DOI:
10.1039/C5QI00025D
(Research Article)
Inorg. Chem. Front., 2015,
2, 550-557
Selective CO2 adsorption in a microporous metal–organic framework with suitable pore sizes and open metal sites†
Received
13th February 2015
, Accepted 7th April 2015
First published on 8th April 2015
Abstract
Microporous metal–organic frameworks, [H2N(Me)2]2[Zn4(L)2(H2O)1.5]·5DMF·H2O (1·DMF) or [H2N(Me)2]2[Zn4(L)2(H2O)1.5]·4DMA·6H2O (1·DMA), have been synthesized under solvothermal conditions using an undeveloped asymmetrical pentacarboxylate ligand, 2,4-di(3′,5′-dicarboxylphenyl)benzoic acid (H5L). Structural analysis demonstrates that 1 is a 3D anionic framework based on two kinds of dinuclear [Zn2(COO)5(H2O)] secondary building units (SBUs), which presents a rare framework constructed from ternary SBUs and shows a new (5,5,5)-connected topology. The desolvated anionic structure of 1·DMF (1a) contains two shapes of 1D channel with suitable pore sizes and a highly polar pore system decorated with open metal sites, diethylammonium cations and carboxylic oxygen atoms, exhibiting multipoint interactions between CO2 molecules and the framework, resulting in high CO2 uptake and significant selectivity for CO2 over CH4 and N2. In addition, 1·DMF and 1·DMA display solid-state photoluminescence stemming from the ligand-centered fluorescent emissions of H5L.
Introduction
The effective capture and removal of CO2 from industrial flue gas is becoming an important environmental issue due to escalated global warming.1 Porous coordination polymers (PCPs) or metal–organic frameworks (MOFs) are rapidly emerging as a new class of porous materials, which show promising application for CO2 capture and separation, and have attracted intense attention owing to their well-defined chemical structures and facile pore modification.2,3 To be an optimal CO2 capture material, a MOF should have high thermal stability, permanent porosity and high adsorption capacity for CO2. A numbers of MOF-based materials showing selective adsorption of CO2 over CH4 and N2 have been reported in the pastfew years,4–6 but highly efficient materials are still rare. So far, how to enhance the separation and storage abilities of these materials from a structural modification point of view remains a great challenge. In an effort to develop porous MOFs for such applications, some strategies, for example generation of open metal sites (OMSs), forming charged frameworks, and the incorporation of specific polar functional groups (–NH2, –OH, –CF3, etc.) within the porous frameworks, have been investigated to augment the CO2 adsorptive affinity and selectivity.7 Moreover, the pore-size-exclusive effect plays an important role in selective gas adsorption and separation in MOFs.8 Tuning/limiting the pores of MOFs to a certain size for selective adsorption and separation of specific gas molecules is an effective method.9 However, MOFs with ideal pore size are less reported because it still remains difficult to pre-design and target such made-to-order porous materials at the molecular level.
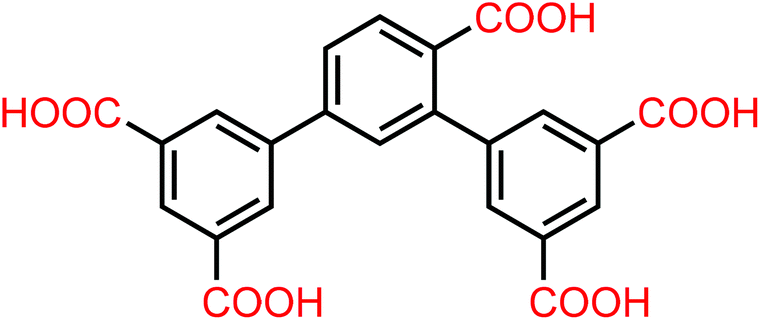
In the construction of MOFs, the judicious selection of appropriate organic ligands is proven to be one of the most crucial aspects.10 Herein, an unexploited 2,4-di(3′5′-dicarboxylphenyl)benzoic acid (H5L) ligand (Scheme 1) is employed to build appropriate MOFs containing optimized pore sizes and OMSs based on the following advantages: (i) the isophthalate-containing linkers easily form porous frameworks based on paddlewheel SBUs,11 and OMSs are often generated from the removal of terminally coordinated solvent molecules to the metal centers; (ii) compared with symmetrical 1,4-benzenedicarboxylate or 1,3,5-benzenetricarboxylate, which have been proven to be excellent organic linkers for constructing MOFs of high porosity and open large pore size,12,13 H5L possesses five carboxylic groups with multiple coordination sites, which is more conducive to forming a multiple-pore system with small pores. Such pores may result in a relative increase in the charge density surrounding the adsorbed CO2 molecules and favor faster CO2 adsorption kinetics in comparison with CH4 and N2, and in turn enhance high CO2 uptake and selectivity.14 In addition, a new synthesis strategy that introduces types of building ligands with reduced symmetry to construct MOFs has been recently proposed,15 which offers a largely unexploited direction towards MOF synthesis by increasing the topological complexity and driving a new mode of framework assembly. In this contribution, we use the asymmetrical H5L ligand to construct microporous MOFs, [H2N(Me)2]2[Zn4(L)2(H2O)1.5]·5DMF·H2O (1·DMF) and [H2N(Me)2]2[Zn4(L)2(H2O)1.5]·4DMA·6H2O (1·DMA), which exhibit high adsorption selectivity and storage capacity for CO2 over CH4 and N2 under ambient conditions.
 |
| | Scheme 1 Schematic structure of the organic linker H5L. | |
Experimental section
Materials and general methods
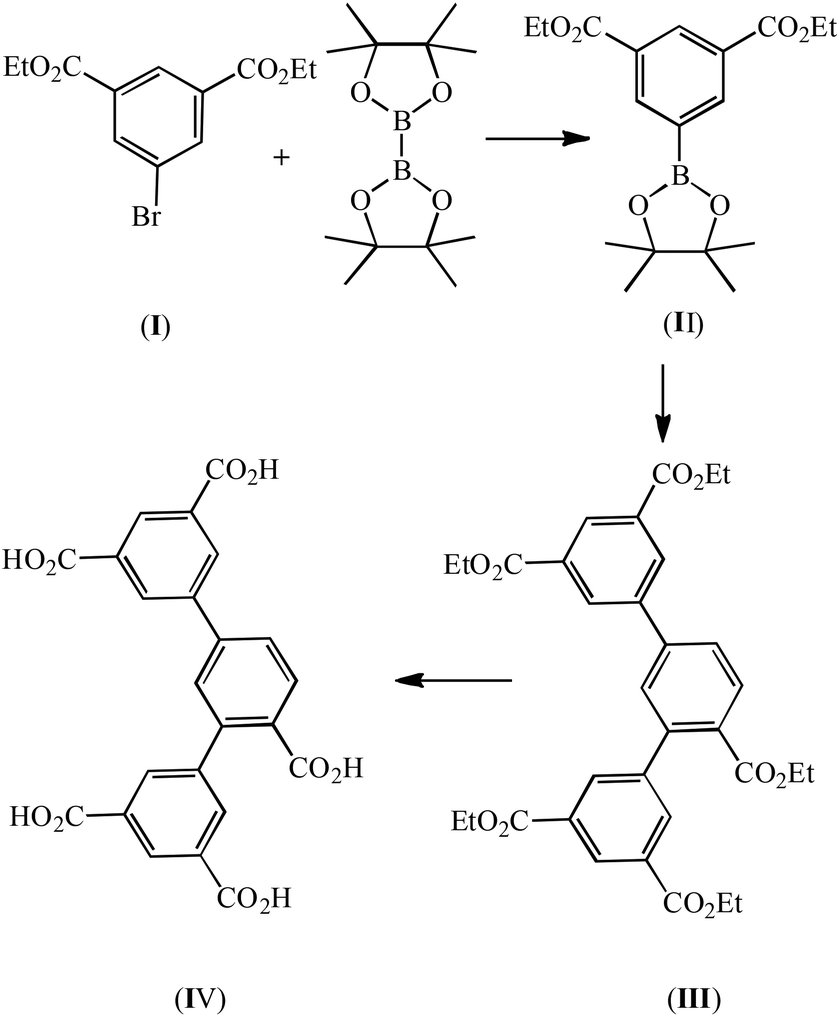
The chemicals diethyl 5-bromoisophthalate (I), ethyl 2,4-dibromobenzoate, bis(pinacolato)diborane and Pd(PPh3)4 were purchased and used without further purification. Infrared spectra were obtained using KBr discs on a Nicolet Avatar 360 FTIR spectrometer in the 400–4000 cm−1 region. Elemental analyses of C, H, and N were performed with a Perkin Elmer 2400C Elemental Analyzer. Thermogravimetric analyses (TGAs) were carried out in a nitrogen stream using a Netzsch TG209F3 instrument at a heating rate of 5 °C min−1. Powder X-ray diffraction (PXRD) data were recorded on a Bruker D8 ADVANCE X-ray powder diffractometer (Cu Kα, 1.5418 Å). The sorption isotherms were measured with an automatic volumetric adsorption apparatus (Micromeritics ASAP 2020M).
