
Pt–Ru catalyzed hydrogen oxidation in alkaline media: oxophilic effect or electronic effect?†
Ying
Wang
,
Gongwei
Wang
,
Guangwei
Li
,
Bing
Huang
,
Jing
Pan
*,
Qiong
Liu
,
Juanjuan
Han
,
Li
Xiao
,
Juntao
Lu
and
Lin
Zhuang
*
College of Chemistry and Molecular Sciences, Hubei Key Lab of Electrochemical Power Sources, Institute for Advanced Studies, Wuhan University, Wuhan 430072, China. E-mail: jpan@whu.edu.cn; lzhuang@whu.edu.cn
First published on 2nd October 2014
Abstract
A current challenge to alkaline polymer electrolyte fuel cells (APEFCs) is the unexpectedly sluggish kinetics of the hydrogen oxidation reaction (HOR). A recently proposed resolution is to enhance the oxophilicity of the catalyst, so as to remove the Had intermediate through the reaction with OHad, but this approach is questioned by other researchers. Here we report a clear and convincing test on this problem. By using PtRu/C as the HOR catalyst for the APEFC, the peak power density is boosted to 1.0 W cm−2, in comparison to 0.6 W cm−2 when using Pt/C as the anode catalyst. Such a remarkable improvement, however, can hardly be explained as an oxophilic effect, because, as monitored by CO stripping, reactive hydroxyl species can generate on certain sites of the Pt surface at more negative potentials than on the PtRu surface in KOH solution. Rather, the incorporation of Ru has posed an electronic effect on weakening the Pt–Had interaction, as revealed by the voltammetric behavior and from density-functional calculations, which thus benefits the oxidative desorption of Had, the rate determining step of HOR in alkaline media. These findings further our fundamental understanding of the HOR catalysis, and cast new light on the exploration of better catalysts for APEFCs.
Broader contextThe rise of alkaline polymer electrolyte fuel cells (APEFCs), a new class of fuel cells, has stimulated increasing research interest in fuel-cell catalysis in an alkaline environment. A particularly intriguing and urgent problem in APEFC research is why the Pt catalyzed hydrogen oxidation reaction (HOR), a very rapid reaction in acids, becomes sluggish in an alkaline environment, and how to address this issue? This article is a timely contribution to both fundamental electrocatalysis and fuel cell technology. Two significant results are reported: (i) the cell performance of the APEFC based on the PtRu/C anode, 1.0 W cm−2, is thus far the highest record in the literature and is comparable to the performance of the state-of-the-art proton-exchange membrane fuel cells (PEMFCs). This is a milestone achievement, marking that the APEFC can be a practical technology for high power applications. (ii) The current argument about the oxophilic effect on HOR kinetics has been clarified by convincing experimental and computational results. The incorporation of Ru does enhance the HOR kinetics, but most likely due to an electronic effect on weakening the Pt–Had interaction, rather than the richness of reactive OHad in the potential region of the HOR. |
In the past decade, the rise of a new class of fuel cells, alkaline polymer electrolyte fuel cells (APEFCs),1–3 has stimulated increasing research interest in fuel-cell catalysis under alkaline conditions.4–11 In principle, APEFCs would combine the advantages of alkaline fuel cells (AFCs) and polymer electrolyte fuel cells (PEFCs), namely, to maintain the electrocatalytic benefit of alkaline media while having the compact cell structure and no risk of electrolyte leak offered by polyelectrolytes.12 The benefits of alkaline media, as compared with acids, include the enhanced kinetics of some fuel-cell reactions, such as the oxygen reduction reaction (ORR)13 and the methanol oxidation reaction (MOR),14 the alleviated corrosion of Pt,12 and the applicability of nonprecious metal catalysts.1,2,4,5,8–10
