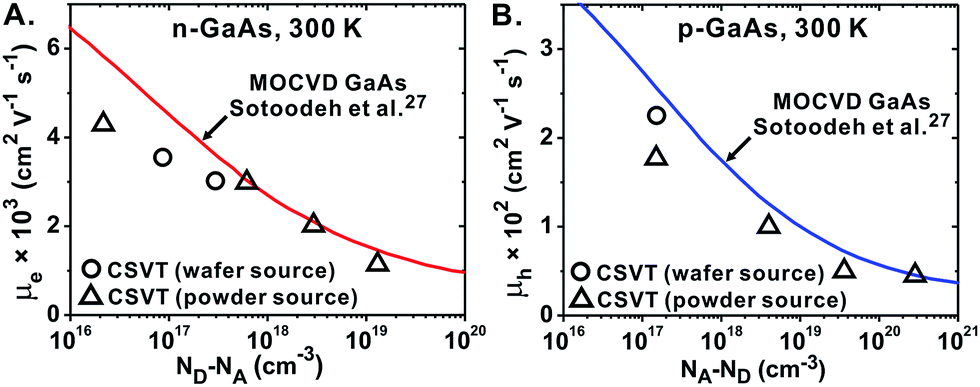 | ||
| Fig. 7 Hall mobilities of n- and p-GaAs films as a function of ND and NA. Solid curves represent the Hall mobility of high-quality epitaxially-grown MOCVD GaAs.27 | ||
As ND and NA are increased, μe and μh decrease due to increased scattering from the ionized dopant atoms in the lattice (Fig. 7).26,48,49 The measured μe and μh of CSVT GaAs films deviate from the MOCVD values more for lightly-doped samples than for highly-doped samples. This is likely because at lower ND/NA, the influence of trace compensating impurities and crystal defects becomes important relative to the dopant atom scattering. Overall, these results indicate that CSVT from GaAs powder sources is competitive with MOCVD in terms of the achievable μe and μh for a wide range of ND/NA.
No significant differences were observed between films deposited from powder and wafer sources. This result is expected because the growth takes place at the interface between substrate and the gas phase. Thus the source's crystalline quality should not affect the CSVT process as long as it does not affect the ability of the surface to be etched by H2O to produce vapor phase As2 and Ga2O. This implies that there is no need for crystalline powder sources, and lower quality powders could potentially be used as sources, for example those made by reaction of Ga and As at low temperatures.58
Conclusions
Epitaxial films of GaAs possessing a wide range of ND and NA were deposited via H2O-mediated CSVT with growth rate > 0.3 μm min−1 at ambient pressure from powdered GaAs precursors. ND and NA were measured and confirmed using complementary analyses which gave equivalent results (impedance spectroscopy, Hall effect measurements, and TOF-SIMS). This is an important step toward the fabrication of more complex device architectures such as p–n junctions.The room temperature mobilities of CSVT GaAs films were similar to those produced in the literature using MOCVD, despite the relatively high growth rates and use of H2O vapor as a transport agent. Due to the high α(λ) of GaAs,55 the LD (2–3 μm for n-GaAs and 5–8 μm for p-GaAs) was sufficient to yield Jsc ≈ 20 mA cm−2 which is near the one-sun limit (22.5 mA cm−2) for specular GaAs with no anti-reflective coating in acetonitrile. For this μe and LD we estimate an electron lifetime of τe ∼ 7 ns for the NA = 1–2 × 1017 cm−3 p-GaAs films. According to previously published simulations28 using this lifetime, η ≈ 25% single-junction photovoltaics could be fabricated if other device-engineering challenges (such as how to create rectifying solid-state junctions and passivate the surfaces) can be solved. Initial efforts to produce GaAs p–n junction PV solar cells using CSVT have produced promising results with Voc > 0.9 V and Φint > 0.9, but they are beyond the scope of this study.59
In addition to the high capital cost associated with MOCVD production of GaAs, lattice-matched substrates are also expensive relative to Si wafers. Tandem architectures which utilize the larger indirect band-gap and smaller lattice constant (closer to Si) of GaAsxP1−x could yield η > 35% devices on Si substrates if challenges associated with the thermal and lattice mismatch can be addressed.60 Growth techniques such as selective area epitaxy12 and the synthesis and use of engineered strain relaxation areas61 may be required for such efforts.