Synthesis of 4,4,5,5-tetramethyl-2-(diethyl-3,5-dicarboxylate phenyl)-1,3-dioxolane (II)
A mixture of I (0.1 mol, 30.0 g), bis(pinacolato)diborane (11.8 mmol, 3.0 g), potassium acetate (0.29 mmol, 28.0 g), Pd (dppf)2Cl2 (7.0 mmol, 5.0 g), and dried 1,4-dioxane (500 mL) was stirred at 100 °C overnight and subsequently extracted with ethyl acetate (200 mL × 3). The organic layer was decolored with activated carbon and dried with anhydrous Na2SO4. The crude product was obtained via concentration under a vacuum and was purified by column chromatography (silica gel, ethylacetate–petroleum ether, 6 v%). Yield 78%. Anal. (%) calcd for C18H25BO6: C, 62.09; H, 7.24. Found: C, 61.92; H, 7.16.
Synthesis of pentaethyl-2,4-di(3′,5′-dicarboxylphenyl)benzoate (III)
A mixture of II (0.1 mol, 34.82 g), ethyl 3,5-bromobenzoate (0.05 mol, 15.4 g), and K3PO4 (0.2 mol, 42.4 g) were mixed in 1,4-dioxane (500 mL), and deaerated with N2 for 10 min. Pd(PPh3)4 (0.9 mmol, 1.0 g) was added to the stirred reaction mixture and the mixture was heated to reflux for ca. one week under N2. The crude product of III was obtained after 1,4-dioxane was removed under a vacuum. Recrystallization from methanol afforded the pure product (Scheme 2). Anal. (%) calcd for C33H34O10: C, 67.11; H, 5.80. Found: C, 67.02; H, 5.65.
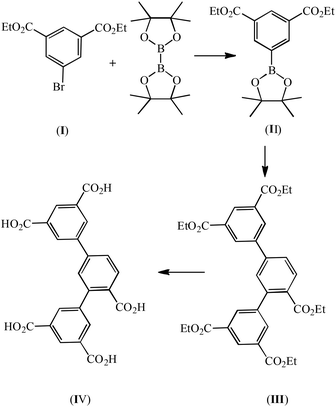 |
| | Scheme 2 The scheme for the synthesis of H5L. | |
Synthesis of 2,4-di(3′,5′-dicarboxylphenyl)benzoic acid (IV)
A mixture of III (0.05 mol, 29.5 g) and 10 g NaOH in 500 mL H2O was refluxed for 2 hours, and then cooled to room temperature. The solution was neutralized with concentrated HCl. White powder was obtained in a yield of 90%. EI-MS: m/z [M − H]−, 449.06 (calcd for C23H14O10, 450.06). Anal. (%) calcd for C23H14O10, C, 61.34; H, 3.13 Found: C, 61.02; H, 2.96.
Synthesis of [H2N(Me)2]2[Zn4(L)2(H2O)1.5]·5DMF·H2O (1·DMF)
A mixture of Zn(NO3)2·6H2O (0.1 mmol, 0.030 g), H5L (0.04 mmol, 0.018 g), DMF (1.5 mL) and water (0.5 mL) was placed in a screw-capped vial, then the vial was capped and placed in an oven at 105 °C for 72 h. The resulting block crystals were washed with DMF three times to give 1·DMF. The yield was ∼24.0 mg (72.6% based on H5L). Anal. Calcd for C65H74N7O27.5Zn4: C, 47.17; H, 4.51; N, 5.92. Found: C, 47.02; H, 4.78; N, 5.63. IR (cm−1): 3424m, 2965w, 2932w, 2807w, 2492w, 2026w, 1660s, 1628s, 1579s, 1450w, 1435w, 1390s, 1255w, 1163w, 1104m, 1061w, 1020w, 920w, 892w, 853w, 781s, 724s, 665w, 579w, 477w.
Synthesis of [H2N(Me)2]2[Zn4(L)2(H2O)1.5]·4DMA·6H2O (1·DMA)
A mixture of Zn(NO3)2·6H2O (0.1 mmol, 0.030 g), H5L (0.04 mmol, 0.018 g), DMA (1.5 mL) and water (1.0 mL) was placed in a screw-capped vial, then the vial was capped and placed in an oven at 105 °C for 72 h. The resulting block crystals were washed with DMA three times to give 1·DMA. The yield was ∼20.6 mg (60.4% based on H5L). Anal. Calcd for C64H85N6O31.5Zn4: C, 45.11; H, 5.03; N, 4.93. Found: C, 45.95; H, 4.88; N, 5.14. IR (cm−1): 3423m, 2964w, 2938w, 2807w, 2487w, 2026w, 1627s, 1506m, 1405s, 1361s, 1261m, 1190w, 1107w, 1020m, 964w, 921w, 891w, 855m, 781s, 724s, 678w, 596w, 478w.
Crystallography
Diffraction data were collected with Mo Kα radiation (λ = 0.71073 Å) on a Bruker-AXS SMART CCD area detector diffractometer. Absorption corrections were carried out utilizing SADABS routine.16 The structures were solved using direct methods and refined using full matrix least squares refinements based on F2. All non-hydrogen atoms were refined anisotropically with the hydrogen atoms added to their geometrically ideal positions and refined isotropically. The contribution of the disordered solvent molecules in 1·DMA was subtracted from the reflection data using the SQUEEZE method as implemented in the PLATON17 program. The final formulas of 1·DMF and 1·DMA were determined by combining the single-crystal structures, elemental microanalyses and TGA data. Selected crystallographic data and structure refinement results are listed in Table 1.
Table 1 Crystal data and structure refinements for 1·DMF and 1·DMA
| Complex no. |
1·DMF |
1·DMA |
|
R
1 = ∑||Fo| − |Fc|)/∑|Fo|; wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2.