Yet the cell performance of APEFCs reported in the literature has hitherto been lower than that of proton-exchange membrane fuel cells (PEMFCs), the acid version of PEFCs. Although alkaline polymer electrolytes (APEs) have been well developed in recent decades15–22 and the OH− conduction in APEs can now be as efficient as the H+ conduction in Nafion under fuel-cell operating conditions,16 the H2 or methanol fueled APEFC still exhibits lower power densities than does PEMFC,23–27 even using higher loading of Pt catalysts. An electrocatalytic defect of the alkaline system was lately identified: the Pt-catalyzed hydrogen oxidation reaction (HOR), a rapid reaction in acids, somehow becomes sluggish in an alkaline environment,6,28 with the exchange current density (i0), a measure of the reaction kinetics at equilibrium potential, being smaller by orders of magnitude than that in acids.11,28
A resolution to this issue was recently proposed by Markovic and co-workers.6 By comparing the HOR behavior on three types of catalyst (Au, Pt, and Ir), they found that the catalytic activity of these catalysts toward the HOR seemed to be in a positive correlation with their oxophilicity, and inferred that the surface hydroxyl species (OHad) can play a reactive role to remove the hydrogen intermediate (Had), thus enhancing the kinetics of the HOR. Albeit inspiring, this observation was strongly questioned by Gasteiger and co-workers,11 who measured the i0 of HOR on Pt/C, Ir/C, and Pd/C, and claimed that the oxophilicity of a catalyst (Ir being more oxophilic than Pt and Pd) does not enhance the HOR activity at high pH.
The proposed reaction between OHad and Had on the catalyst surface seems to resemble the situation of MOR, where OHad is required to remove the COad intermediate on Pt surface.29–31 In this sense, alloying Pt with a more active metal to provide reactive OHad at a more negative potential is an effective strategy for kinetic enhancement, and PtRu bimetallic catalyst is such a typical example.29 Markovic and co-workers did mention that PtRu alloys were the most direct probe of the effects of OHad on the rate of the HOR,6 but it was, again, doubted by Gasteiger and co-workers.11 In the present work, we try to perform a clear and convincing judgment on the ongoing argument about the oxophilic effect on the HOR in alkaline media. We first compare Pt/C and PtRu/C as the anode catalyst in a real APEFC, and then characterize their oxophilic behavior by CO stripping experiments, finally we discuss the impact of Ru on the Pt–Had interaction and the HOR kinetics.
To assemble single cells of APEFC, a high-performance APE, aQAPS-Sx,16,17 was employed as both the membrane and the ionomer in the electrodes. Commercial Pt/C or PtRu/C catalysts (Johnson-Matthey, 60 wt% of metal, the Ru atomic ratio in PtRu alloy was estimated to be 20% based on XRD analysis) served as the HOR catalyst, and Pt/C was used to catalyze the ORR. The metal loadings in both the anode and the cathode were controlled to be 0.4 mg cm−2. Fig. 1 shows the cell performance obtained at 60 °C. The peak power density of the APEFC based on the Pt/C anode approaches 0.6 W cm−2, which is the best record in the literature for APEFCs operated under similar conditions.23–25 Strikingly, upon using PtRu/C as the anode, the peak power density is boosted to 1.0 W cm−2, even though the Pt loading in this anode is actually only 0.26 mg cm−2 (with a Ru loading of 0.14 mg cm−2). Furthermore, when reducing the PtRu loading in the anode to 0.2 mg cm−2 (i.e., the Pt loading is only 0.13 mg cm−2 in anode), the peak power density of the APEFC is still as high as 0.87 W cm−2 (Fig. S1†). Unambiguously, PtRu/C is a much better HOR catalyst than Pt/C for the APEFC.
