
An efficient synthesis of gem-diiodoolefins and (E)-iodoalkenes from propargylic amides with a Cu(I)/Cu(III) cycle†
Shuo
Zhang
a,
Ying
Chen
a,
Jianwu
Wang
*a,
Yue
Pan
a,
Zhenghu
Xu
*ab and
Chen-Ho
Tung
ac
aKey Lab for Colloid and Interface Chemistry of Education Ministry, School of chemistry and Chemical Engineering, Shandong University, Jinan 250100, People's Republic of China. E-mail: xuzh@sdu.edu.cn; jwwang@sdu.edu.cn
bState Key Laboratory of Organometallic Chemistry Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, PR China
cKey Laboratory of Photochemical Conversion and Optoelectronic Materials, Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Beijing 100190, PR China
First published on 27th March 2015
Abstract
gem-Dihaloalkenes are very important building blocks widely used in organic synthesis. However, the synthetic methods available for this functional group are very limited. Starting from an unexpected copper-catalyzed cyclization of propargylic amides, a new route for the synthesis of gem-dihaloalkenes was developed. According to the plausible Cu(I)/Cu(III) catalytic cycle, another oxidative iodination protocol was developed. Through this improved method, a series of gem-diiodoolefins were synthesized in high yields. In addition, (E)-iodoalkenes could also be synthesized efficiently using the same approach, which is complementary to the gold-catalyzed reaction giving (Z)-iodoalkenes. These iodoolefins could be further transformed into various trisubstituted or tetrasubstituted alkenes, enynes, dienes and trienes with palladium-catalyzed coupling reactions.
1. Introduction
1,1-Dihalo-1-alkenes represent an important type of organic compounds. They exist in many bioactive natural products as well as pharmaceuticals (Scheme 1).1 For example, deltamethrin1a products bearing a gem-dibromoalkene functional group are among the most popular and widely used insecticides in the world. More importantly, 1,1-dihalo-1-alkenes are very valuable synthetic intermediates used in synthetic chemistry.2 In the presence of a base or metal reagent, 1,1-dihalo-1-alkenes went through α-elimination generating vinylidene