|
| Empirical formula |
C65H74N7O27.5Zn4 |
C46H21O21.5Zn4 |
| Temperature [K] |
293(2) |
296(2) |
| Crystal system |
Monoclinic |
Monoclinic |
| Space group |
P21/n |
P21/n |
|
a [Å] |
14.123(4) |
14.1192(17) |
|
b [Å] |
33.380(9) |
33.389(4) |
|
c [Å] |
16.833(3) |
16.938(2) |
|
β [°] |
111.943(16) |
111.664(2) |
|
V [Å3] |
7361(3) |
7421.0(16) |
|
Z
|
2 |
2 |
|
D
calcd [g cm−3] |
1.493 |
1.055 |
|
μ [mm−1] |
1.373 |
1.330 |
| GOF on F2 |
1.122 |
1.142 |
| Reflns collected/unique |
38![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 297/13772 297/13772 |
46438/18188 |
|
R
int
|
0.0440 |
0.0903 |
| Final Ra |
R
1 = 0.0684 |
R
1 = 0.0706 |
| Indices [I > 2σ(I)] |
wR2 = 0.2208 |
wR2 = 0.1757 |
Results and discussion
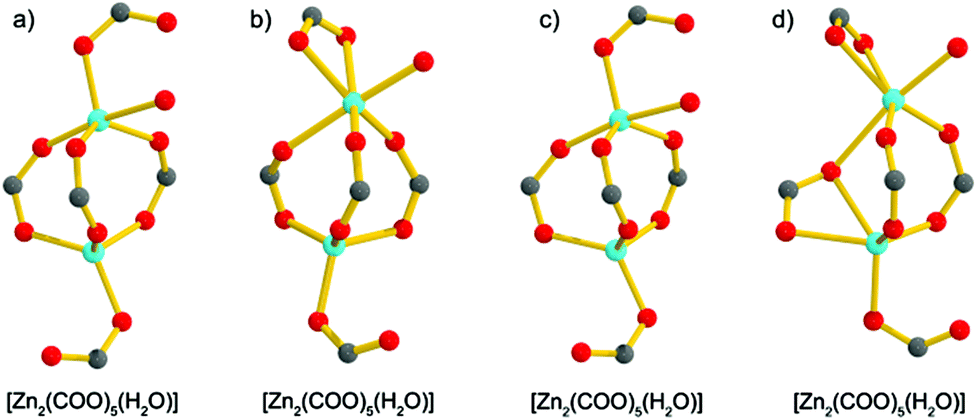
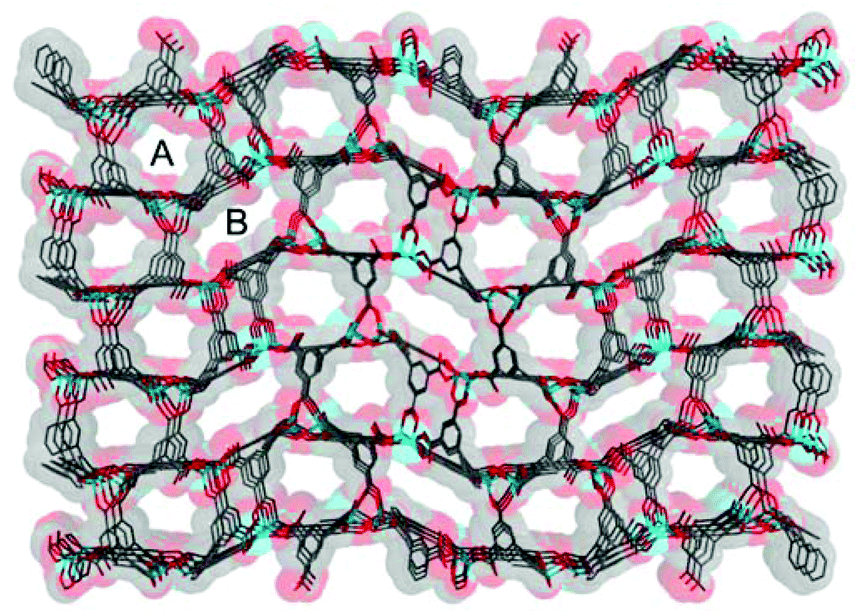
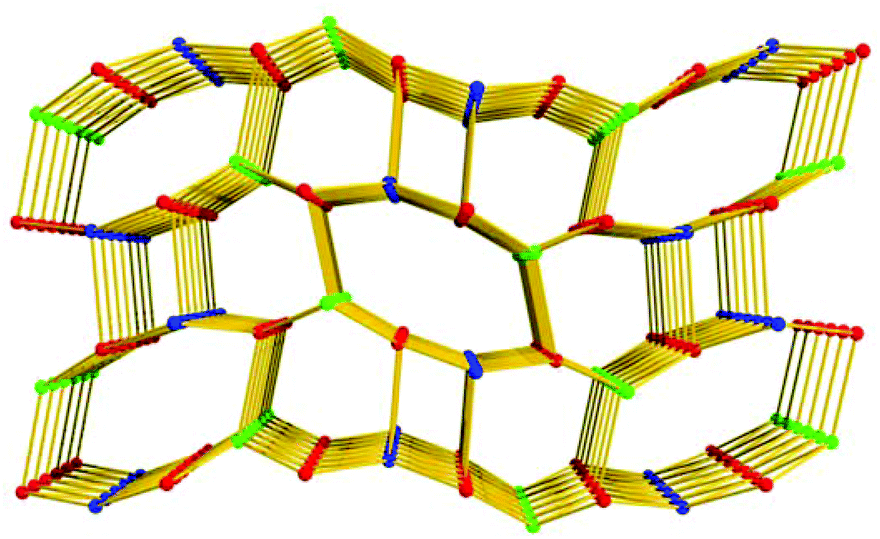
Compound 1·DMF was solvothermally synthesized by heating a solution of Zn(NO3)2·6H2O and H5L in DMF at 105 °C for three days. Single-crystal X-ray diffraction reveals that 1·DMF crystallizes in the monoclinic space group P21/n and shows a 3D microporous framework. The asymmetric unit consists of four independent Zn2+ ions, two fully-deprotonated L5− ligands, and one and half coordinated H2O molecules (Fig. S1†). Zn1 and Zn2 are bridged by three μ2-carboxylate groups and the axial position is further coordinated by two monodentate carboxylate groups to from a [Zn2(COO)5(H2O)] SBU, where one coordinated H2O molecule is bound to the Zn1 center (Fig. 1a). Similarly, Zn3 and Zn4 are bridged by three μ2-carboxylate groups, one monodentate carboxylate group and one chelating carboxylate group to give a distinct [Zn2(COO)5(H2O)] SBU, in which one coordinated H2O molecule is bound to the Zn4 center (Fig. 1b). All the L5− ligands in 1 join to eight Zinc atoms but show two distinct kinds of coordination mode: one exhibits μ1–η1, syn–syn-μ1–η1:η1, syn–syn-μ2–η1:η1, and syn–anti-μ2–η1:η1, while the other adopts μ1–η1, and syn–anti-μ2–η1:η1 (Fig. S2†). The two kinds of SBU and L5− ligand are alternately connected with each other to generate a 3D framework. This arrangement leads to the formation of 1D pore channels with two different shapes along the a-axis, labeled A and B in Fig. 2, with effective aperture sizes of 6.0 × 6.4 Å2 and 4.8 × 11.7 Å2, respectively. The void volume calculated by the PLATON program is 54.4% of the total crystal volume after excluding H2N(Me)2 cations, DMF and H2O molecules.
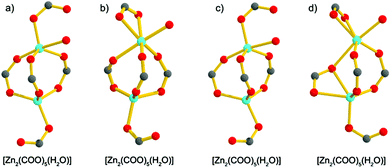 |
| | Fig. 1 The [Zn2(COO)5(H2O)] SBUs in 1·DMF (a) and (b), and the [Zn2(COO)5(H2O)] SBUs in 1·DMA (c) and (d). | |
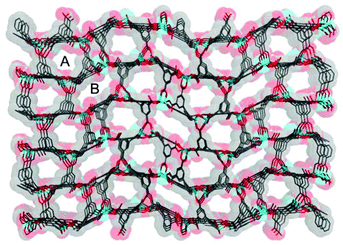 |
| | Fig. 2 The 3D microporous framework of 1; A and B present different shapes of channels. | |
Notably, when DMA was used to replace DMF in the reaction system, 1·DMA was obtained, which is almost the same as the structure of 1·DMF. In contrast to the structure of 1·DMF, the main difference is the coordination modes of the zinc atoms, as shown in Fig. S3,† which shows the two kinds of SBU to have the same component [Zn2(COO)5(H2O)] but show different ways of bridging with the carboxylate groups (Fig. 1c and d). It should be pointed out that many examples of MOFs can be obtained using different solvents, in which the metal centres and organic ligands have the same coordination and bridging modes,18 excluding the guest solvents. To the best of our knowledge, 1·DMF and 1·DMA present a unique example of having the same framework based on different building units. Moreover, a large number of MOFs based on various kinds of Zn2(COO)4 SBU have been reported,19 and a few MOFs constructed from Zn2(COO)3 SBUs have also been obtained,20 while the [Zn2(COO)5] SBUs are only found in MOAAF-2–MOAAF-6 according to the latest Cambridge Structural Database.21 The [Zn2(COO)5(H2O)] SBU in 1 is obtained for the first time among the reported MOFs.
Topologically, in the structure of 1, all the [Zn2(COO)5H2O] SBUs and L5− ligands act as five-connected nodes, and thus 1 shows a rare (5,5,5)-connected topology (Fig. 3). Although binary SBUs (with one kind of inorganic SBU and one kind of organic SBU) are common in the construction of MOFs, such as MOF-5,12a HKUST-1,13a MIL-10122a and UiO-66,22b MOFs based on ternary SBUs are unique and extremely difficult to form. To date, there have been only a few MOFs built from ternary SBUs: UMCM-150, CPM-5, MCF-39, USF-3 and -4, asc-1, -2 and -3, [Cu6O(TZI)3(H2O)9(NO3)]n, and JLU-Liu5 and -Liu6.23 In 1, the two kinds of [Zn2(COO)5(H2O)] unit act as inorganic SBUs and the L5− as an organic SBU, and herein 1 gives the first example of a (5,5,5)-connected framework based on ternary SBUs.