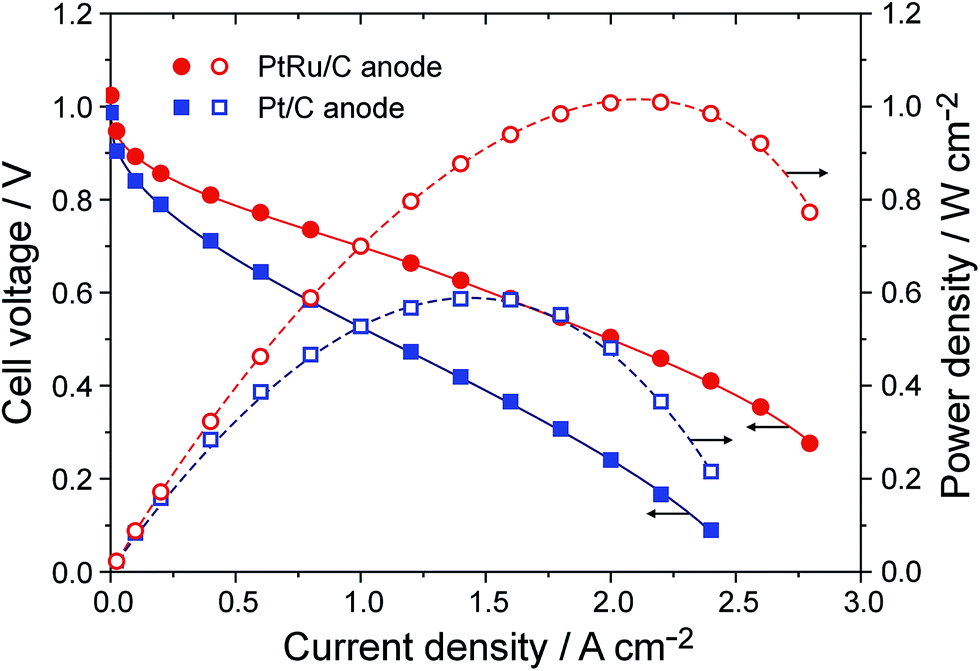 | ||
| Fig. 1 Cell performance of the APEFC using the PtRu anode or Pt anode. Single cells were operated at 60 °C. The APE membrane was aQAPS-S8 (IEC = 1.0 mmol g−1, 50 ± 5 μm in thickness) and the APE ionomer in electrodes was aQAPS-S14 (IEC = 1.0 mmol g−1, 20 wt% in the catalyst layer).16 Pt/C was used as the cathode catalyst. Metal loadings in both the anode and the cathode were all controlled to be 0.4 mg cm−2. Fully humidified H2 and O2 were fed in a flow rate 400 mL min−1 with a backpressure of 0.1 MPa. | ||
The superiority of PtRu/C over Pt/C as the anode catalyst for the APEFC seems to be a favorable proof of the oxophilic effect on the rate of the HOR, since Ru was known to have a greater oxophilicity than does Pt. But our knowledge about PtRu was mostly obtained from the previous studies of the MOR in acids, the surface hydroxyl species in the alkaline environment could behave quite differently, and the specifically adsorbed OH− may also be reactive. Therefore, careful examinations on the reactive hydroxyl species in alkaline media are necessary. Considering that the reaction between Had and OHad involves no electron transfer and cannot be observed in an electrochemical fashion, we decided to use the CO stripping process to monitor the generation of reactive hydroxyl species, since the anodic current of CO oxidation has to be triggered by the reactive hydroxyl species.31 As demonstrated in Fig. 2, in 0.1 M H2SO4 solution, the CO stripping on Pt/C takes on a single sharp peak at 0.85 V, and, upon alloying with Ru, the CO stripping peak is somewhat broadened and moves negatively by 0.3 V, showing that Ru does accelerate the formation of OHad in an acidic environment. However, the CO stripping on Pt/C behaves rather differently in 0.1 M KOH solution: multiple anodic peaks appear and the onset potential shifts to ∼0.2 V, a potential even more negative than the onset of CO stripping on PtRu/C (∼0.35 V) in either acid or alkaline media. Such a surprising finding indicates that, in an alkaline environment, the reactive hydroxyl species, be it OHad or OHad−, can generate on certain sites of the Pt surface more favorably than on the PtRu surface; but when alloyed with Ru, the surface reactivity of Pt is suppressed, thereby no reactive hydroxyl species appearing in the potential region negative to 0.35 V.