Acknowledgements
This work was supported by the Department of Energy (DOE) SunShot Initiative BRIDGE program (DE-EE0005957). AR acknowledges support from the Julie and Rocky Dixon Graduate Innovation Fellowship. AG acknowledges support from a NSF Graduate Research Fellowship (DGE-0829517). SB acknowledges support from Research Corporation for Science Advancement as a Cottrell Scholar. We acknowledge support from the user program at the Molecular Foundry, Lawrence Berkeley National Laboratory, with funding from the U.S. DOE Office of Science, Office of Basic Energy Sciences under Contract DE-AC02-05CH1123. The CAMCOR shared instrument facilities are supported by grants from the W.M Keck Foundation, the M.J. Murdock Charitable Trust, ONAMI, and NSF. The SUNRISE Photovoltaic Laboratory is supported by Oregon Built Environment and Sustainable Technologies. Assistance from Fuding Lin, Adam Smith, Stephen Golledge, Fred Stevie, Kris Johnson, Jeff Garmin, John Boosinger, Cliff Dax, Alex Negrete, Richard Cramer, Solomon Levinrad, Athavan Nadarajah, Lena Trotochaud, Matthew Kast, Sangeun Chun, Michaela Burke, T.J. Mills, Adam Batchellor and David Senkovich is acknowledged.Notes and references
- O. D. Miller, E. Yablonovitch and S. R. Kurtz, IEEE J. Photovolt., 2012, 2, 303–311 CrossRef.
- W. Xufeng, M. R. Khan, J. L. Gray, M. A. Alam and M. S. Lundstrom, IEEE J. Photovolt., 2013, 3, 737–744 CrossRef.
- L. S. Mattos, S. R. Scully, M. Syfu, E. Olson, Y. Linlin, C. Ling, B. M. Kayes and H. Gang, Proc. 38th IEEE Photovolt. Spec. Conf., 2012, pp. 3187–3190 Search PubMed.
- M. A. Green, K. Emery, Y. Hishikawa, W. Warta and E. D. Dunlop, Prog. Photovoltaics, 2014, 22, 1–9 Search PubMed.
- E. Yablonovitch, O. D. Miller and S. R. Kurtz, Proc. 38th IEEE Photovolt. Spec. Conf., 2012, pp. 1556–1559 Search PubMed.
- M. Bosi and C. Pelosi, Prog. Photovoltaics, 2007, 15, 51–68 CAS.
- G. Perrier, R. Philippe and J. P. Dodelet, J. Mater. Res., 1988, 3, 1031–1042 CrossRef CAS.
- F. H. Nicoll, J. Electrochem. Soc., 1963, 110, 1165–1167 CrossRef CAS PubMed.
- E. Sirtl, J. Phys. Chem. Solids, 1963, 24, 1285–1289 CrossRef.
- P. Robinson, RCA Rev., 1963, 24, 574–584 CAS.
- M. Hammadi, J. C. Bourgoin and H. Samic, J. Mater. Sci.: Mater. Electron., 1999, 10, 399–402 CrossRef.
- M. G. Mauk, B. W. Feyock and J. E. Cotter, J. Cryst. Growth, 2001, 225, 528–533 CrossRef CAS.
- M. G. Mauk, A. N. Tata and B. W. Feyock, J. Cryst. Growth, 2001, 225, 359–365 CrossRef CAS.
- S. R. Kurtz and R. McConnell, AIP Conf. Proc., 1997, 404, 191–205 CrossRef CAS PubMed.