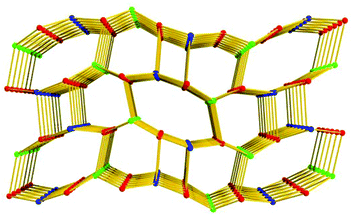 |
| | Fig. 3 The unique (5,5,5)-connected topology of 1 (the colored balls represent: L5−, red; SBU1, blue; and SBU2, green.). | |
The experimental PXRD patterns of 1·DMF and 1·DMA match the simulated ones from the respective single crystal structures (Fig. S4†), confirming the phase purity. Thermogravimetric analysis (TGA) of 1·DMF and 1·DMA was performed (Fig. S6†). The TGA curve of 1·DMF indicates a weight loss of 24.9% in the temperature range of 30–310 °C, which is consistent with the release of five DMF molecules, one lattice H2O molecule, and one and half coordinated H2O molecules per formula unit (calc. 24.8%). For 1·DMA, a weight loss of 28.1% between 30 and 305 °C was observed, corresponding to four DMA molecules, six lattice H2O molecules, and one and half coordinated H2O molecules per formula unit (calc. 27.6%). The desolvated sample (1a) was prepared by heating an as-synthesized sample of 1·DMF at 160 °C for 6 h and then 180 °C for 3 h under vacuum, which was verified by TGA and IR analysis (Fig. S7†). The PXRD of 1a exhibits a shift and weakening of some peaks, indicating a possible framework change due to the numerous solvent releases (Fig. S5†).
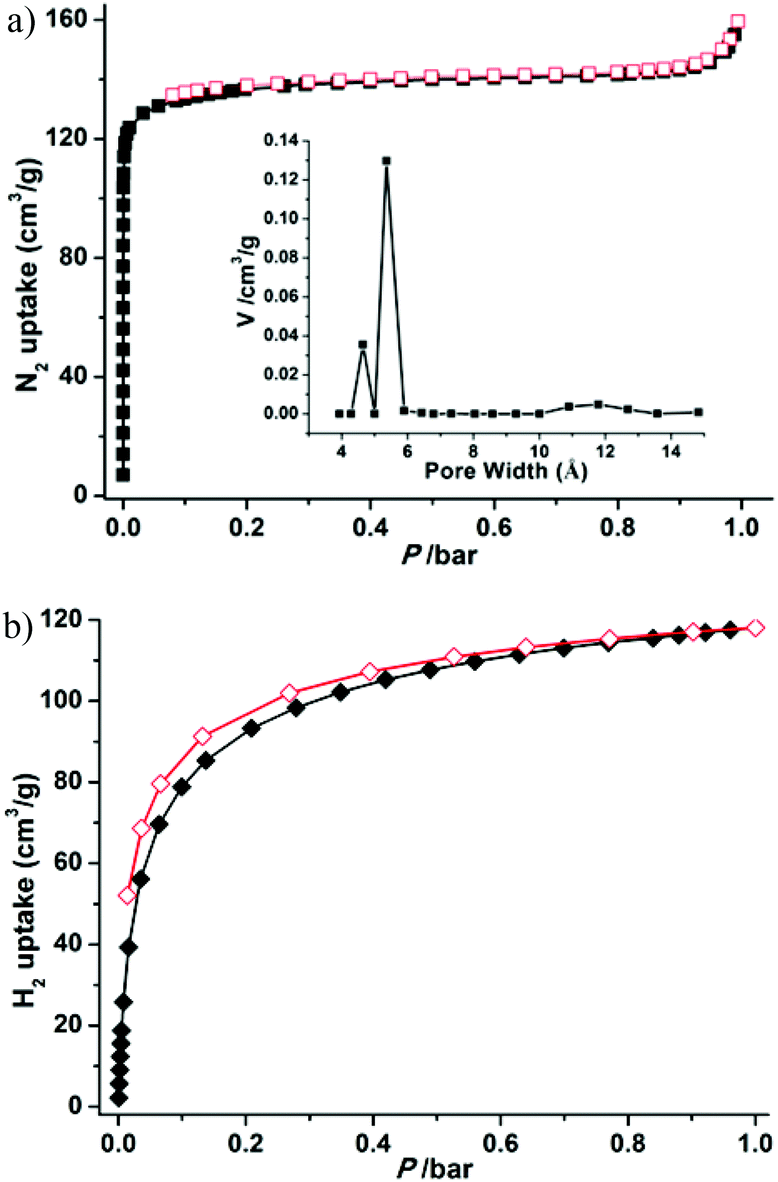
To confirm the permanent porosity of the framework, gas-adsorption measurements (N2, H2, CO2, and CH4) of 1a were performed. The reversible N2 adsorption isotherm at 77 K shows the characteristics of a microporous material with a type-I adsorption isotherm behavior (Fig. 4a). The Brunauer–Emmett–Teller (BET) and Langmuir surface areas of 1a are evaluated to be 544 and 589 m2 g−1, respectively, and the total micropore volume was estimated using the Dubinin–Astakhov (DA) method to be 0.205 cm3 g−1. The saturated loading of 160 cm3 g−1 at 1 bar is close to the calculated value of 196 cm3 g−1 from the crystal density (1.123 g cm−3), free void (34%) and liquid N2 density (0.808 g cm−3). The experimental pore size distribution (PSD) curve based on the NLDFT model demonstrates two kinds of pore in the material, with pore sizes of about 4.6 and 5.4 Å, respectively (inset Fig. 4a), corresponding to the two shapes of channel. The H2 adsorption experiment at 77 K exhibits an uptake of 118.1 cm3 g−1 (1.1 wt%) at 1 bar (Fig. 4b), which is slightly lower than those of MOF-5 (127 cm3 g−1) and MOF-177 (123 cm3 g−1),24 but is superior to that of the best zeolite ZSM-5 (0.7 wt%).25 Notably, the H2 adsorption isotherm is steep in the low pressure region, showing the strong interactions between the relatively small pores and H2 molecules (2.8 Å).26
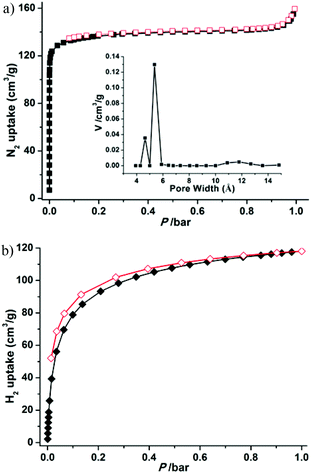 |
| | Fig. 4 N2 (a) and H2 (b) adsorption and desorption isotherms at 77 K and pore size distribution (inset) of 1a. | |
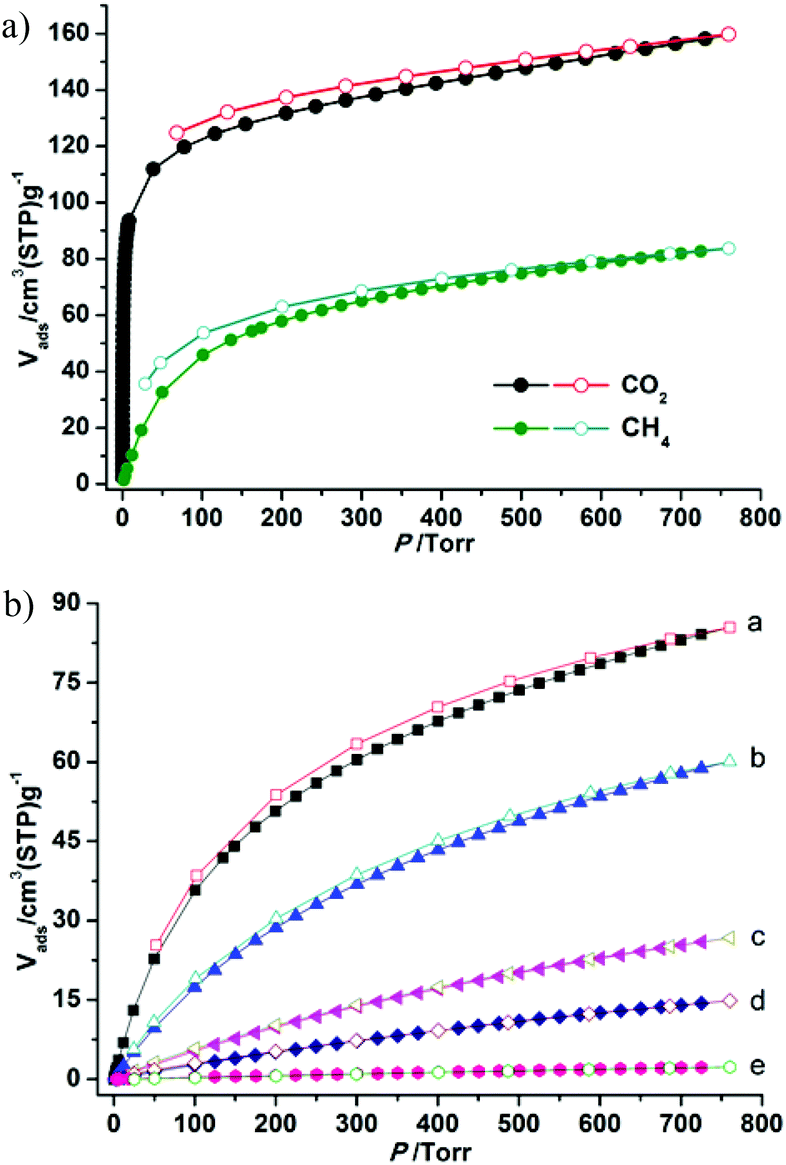
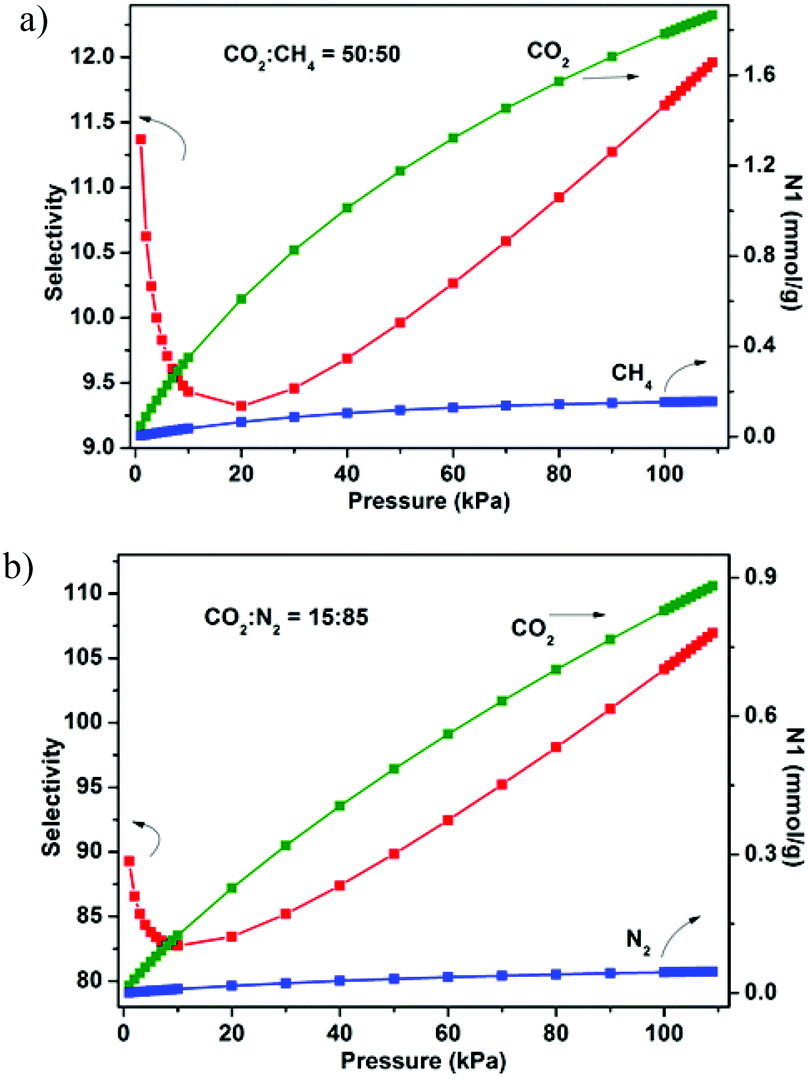