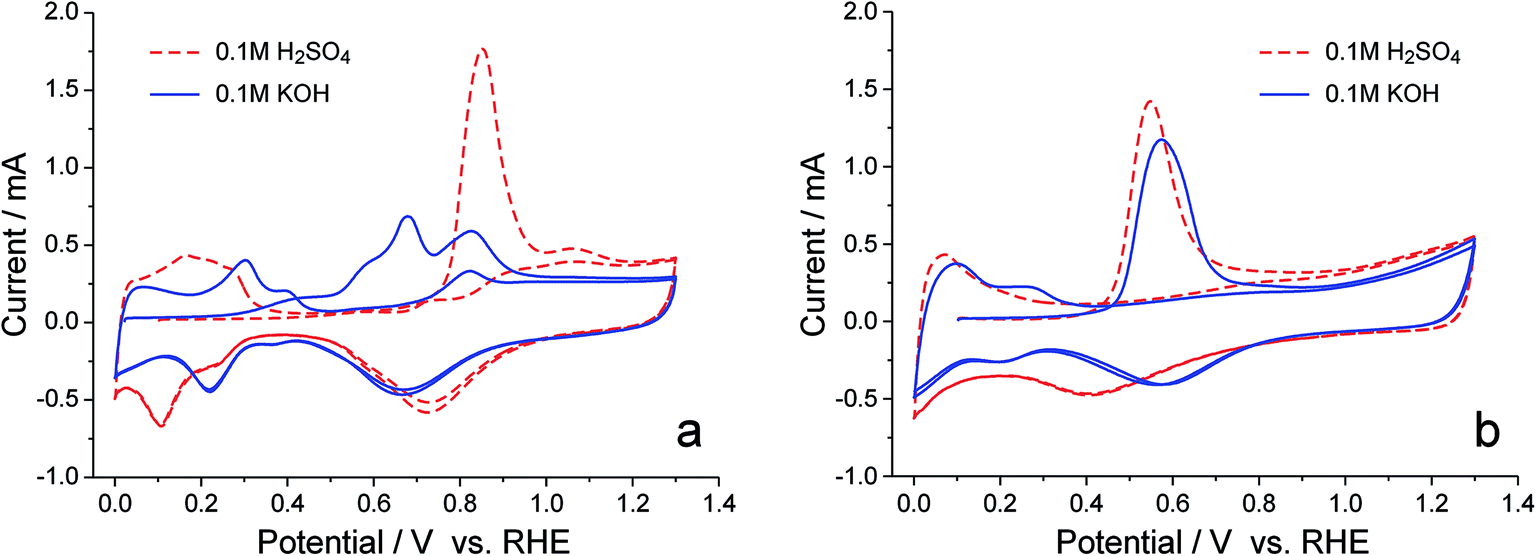 | ||
| Fig. 2 CO stripping on Pt/C (a) and PtRu/C (b) in 0.1 M H2SO4 and 0.1 M KOH solutions. The potential was held at 0.1 V in CO-saturated solutions for 10 min (in the case of Pt/C in KOH solution, the starting potential was extended to 0.02 V for better observation on the early onset of CO stripping), and then, after purging the solution with Ar for 10 min, cycled twice in 20 mV s−1. | ||
On the basis of the above observations, the promotion effect of Ru on catalyzing the HOR in alkaline media can hardly be explained as an oxophilic effect. The existence of reactive hydroxyl species on either the Pt or PtRu surface at potentials negative to 0.2 V also seems unlikely, as revealed in Fig. 2. On the other hand, the Ru has posed an obvious effect on weakening the Pt–Had interaction, as a consequence of the suppressed surface reactivity of Pt. As illustrated in Fig. 3a, in KOH solution, the hydrogen underpotential deposition (H-UPD) and subsequent desorption behavior on PtRu/C is clearly different from that on Pt/C: whereas strong Had peaks are the major signal for Pt/C, the weak Had peak is dominant for PtRu/C. Such an electronic effect of Ru is independent of the electrolyte, similar results were also observed in H2SO4 solution (Fig. S2†). These experimental observations turn out to be consistent with the DFT-calculated adsorption energies of H on Pt(111) and Pt3Ru(111), −0.33 eV and −0.19 eV, respectively (Fig. 3b). If the Ru site on Pt3Ru(111) is covered with OHad, the Pt–Had bonding will be further reduced to −0.12 eV. These adsorption energies of H are relative to a half of the energy of H2, and the negative values indicate that H2 dissociation is thermodynamically spontaneous on these surfaces. It has also been known that the kinetics of the HOR in alkaline media is limited by the Volmer step (i.e., the oxidative desorption of Had),11 it is therefore reasonable that weakening the Pt–Had interaction can lead to an enhancement in the HOR kinetics.32