- R. Venkatasubramanian, B. O'Quinn and E. Siivola, AIP Conf. Proc., 1997, 394, 259–270 CrossRef CAS PubMed.
- S. M. Vernon, S. P. Tobin, V. E. Haven, L. M. Geoffroy and M. M. Sanfacon, Proc. 22nd IEEE Photovolt. Spec. Conf., 1991, vol. 351, pp. 353–357 Search PubMed.
- M. G. Mauk, J. R. Balliet and B. W. Feyock, J. Cryst. Growth, 2003, 250, 50–56 CrossRef CAS.
- M. G. Astles, Liquid-Phase Epitaxial Growth of III–V Compound Semiconductor Materials and their Device Applications, Wiley-VCH Verlag GmbH, 1990 Search PubMed.
- M. G. Mauk, B. W. Feyock, R. B. Hall, K. D. Cavanaugh and J. E. Cotter, Proc. 26th IEEE Photovolt. Spec. Conf., 1997, pp. 511–514 Search PubMed.
- F. Silva-Andrade, A. Ilinskii, F. Chavez, T. Prutskij, F. Tenorio, Y. Bravo-Garcia and R. Pena-Sierra, Mod. Phys. Lett. B, 2001, 15, 752–755 CrossRef CAS.
- P. A. Hoss, L. Murray and J. Rivera, J. Electrochem. Soc., 1968, 115, 553–556 CrossRef CAS PubMed.
- A. J. Ritenour and S. W. Boettcher, Proc. 38th IEEE Photovolt. Spec. Conf., 2012, pp. 913–917 Search PubMed.
- A. J. Ritenour, R. C. Cramer, S. Levinrad and S. W. Boettcher, ACS Appl. Mater. Interfaces, 2012, 4, 69–73 CAS.
- C. M. Gronet and N. S. Lewis, Appl. Phys. Lett., 1983, 43, 115–117 CrossRef CAS PubMed.
- R. Purohit, J. Mater. Sci., 1968, 3, 330–332 CrossRef CAS.
- W. Walukiewicz, L. Lagowski, L. Jastrzebski, M. Lichtensteiger and H. C. Gatos, J. Appl. Phys., 1979, 50, 899–908 CrossRef CAS PubMed.
- M. Sotoodeh, A. Khalid and A. Rezazadeh, J. Appl. Phys., 2000, 87, 2890–2900 CrossRef CAS PubMed.
- J. W. Boucher, A. J. Ritenour and S. W. Boettcher, Proc. SPIE, 2013, 87250Z CrossRef PubMed.
- Crystal Growth Technology, ed. H. J. Scheel and P. Capper, Wiley-VCH, 2008 Search PubMed.
- V. Rideout, Solid-State Electron., 1975, 18, 541–550 CrossRef CAS.
- L. G. Casagrande, A. Juang and N. S. Lewis, J. Phys. Chem. B, 2000, 104, 5436–5447 CrossRef CAS.
- F. R. F. Fan and A. J. Bard, J. Am. Chem. Soc., 1980, 102, 3677–3683 CrossRef CAS.
- P. A. Kohl and A. J. Bard, J. Electrochem. Soc., 1979, 126, 59–67 CrossRef CAS PubMed.
- A. J. Ritenour, S. Levinrad, C. Bradley, R. C. Cramer and S. W. Boettcher, ACS Nano, 2013, 6840–6849 CrossRef CAS PubMed.
- K. E. Pomykal, A. M. Fajardo and N. S. Lewis, J. Phys. Chem., 1995, 99, 8302–8310 CrossRef CAS.
- K. Gelderman, L. Lee and S. Donne, J. Chem. Educ., 2007, 84, 685 CrossRef CAS.
- S. M. Sze, Physics of Semiconductor Devices, John Wiley & Sons, New York, 1981 Search PubMed.
- L. Van der Pauw, Philips Tech. Rev., 1958, 20, 220–224 Search PubMed.