The establishment of the permanent porosity of 1a encourages us to examine its potential application for CO2/CH4 and CO2/N2 separation. As shown in Fig. 5, the CO2 and CH4 adsorption properties on 1a at 195, 273, and 298 K were investigated. At low temperature (195 K), the CO2 uptake reaches 159.7 cm3 g−1 (31.4 wt%) at 1 bar and displays a type-I isotherm. The CH4 uptake of 83.7 cm3 g−1 (6.0 wt%) is much lower than the value of CO2 uptake under the same conditions. Such adsorption selectivity is more significant at high temperatures. The CO2 uptakes are 85.5 cm3 g−1 (16.8 wt%) at 273 K and 60.1 cm3 g−1 (11.8 wt%) at 298 K and 1 bar. The CO2 uptake of 1a at 298 K and 1 bar is inferior to the recently reported zeolite-like zinc–tetrazole framework [Zn(btz)] (22.0 wt%),27 being comparable to MAF-23, which possesses multiple strong adsorption sites (11.0 wt%),28 but is superior to some Zn4O-based frameworks with large pore dimensions (9.0–32.0 Å), such as UMCM-1 (3.8 wt%),29 MOF-177 (3.5 wt%),2a and SNU-70 (3.5 wt%),8c implying that the small pore size may be more attractive for CO2 under low pressure. Meanwhile, the CH4 uptakes are 26.7 cm3 g−1 (1.9 wt%) and 14.8 (1.1 wt%) at 273 and 298 K, respectively. In addition, the N2 sorption isotherm at 298 K was also measured, which shows only a trace of uptake (2.3 cm3 g−1) at 1 bar. To evaluate the CO2/CH4 and CO2/N2 selectivities of 1a under mixture gas conditions with single component isotherms at 298 K, the ideal adsorbed solution theory30 (IAST) can be used to predict the multicomponent adsorption behaviors (Fig. S8†). Fig. 6 shows the predicated selectivities for CO2/CH4 and CO2/N2 as a function of pressure when the gas phase mole fractions are 50/50 and 15/85, which are typical feed compositions for landfill gas and flue gas, respectively. It is worth noting that very high selectivities for both the CO2/CH4 and CO2/N2 are obtained. The initial values of selectivities for CO2/CH4 and CO2/N2 are 11.4 and 95.2 and the selectivities are 12.0 and 107.0 at 1 bar, respectively. The values of the selectivities for CO2/CH4 outperform many reported MOFs and carbon materials under the same conditions,31 and are comparable to the high selectivities of MOFs functionalized with OMSs or polar groups.32 The highly selective adsorption of CO2 rather than CH4 and N2 suggests the possible application of 1a in capturing CO2 from upgrading natural gas or flue gas. In addition, both the CO2/CH4 and CO2/N2 selectivities show a small increasing trend under relatively high pressure, which is rare in MOFs. Taking into account the PXRD pattern of the samples after gas adsorption, the increased selectivity should be attributed to the slight flexibility of the framework; as the pressure increases and gas molecules are adsorbed, a distortion of the framework may occur, which results in enhanced sieve effects and a corresponding higher selectivity. The increased selectivity of 1a indicates that the enhanced sieve effect of the pores might be a key effect that could be of benefit for CO2 capture under relatively high pressure.