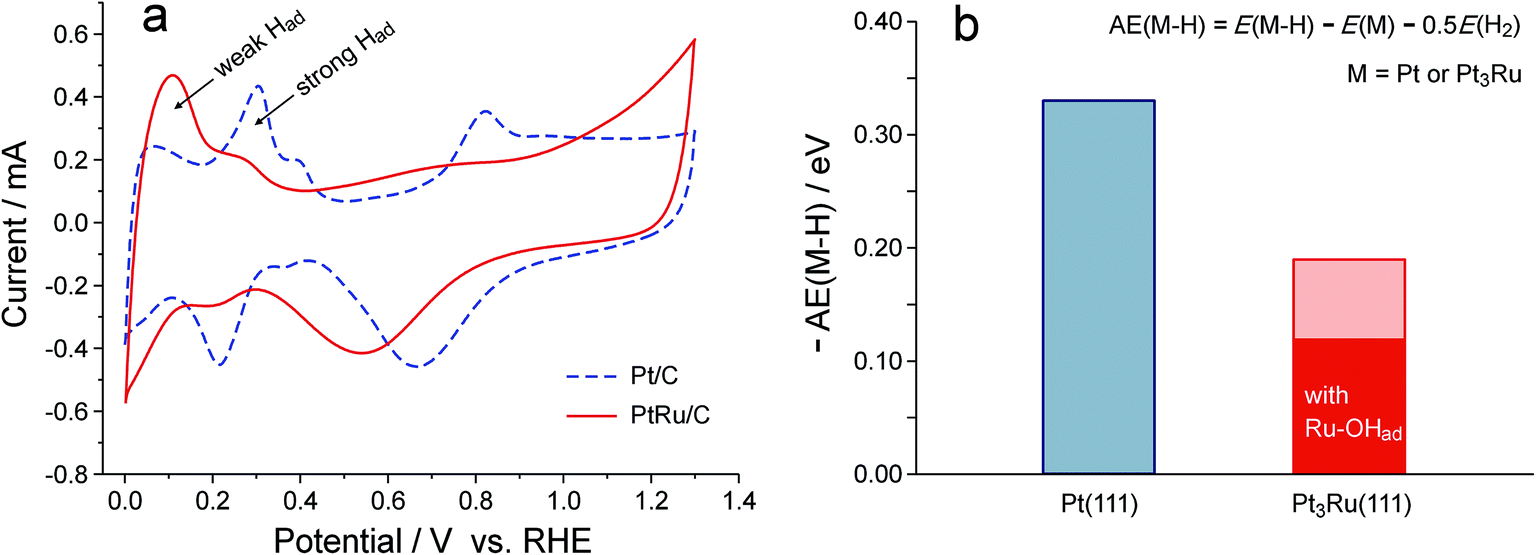 | ||
| Fig. 3 The impact of Ru on the Pt–Had interaction. (a) Cyclic voltammetry of Pt/C and PtRu/C in deaerated 0.1 M KOH solution. (b) DFT-calculated adsorption energy of H on Pt(111) and Pt3Ru(111) with or without OHad on Ru. The adsorption energy is relative to half of the energy of H2, such that the negative value indicates that the H2 dissociation is thermodynamically spontaneous on the studied surface. | ||
To make a quantitative evaluation on the improvement in the rate of the HOR catalyzed by PtRu/C, rotating disk electrode (RDE) measurements were conducted with the catalyst loading on the electrode being reduced to such a level that the slope of the polarization curve at zero current is independent of the rotation rate (Fig. 4a),33,34 and the specific surface area of Pt for the PtRu/C catalyst, which cannot be accurately obtained from either the H-UPD charge or the CO-stripping charge,35 was determined by the Cu stripping experiment (Fig. 4b), following the methodology reported by Kucernak and co-workers.36 Based on these experiments, the i0 of HOR on PtRu/C in 0.1 M KOH solution is calculated to be 0.7 mA cmPt−2, more than two times that for Pt/C (0.3 mA cmPt−2) under the same conditions. Such a difference in i0 would cause a shift in overpotential by ca. 30 mV in the polarization curve (if the Tafel slopes were always the same for PtRu/C and Pt/C), which appears to be consistent with the APEFC data at low polarization (Fig. 1). At high current densities, however, the cell performance improvement is further boosted (e.g., ca. 180 mV of voltage difference at 1.0 A cm−2), which may result from a change in the Tafel slope or in the ionic conductivity of APE (whose hydration level changes with the current density).