- R. G. Wilson, F. A. Stevie and C. W. Magee, Secondary ion mass spectrometry: a practical handbook for depth profiling and bulk impurity analysis, WileyNew York, 1989 Search PubMed.
- B. Akamatsu, J. Henoc and P. Henoc, J. Appl. Phys., 1981, 52, 7245–7250 CrossRef CAS PubMed.
- B. J. Skromme, C. J. Sandroff, E. Yablonovitch and T. Gmitter, Appl. Phys. Lett., 1987, 51, 2022–2024 CrossRef CAS PubMed.
- J. Bourgoin, D. Stievenard, D. Deresmes and J. Mimila Arroyo, J. Appl. Phys., 1991, 69, 284–290 CrossRef CAS PubMed.
- C. Le Bel, D. Cossement, J. P. Dodelet, R. Leonelli, Y. DePuydt and P. Bertrand, J. Appl. Phys., 1993, 73, 1288–1296 CrossRef CAS PubMed.
- E. Koskiahde, D. Cossement, R. Paynter, J. P. Dodelet, A. Jean and B. A. Lombos, Can. J. Phys., 1989, 67, 251–258 CrossRef CAS.
- G. Gottlieb, J. Electrochem. Soc., 1965, 112, 192–196 CrossRef CAS PubMed.
- D. Cossement, J. P. Dodelet, T. Bretagnon, A. Jean and B. A. Lombos, J. Electrochem. Soc., 1991, 138, 830–834 CrossRef CAS PubMed.
- L. G. Casagrande, B. J. Tufts and N. S. Lewis, J. Phys. Chem., 1991, 95, 1373–1380 CrossRef CAS.
- D. L. Rode and S. Knight, Phys. Rev. B: Solid State, 1971, 3, 2534–2541 CrossRef.
- E. F. Schubert, Doping in III–V semiconductors, Cambridge Univ Press, 1993 Search PubMed.
- G. K. Schweitzer and L. L. Pesterfield, The Aqueous Chemistry of the Elements, Oxfort University Press, 2010 Search PubMed.
- W. W. Gärtner, Phys. Rev., 1959, 116, 84 CrossRef.
- B. J. Tufts, I. L. Abrahams, L. G. Casagrande and N. S. Lewis, J. Phys. Chem., 1989, 93, 3260–3269 CrossRef CAS.
- C. Xiang, G. M. Kimball, R. L. Grimm, B. S. Brunschwig, H. A. Atwater and N. S. Lewis, Energy Environ. Sci., 2011, 4, 1311–1318 CAS.
- S. Adachi, Physical Properties of III–V Semiconductor Compounds, John Wiley & Sons, 1st edn, 1992 Search PubMed.
- Handbook of Optical Constants of Solids, ed. E. D. Palik, Academic Press, Inc., 1985 Search PubMed.
- S. R. Lunt, G. N. Ryba, P. G. Santangelo and N. S. Lewis, J. Appl. Phys., 1991, 70, 7449–7467 CrossRef CAS PubMed.
- M. R. Brozel and G. Stillman, Properties of gallium arsenide, IET, 1996 Search PubMed.
- E. Fahrenkrug, J. Gu and S. Maldonado, J. Am. Chem. Soc., 2012, 135, 330–339 CrossRef PubMed.
- J. W. Boucher, A. J. Ritenour, A. L. Greenaway, S. Aloni and S. W. Boettcher, Proc. 40th IEEE Photovolt. Spec. Conf., 2014 Search PubMed.
- J. Faucher, A. Gerger, S. Tomasulo, C. Ebert, A. Lochtefeld, A. Barnett and M. Lee, Appl. Phys. Lett., 2013, 103, 191901 CrossRef PubMed.
- K. J. Schmieder, A. Gerger, M. Diaz, Z. Pulwin, C. Ebert, A. Lochtefeld, R. Opila and A. Barnett, Proc. 38th IEEE Photovolt. Spec. Conf., 2012, pp. 968–973 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ee01943a |
| This journal is © The Royal Society of Chemistry 2015 |