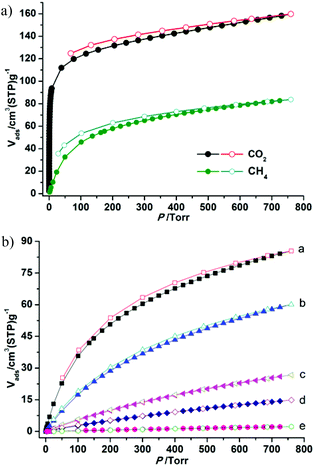 |
| | Fig. 5 The adsorption isotherms of 1a: (a) CO2 and CH4 at 195 K; (b) CO2 adsorption and desorption at 273 K (a) and 298 K (b), CH4 adsorption and desorption at 273 K (c) and 298 K (d), N2 adsorption and desorption at 298 K (e). | |
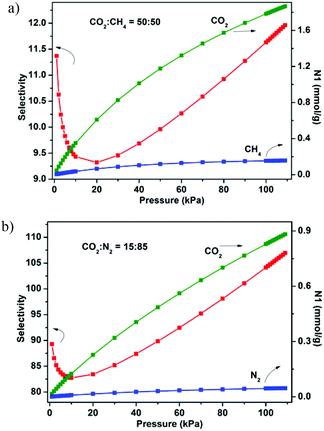 |
| | Fig. 6 IAST adsorption selectivities of 1a for equimolar mixtures of CO2 and CH4 (a) and a mixture of CO2 (15%) and N2 (85%) (b) at 298 K. | |
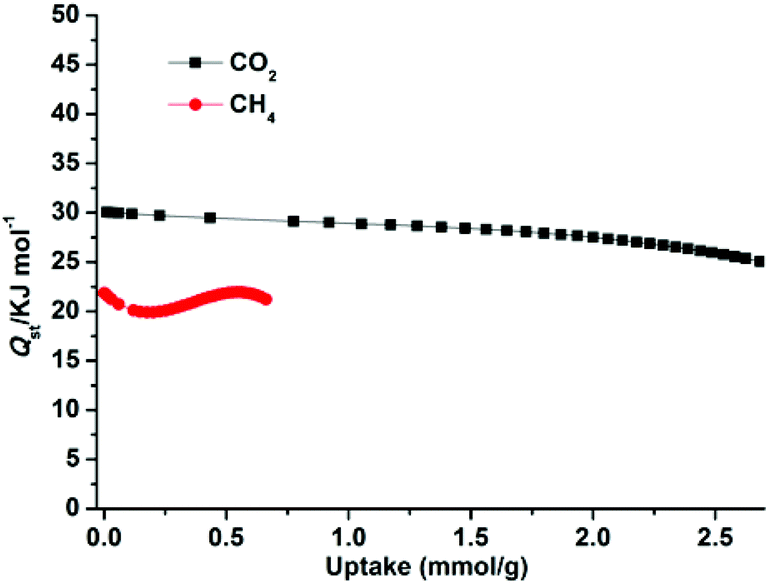
The higher selectivity for CO2 may be attributed to the effect of the OMSs (the exposed Zn2+ sites, stemming from the release of coordinated H2O), the cationic framework, the dimethylammonium cationic guests, and the numerous uncoordinated O donors (located within the wall of the porous surface), which provide an additional electric field and make the framework highly polar,8c,23g and effectively improve the interaction between highly quadrupolar CO2 molecules and the host framework. Moreover, the narrow pore sizes of 1a and the small kinetic diameter of CO2 (3.30 Å) compared to CH4 (3.80 Å) and N2 (3.64 Å) enable CO2 molecules to easily enter the pores. To further check the affinity of 1a for CO2, the CO2 adsorption enthalpies (Qst) were calculated according to the virial equation from the sorption isotherms at 273 and 298 K (Fig. S9†). At initial coverage, Qst exhibits a maximum of 30.1 kJ mol−1 (Fig. 7); this value is similar to that of dia-7i-1-Co (30 kJ mol−1)8a and comparable to those of MOFs decorated with typical active sites such as Lewis basic sites (LBSs) and OMSs, for example Cu(bcppm) (29 kJ mol−1),33a Cu-btc (28 kJ mol−1),33b and PCN-88 (27 kJ mol−1),9a but lower than those of some high polarity-functionalized MOFs (45–90 kJ mol−1).3a,34 This intermediate adsorption enthalpy implies a low-energy consumption for adsorbent regeneration, a desired property for separation applications. For comparison, the Qst values for CH4 were also calculated from adsorption data collected at 273 and 298 K for 1a (Fig. S10†). The maximum value of CH4 is 21.9 kJ mol−1 and is much smaller than that of CO2, showing a weaker CH4 binding affinity than CO2 throughout the adsorption process.
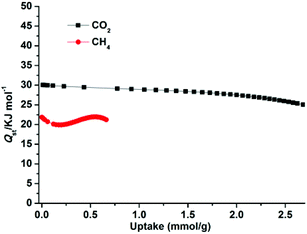 |
| | Fig. 7 The isosteric heats of CO2 and CH4 adsorption for 1a estimated using the virial equation from the adsorption isotherms at 273 and 298 K. | |
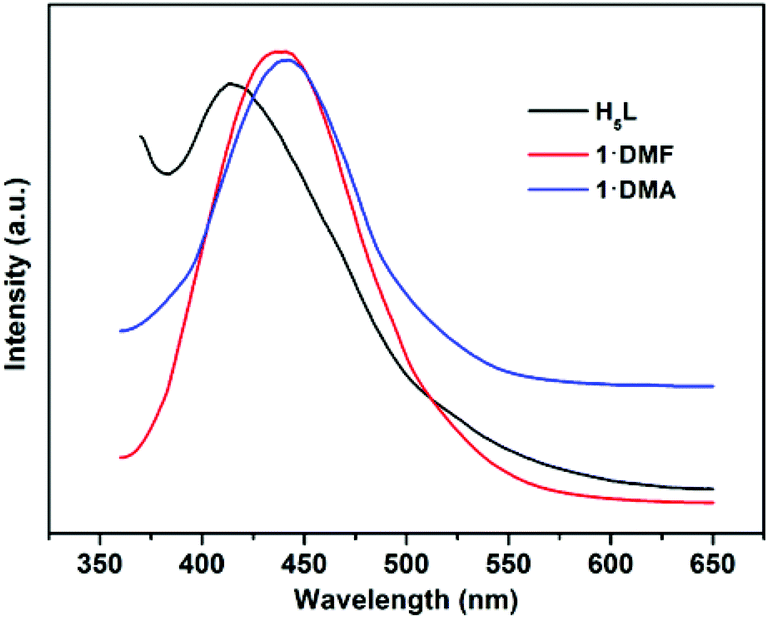
In addition, the solid-state luminescence properties of 1·DMF and 1·DMA, as well as the free ligand H5L, are investigated at room temperature. As shown in Fig. 8 and S11,†1·DMF and 1·DMA display fluorescence emission bands at 441 nm and 443 nm upon excitation at 344 nm and 341 nm, respectively. These bands can probably be assigned to the π–π* intra-ligand luminescent emission, since similar emission is observed at 414 nm upon excitation at 343 nm for the free ligand. In comparison with the emission of H5L, a slight red-shift of ∼18 nm occurs in the maximum emission peaks of the compounds, which can presumably be associated with the coordinative environment around the ligand.
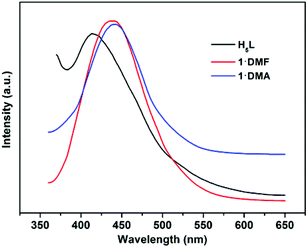 |
| | Fig. 8 Fluorescence emission spectra of the ligand H5L (black), 1·DMF (red) and 1·DMA (blue) in solid state at room temperature. | |
Conclusions
In summary, by utilizing an asymmetrical pentacarboxylate ligand, a novel microporous MOF based on uncommon ternary SBUs has been successfully assembled. Interestingly, all the SBUs act as five-connected nodes to form a rare (5,5,5)-connected topology. The desolvated framework shows permanent porosity with suitable pore spaces and high polarity porous surfaces decorated with OMSs and LBSs, which enable this material to exhibit higher uptake capacities for CO2 and high selectivity for CO2 over N2 and CH4 gases. The unique structure and excellent gas adsorption properties further reveal that unsymmetrical polycarboxylate linkers can be used as a good building block to construct functional MOF materials.
Acknowledgements
We are grateful for financial support from the NSF of China (grants 21371142, 91022004, 21201139) and the NSF of Shaanxi, China (grants 2013JQ2016, 2013KJXX-26).
Notes and references
-
J. K. Casper, Greenhouse Gases: Worldwide Impacts, Infobase Publishing, New York, 2010 Search PubMed.
-
(a) A. R. Millward and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 17998–17999 CrossRef CAS PubMed;
(b) D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed;
(c) Y. S. Bae and R. Q. Snurr, Angew. Chem., Int. Ed., 2011, 50, 11586–11596 CrossRef CAS PubMed;
(d) J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed;
(e) J. Liu, P. K. Thallapally, B. P. McGrail, D. R. Brown and J. Liu, Chem. Soc. Rev., 2012, 41, 2308–2322 RSC;
(f) K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed;
(g) S. Xiang, Y. He, Z. Zhang, H. Wu, W. Zhou, R. Krishna and B. Chen, Nat. Commun., 2012, 3, 954 CrossRef PubMed;
(h) P. Nugent, Y. Belmabkhout, S. D. Burd, A. J. Cairns, R. Luebke, K. Forrest, T. Pham, S. Ma, B. Space, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, Nature, 2013, 495, 80–84 CrossRef CAS PubMed.