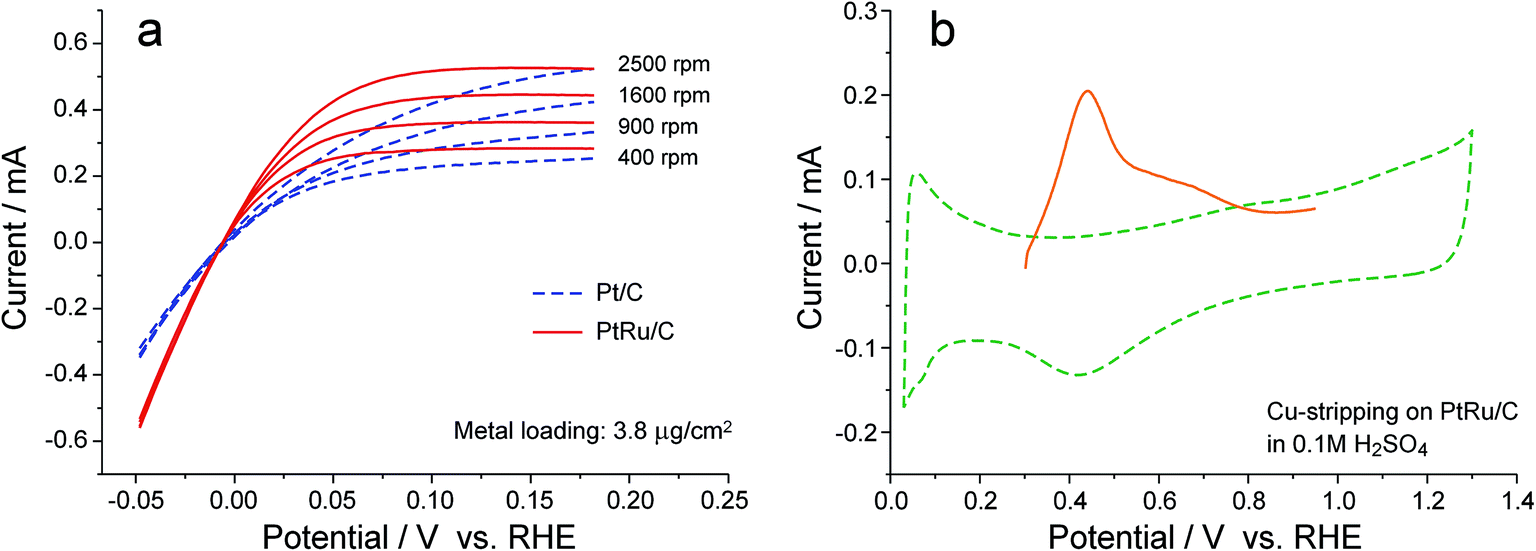 | ||
| Fig. 4 Determination of the exchange current density of HOR on Pt/C and PtRu/C. (a) Rotating disk electrode (RDE) measurements in deaerated 0.1 M KOH solution with a potential scan rate 2 mV s−1. The metal loading on glassy carbon substrate was kept in a sufficiently low level through diluting the catalyst with blank carbon powder (Vulcan XC72),34 such that the curve slope at zero current is independent of the rotation rate. Data were averaged over the forward and backward scans so as to cancel out charging currents. (b) Cu stripping on PtRu/C for determining the specific surface area of Pt.36 The electrolyte was a deaerated 0.1 M H2SO4 solution containing 2 mM CuSO4. The electrode potential was first held at 0.95 V to oxidize all Ru sites, and then jumped to 0.3 V and held for 10 min, followed by a linear scan to 0.95 V in 5 mV s−1. Background cyclic voltammogram (dash line) was obtained in the electrolyte without CuSO4. | ||
In summary, PtRu/C catalyzes the HOR in alkaline media at a rate more than two times that of Pt/C. The peak power density of the APEFC using the PtRu/C anode reaches 1.0 W cm−2 at 60 °C, comparable to the cell performance of the state-of-the-art PEMFC. To compare the oxophilicity between Pt/C and PtRu/C in alkaline media, CO stripping was employed as a probe experiment, showing that reactive hydroxyl species can generate on certain sites of the Pt surface at more negative potentials than on the PtRu surface in KOH solution. Therefore the enhancement in the kinetics of the HOR catalyzed by PtRu/C can hardly be ascribed to an oxophilic effect, but rather, the weakening in the Pt–Had interaction, caused by the electronic effect of Ru alloying, is most probably responsible, as it benefits the oxidative desorption of Had, the rate-determining step of the HOR in alkaline media.