-
(a) T. M. McDonald, W. R. Lee, J. A. Mason, B. M. Wiers, C. S. Hong and J. R. Long, J. Am. Chem. Soc., 2012, 134, 7056–7065 CrossRef CAS PubMed;
(b) L. Du, Z. Lu, K. Zheng, J. Wang, X. Zheng, Y. Pan, X. You and J. Bai, J. Am. Chem. Soc., 2013, 135, 562–565 CrossRef CAS PubMed;
(c) A. M. Fracaroli, H. Furukawa, M. Suzuki, M. Dodd, S. Okajima, F. Gándara, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 8863–8866 CrossRef CAS PubMed;
(d) Z. Zhang, Z.-Z. Yao, S. Xiang and B. Chen, Energy Environ. Sci., 2014, 7, 2868–2899 RSC.
-
(a) M. Du, C.-P. Li, M. Chen, Z.-W. Ge, X. Wang, L. Wang and C.-S. Liu, J. Am. Chem. Soc., 2014, 136, 10906–10909 CrossRef CAS PubMed;
(b) D. H. Hong and M. P. Suh, Chem. Commun., 2012, 48, 9168–9170 RSC;
(c) Q. Lin, T. Wu, S.-T. Zheng, X. Bu and P. Feng, Chem. Commun., 2011, 47, 11852–11854 RSC;
(d) M. Y. Masoomi, K. C. Stylianou, A. Morsali, P. Retailleau and D. Maspoch, Cryst. Growth Des., 2014, 14, 2092–2096 CrossRef CAS.
-
(a) Y. Inubushi, S. Horike, T. Fukushima, G. Akiyama, R. Matsuda and S. Kitagawa, Chem. Commun., 2010, 46, 9229–9231 RSC;
(b) R.-J. Li, M. Li, X.-P. Zhou, D. Li and M. O'Keeffe, Chem. Commun., 2014, 50, 4047–4049 RSC;
(c) B. Liu, Y. Li, L. Hou, G. Yang, Y.-Y. Wang and Q.-Z. Shi, J. Mater. Chem. A, 2013, 1, 6535–6538 RSC;
(d) Y.-X. Tan, Y.-P. He and J. Zhang, Inorg. Chem., 2011, 50, 11527–11531 CrossRef CAS PubMed.
-
(a) X.-J. Wang, P.-Z. Li, Y. Chen, Q. Zhang, H. Zhang, X. X. Chan, R. Ganguly, Y. Li, J. Jiang and Y. Zhao, Sci. Rep., 2013, 3, 1149 Search PubMed;
(b) J. Qian, F. Jiang, D. Yuan, M. Wu, S. Zhang, L. Zhang and M. Hong, Chem. Commun., 2012, 48, 9696–9698 RSC;
(c) N. Sikdar, A. Hazra and T. K. Maji, Inorg. Chem., 2013, 52, 11385–11397 CrossRef PubMed;
(d) B. Liu, W.-P. Wu, L. Hou and Y.-Y. Wang, Chem. Commun., 2014, 50, 8731–8734 RSC;
(e) Z.-H. Xuan, D.-S. Zhang, Z. Chang, T.-L. Hu and X.-H. Bu, Inorg. Chem., 2014, 53, 8985–8990 CrossRef CAS PubMed.
-
(a) P. S. Nugent, V. L. Rhodus, T. Pham, K. Forrest, L. Wojtas, B. Space and M. J. Zaworotko, J. Am. Chem. Soc., 2013, 135, 10950–10953 CrossRef CAS PubMed;
(b) M.-H. Choi, H. J. Park, D. H. Hong and M. P. Suh, Chem. – Eur. J., 2013, 19, 17432–17438 CrossRef CAS PubMed;
(c) B. Wang, H. Huang, X.-L. Lv, Y. Xie, M. Li and J.-R. Li, Inorg. Chem., 2014, 53, 9254–9259 CrossRef CAS PubMed;
(d) X. Lv, L. Li, S. Tang, C. Wang and X. Zhao, Chem. Commun., 2014, 50, 6886–6889 RSC;
(e) B. Liu, J. Shi, K.-F. Yue, D.-S. Li and Y.-Y. Wang, Cryst. Growth Des., 2014, 14, 2003–2008 CrossRef CAS;
(f) B. Zheng, J. Bai, J. Duan, L. Wojtas and M. J. Zaworotko, J. Am. Chem. Soc., 2011, 133, 748–751 CrossRef CAS PubMed.
-
(a) S. K. Elsaidi, M. H. Mohamed, L. Wojtas, A. Chanthapally, T. Pham, B. Space, J. J. Vittal and M. J. Zaworotko, J. Am. Chem. Soc., 2014, 136, 5072–5077 CrossRef CAS PubMed;
(b) R. Wang, Q. Meng, L. Zhang, H. Wang, F. Dai, W. Guo, L. Zhao and D. Sun, Chem. Commun., 2014, 50, 4911–4914 RSC;
(c) T. K. Prasad and M. P. Suh, Chem. – Eur. J., 2012, 18, 8673–8680 CrossRef CAS PubMed.
-
(a) J.-R. Li, J. Yu, W. Lu, L.-B. Sun, J. Sculley, P. B. Balbuena and H.-C. Zhou, Nat. Commun., 2013, 4, 1538 CrossRef PubMed;
(b) L. Pan, D. H. Olson, L. R. Ciemnolonski, R. Heddy and J. Li, Angew. Chem., Int. Ed., 2006, 45, 616–619 CrossRef CAS PubMed;
(c) R.-B. Lin, D. Chen, Y.-Y. Lin, J.-P. Zhang and X.-M. Chen, Inorg. Chem., 2012, 51, 9950–9955 CrossRef CAS PubMed.
- W. Lu, Z. Wei, Z.-Y. Gu, T.-F. Liu, J. Park, J. Park, J. Tian, M. Zhang, Q. Zhang, T. Gentle III, M. Bosch and H.-C. Zhou, Chem. Soc. Rev., 2014, 43, 5561–5593 RSC.
- Y. He, B. Li, M. O'Keeffe and B. Chen, Chem. Soc. Rev., 2014, 43, 5618–5656 RSC.
-
(a) H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–278 CrossRef CAS PubMed;
(b) M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed.
-
(a) S. S.-Y. Chui, S. M.-F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS;
(b) G. Férey, C. Serre, C. Mellot-Draznieks, F. Millange, S. Surblé, J. Dutour and I. Margiolaki, Angew. Chem., Int. Ed., 2004, 43, 6296–6301 CrossRef PubMed.
- O. Shekhah, Y. Belmabkhout, Z. Chen, V. Guillerm, A. Cairns, K. Adil and M. Eddaoudi, Nat. Commun., 2014, 5, 4228 CAS.
-
(a) Y. Liu, J.-R. Li, W. M. Verdegaal, T.-F. Liu and H.-C. Zhou, Chem. – Eur. J., 2013, 19, 5637–5643 CrossRef CAS PubMed;
(b) A. G. Wong-Foy, O. Lebel and A. J. Matzger, J. Am. Chem. Soc., 2007, 129, 15740–15741 CrossRef CAS PubMed;
(c) Y. Xie, H. Yang, Z. U. Wang, Y. Liu, H.-C. Zhou and J.-R. Li, Chem. Commun., 2014, 50, 563–565 RSC.
-
Bruker, SADABS, SMART and SAINT, Bruker AXS Inc, Madison, Wisconson, USA, 2002 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
-
(a) W. Cho, H. J. Lee and M. Oh, J. Am. Chem. Soc., 2008, 130, 16943–16946 CrossRef CAS PubMed;
(b) B. Liu, H. Miao, L.-Y. Pang, L. Hou, Y.-Y. Wang and Q.-Z. Shi, CrystEngComm, 2012, 14, 2954–2958 RSC;
(c) C.-P. Li and M. Du, Chem. Commun., 2011, 47, 5958–5972 RSC.