Acknowledgements
This work was financially supported by the National Basic Research Program (2012CB215500, 2012CB932800), the National Natural Science Foundation of China (21125312, 21303124, 21203142), the Doctoral Fund of Ministry of Education of China (20110141130002), and the Fundamental Research Funds for the Central Universities.References
- S. Lu, J. Pan, A. Huang, L. Zhuang and J. Lu, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20611 CrossRef CAS.
- K. Asazawa, K. Yamada, H. Tanaka, A. Oka, M. Taniguchi and T. Kobayashi, Angew. Chem., Int. Ed., 2007, 46, 8024 CrossRef CAS PubMed.
- J. R. Varcoe and R. C. T. Slade, Fuel Cells, 2005, 5, 187 CrossRef CAS.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760 CrossRef CAS PubMed.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 546 CrossRef CAS PubMed.
- D. Strmcnik, M. Uchimura, C. Wang, R. Subbaraman, N. Danilovic, D. van der Vliet, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Nat. Chem., 2013, 5, 300 CrossRef PubMed.
- S. M. Alia, B. S. Pivovar and Y. Yan, J. Am. Chem. Soc., 2013, 135, 13473 CrossRef CAS PubMed.
- C. Yang, B. Huang, L. Xiao, Z. Ren, Z. Liu, J. Lu and L. Zhuang, Chem. Commun., 2013, 49, 11023 RSC.
- Y. J. Sa, C. Park, H. Y. Jeong, S.-H. Park, Z. Lee, K. T. Kim, G.-G. Park and S. H. Joo, Angew. Chem., Int. Ed., 2014, 126, 4186 CrossRef.
- W. Sheng, A. P. Bivens, M. Myint, Z. Zhuang, R. V. Forest, Q. Fang, J. G. Chen and Y. Yan, Energy Environ. Sci., 2014, 7, 1719 CAS.
- J. Durst, A. Siebel, C. Simon, F. Hasché, J. Herranz and H. A. Gasteiger, Energy Environ. Sci., 2014, 7, 2255 CAS.
- D. Tang, J. Pan, S. Lu, L. Zhuang and J. Lu, Sci. China: Chem., 2010, 53, 357 CrossRef CAS PubMed.
- J. S. Spendelow and A. Wieckowski, Phys. Chem. Chem. Phys., 2007, 9, 2654 RSC.
- Y. Wang, L. Li, L. Hu, L. Zhuang, J. Lu and B. Xu, Electrochem. Commun., 2003, 5, 662 CrossRef CAS.