-
(a) H. Chun, J. Am. Chem. Soc., 2008, 130, 800–801 CrossRef CAS PubMed;
(b) M. Wu, F. Jiang, W. Wei, Q. Gao, Y. Huang, L. Chen and M. Hong, Cryst. Growth Des., 2009, 9, 2559–2561 CrossRef CAS;
(c) Y.-P. He, Y.-X. Tan and J. Zhang, Inorg. Chem., 2012, 51, 11232–11234 CrossRef CAS PubMed;
(d) X. Zhao, X. Wang, S. Wang, J. Dou, P. Cui, Z. Chen, D. Sun, X. Wang and D. Sun, Cryst. Growth Des., 2012, 12, 2736–2739 CrossRef CAS;
(e) Z.-J. Lin, T.-F. Liu, X.-L. Zhao, J. Lü and R. Cao, Cryst. Growth Des., 2011, 11, 4284–4287 CrossRef CAS.
-
(a) X. Zhao, H. He, F. Dai, D. Sun and Y. Ke, Inorg. Chem., 2010, 49, 8650–8652 CrossRef CAS PubMed;
(b) T.-F. Liu, J. Lü, Z. Guo, D. M. Proserpio and R. Cao, Cryst. Growth Des., 2010, 10, 1489–1491 CrossRef CAS;
(c) B. Liu, D.-S. Li, L. Hou, G.-P. Yang, Y.-Y. Wang and Q.-Z. Shi, Dalton Trans., 2013, 42, 9822–9825 RSC;
(d) X.-S. Wang, M. Chrzanowski, W.-Y. Gao, L. Wojtas, Y.-S. Chen, M. J. Zaworotko and S. Ma, Chem. Sci., 2012, 3, 2823–2827 RSC.
- M. J. Manos, E. E. Moushi, G. S. Papaefstathiou and A. J. Tasiopoulos, Cryst. Growth Des., 2012, 12, 5471–5480 CAS.
-
(a) G. Férey, C. Mellot Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040–2042 CrossRef PubMed;
(b) J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
-
(a) A. G. Wong-Foy, O. Lebeland and A. J. Matzger, J. Am. Chem. Soc., 2007, 129, 15740–15741 CrossRef CAS PubMed;
(b) S.-T. Zheng, J. T. Bu, Y. Li, T. Wu, F. Zuo, P. Feng and X. Bu, J. Am. Chem. Soc., 2010, 132, 17062–17064 CrossRef CAS PubMed;
(c) Q. Chen, W. Xue, B.-Y. Wang, M.-H. Zeng and X.-M. Chen, CrystEngComm, 2012, 14, 2009–2014 RSC;
(d) A. Schoedel, A. J. Cairns, Y. Belmabkhout, L. Wojtas, M. Mohamed, Z. Zhang, D. M. Proserpio, M. Eddaoudi and M. J. Zaworotko, Angew. Chem., Int. Ed., 2013, 52, 2902–2905 CrossRef CAS PubMed;
(e) F. Nouar, J. F. Eubank, T. Bousquet, L. Wojtas, M. J. Zaworotko and M. Eddaoudi, J. Am. Chem. Soc., 2008, 130, 1833–1835 CrossRef CAS PubMed;
(f) X.-R. Hao, X.-L. Wang, K.-Z. Shao, G.-S. Yang, Z.-M. Su and G. Yuan, CrystEngComm, 2012, 14, 5596–5603 RSC;
(g) D. Wang, T. Zhao, Y. Cao, S. Yao, G. Li, Q. Huo and Y. Liu, Chem. Commun., 2014, 50, 8648–8650 RSC.
- J. L. C. Rowsell, A. R. Millward, K. S. Park and O. M. Yaghi, J. Am. Chem. Soc., 2004, 126, 5666–5667 CrossRef CAS PubMed.
- L. Schlapbach and A. Züttel, Nature, 2001, 414, 353–358 CrossRef CAS PubMed.
-
(a) B. Chen, N. W. Ockwig, A. R. Millward, D. S. Contreras and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4745–4749 CrossRef CAS PubMed;
(b) S.-S. Chen, M. Chen, S. Takamizawa, M.-S. Chen, Z. Sua and W.-Y. Sun, Chem. Commun., 2011, 47, 752–754 RSC;
(c) P. Song, Y. Li, B. He, J. Yang, J. Zheng and X. Li, Microporous Mesoporous Mater., 2011, 142, 208–213 CrossRef CAS PubMed.
- P. Cui, Y.-G. Ma, H.-H. Li, B. Zhao, J.-R. Li, P. Cheng, P. B. Balbuena and H.-C. Zhou, J. Am. Chem. Soc., 2012, 134, 18892–18895 CrossRef CAS PubMed.
- P.-Q. Liao, D.-D. Zhou, A.-X. Zhu, L. Jiang, R.-B. Lin, J.-P. Zhang and X.-M. Chen, J. Am. Chem. Soc., 2012, 134, 17380–17383 CrossRef CAS PubMed.
- A. Ö. Yazaydin, R. Q. Snurr, T.-H. Park, K. Koh, J. Liu, M. D. LeVan, A. I. Benin, P. Jakubczak, M. Lanuza, D. B. Galloway, J. J. Low and R. R. Willis, J. Am. Chem. Soc., 2009, 131, 18198–18199 CrossRef CAS PubMed.
- A. L. Myers and J. M. Prausnitz, AIChE J., 1965, 11, 121 CrossRef CAS PubMed.
-
(a) Z.-H. Xiang, X. Peng, X. Cheng, X.-J. Li and D.-P. Cao, J. Phys. Chem. C, 2011, 115, 19864–19871 CrossRef CAS;
(b) R. Babarao, Z.-Q. Hu, J.-W. Jiang, S. Chempath and S. I. Sandler, Langmuir, 2007, 23, 659–666 CrossRef CAS PubMed;
(c) B. Zheng, R. Yun, J. Bai, Z. Lu, L. Du and Y. Li, Inorg. Chem., 2013, 52, 2823–2829 CrossRef CAS PubMed;
(d) Z.-J. Lin, Y.-B. Huang, T.-F. Liu, X.-Y. Li and R. Cao, Inorg. Chem., 2013, 52, 3127–3132 CrossRef CAS PubMed.
-
(a) R. Banerjee, H. Furukawa, D. Britt, C. Knobler, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 3875–3877 CrossRef CAS PubMed;
(b) Z. Zhang, S. Xiang, Y.-S. Chen, S. Ma, Y. Lee, T. Phely-Bobin and B. Chen, Inorg. Chem., 2010, 49, 8444–8448 CrossRef CAS PubMed;
(c) Y.-N. Gong, Y.-L. Huang, L. Jiang and T.-B. Lu, Inorg. Chem., 2014, 53, 9457–9459 CrossRef CAS PubMed;
(d) I. Spanopoulos, P. Xydias, C. D. Malliakas and P. N. Trikalitis, Inorg. Chem., 2013, 52, 855–862 CrossRef CAS PubMed;
(e) Y. Li, Z. Ju, B. Wu and D. Yuan, Cryst. Growth Des., 2013, 13, 4125–4130 CrossRef CAS.
-
(a) W. M. Bloch, R. Babarao, M. R. Hill, C. J. Doonan and C. J. Sumby, J. Am. Chem. Soc., 2013, 135, 10441–10448 CrossRef CAS PubMed;
(b) Q. Yan, Y. Lin, C. Kong and L. Chen, Chem. Commun., 2013, 49, 6873–6875 RSC.
-
(a) R. Vaidhyanathan, S. S. Iremonger, G. K. H. Shimizu, P. G. Boyd, S. Alavi and T. K. Woo, Science, 2010, 330, 650–653 CrossRef CAS PubMed;
(b) J. Luo, J. Wang, G. Li, Q. Huo and Y. Liu, Chem. Commun., 2013, 49, 11433–11435 RSC;
(c) D.-D. Zhou, C.-T. He, P.-Q. Liao, W. Xue, W.-X. Zhang, H.-L. Zhou, J.-P. Zhang and X.-M. Chen, Chem. Commun., 2013, 49, 11728–11730 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures, TGA, IR, PXRD, excitation spectra and additional sorption patterns. CCDC 1033728 and 1033729. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qi00025d |
|
| This journal is © the Partner Organisations 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.