- J. R. Varcoe, P. Antanassov, D. R. Dekel, A. M. Herring, M. A. Hickner, P. A. Kohl, A. R. Kucernak, W. E. Mustain, K. Nijmeijer, K. Scott, T. Xu and L. Zhuang, Energy Environ. Sci., 2014, 7, 3135 Search PubMed.
- J. Pan, C. Chen, Y. Li, L. Wang, L. Tan, G. Li, X. Tang, L. Xiao, J. Lu and L. Zhuang, Energy Environ. Sci., 2014, 7, 354 CAS.
- J. Pan, C. Chen, L. Zhuang and J. Lu, Acc. Chem. Res., 2012, 45, 473 CrossRef CAS PubMed.
- Y.-J. Wang, J. Qiao, R. Baker and J. Zhang, Chem. Soc. Rev., 2013, 42, 5768 RSC.
- S. Gu, R. Cai, T. Luo, Z. Chen, M. Sun, Y. Liu, G. He and Y. Yan, Angew. Chem., Int. Ed., 2009, 48, 6499 CrossRef CAS PubMed.
- N. J. Robertson, H. A. Kostalik IV, T. J. Clark, P. F. Mutolo, H. D. Abruña and G. W. Coates, J. Am. Chem. Soc., 2010, 132, 3400 CrossRef CAS PubMed.
- N. Li, Y. Leng, M. A. Hickner and C.-Y. Wang, J. Am. Chem. Soc., 2013, 135, 10124 CrossRef CAS PubMed.
- J. Pan, Y. Li, J. Han, G. Li, L. Tan, C. Chen, J. Lu and L. Zhuang, Energy Environ. Sci., 2013, 6, 2912 CAS.
- G. Li, J. Pan, J. Han, C. Chen, J. Lu and L. Zhuang, J. Mater. Chem. A, 2013, 1, 12497 CAS.
- L. Zeng and T. Zhao, Electrochem. Commun., 2013, 34, 278 CrossRef CAS PubMed.
- Y. Zhao, H. Yu, D. Yang, J. Li, Z. Shao and B. Yi, J. Power Sources, 2013, 221, 247 CrossRef CAS PubMed.
- M. Mamlouk, J. A. Horsfall, C. Willams and K. Scott, Int. J. Hydrogen Energy, 2012, 37, 11912 CrossRef CAS PubMed.
- X. Sun, Y. Zhang, P. Song, J. Pan, L. Zhuang, W. Xu and W. Xing, ACS Catal., 2013, 3, 1726 CrossRef CAS.
- W. Sheng, H. A. Gasteiger and Y. Shao-Horn, J. Electrochem. Soc., 2010, 157, B1529 CrossRef CAS PubMed.
- H. A. Gasteiger, N. Markovic, P. N. Ross and E. J. Cairns, J. Phys. Chem., 1994, 98, 617 CrossRef CAS.
- C. Roth, N. Benker, T. Buhrmester, M. Mazurek, M. Loster, H. Fuess, D. C. Koningsberger and D. E. Ramaker, J. Am. Chem. Soc., 2005, 127, 14607 CrossRef CAS PubMed.
- L. Zhuang, J. Jin and H. D. Abruña, J. Am. Chem. Soc., 2007, 129, 11033 CrossRef CAS PubMed.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23 CrossRef PubMed.
- D. Tang, J. Lu, L. Zhuang and P. Liu, J. Electroanal. Chem., 2010, 644, 144 CrossRef CAS PubMed.
- Y. Sun, J. Lu and L. Zhuang, Electrochim. Acta, 2010, 55, 844 CrossRef CAS PubMed.
- C. L. Green and A. Kucernak, J. Phys. Chem. B, 2002, 106, 1036 CrossRef CAS.
- C. L. Green and A. Kucernak, J. Phys. Chem. B, 2002, 106, 11446 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 & S2, experimental and computational details. See DOI: 10.1039/c4ee02564d |
| This journal is © The Royal Society of Chemistry 2015 |