type intermediates, and the subsequent migration of H atoms or the alkyl group formed alkynes (Scheme 2). Thus 1,1-dihalo-1-alkenes are widely used as alkyne equivalents, such as the well-known Corey–Fuchs reaction.3 By using this method, the Kibayashi group realized the total synthesis of pumiliotoxin A and 225F from gem-dibromoalkenes.4 Recently, the blossoming of transition-metal catalyzed cross coupling reactions has made this gem-dihaolo-1-alkenes extremely useful in the synthesis of various important heterocycles (Scheme 2). For example, through the sequential intramolecular and intermolecular coupling reactions, Lautens et al. developed a series of efficient methods for the construction of multisubstituted indoles, benzofurans, and benzothiophenes.5 The Wu group developed an efficient synthetic method for polyfluoroarylpyrrolo[1,2-α]quinolines via palladium-catalyzed double arylation reactions of gem-dibromoalkenes.6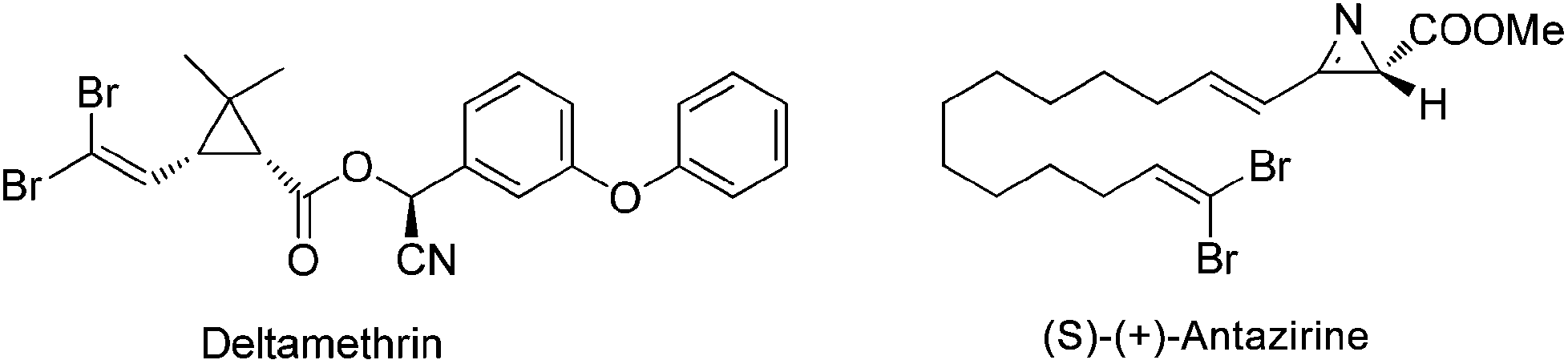 | ||
| Scheme 1 Natural products and pharmaceuticals containing gem-dihaloalkenes. | ||
 | ||
| Scheme 2 Synthesis of gem-dihaloalkenes and their synthetic applications. | ||
1,1-Dihalo-1-alkenes are important building blocks in organic syntheses, however the synthetic methods available for this functional group are very limited. The current methods mostly focus on the Wittig type reaction between carbonyl compounds and tetrahalocarbon (Scheme 2).7 However, this protocol requires a large excess amount of triphenylphosphine (4–6 equiv.), which is not ideal from the point view of atom economy and green chemistry. The development of other practical synthetic methods is highly desirable. We have reported herein a facile synthesis of gem-diiodoolefins with a Cu(I)/Cu(III) catalytic cycle. Following this approach, various vinyl halides with different stereo-configurations could be easily prepared. The obtained gem-diiodoolefins were further transformed into tetrasubstituted olefins with the palladium-catalyzed cross coupling reactions.
2. Results and discussion
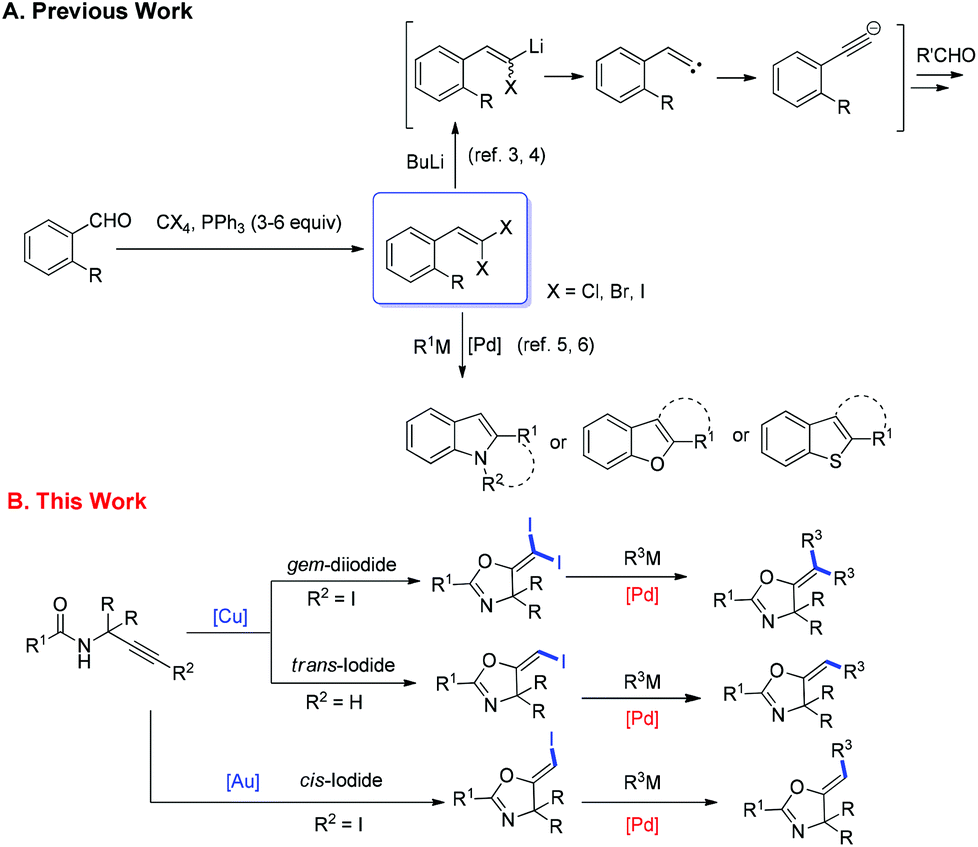
2.1 An unexpected synthesis of gem-diiodoalkenes from iodoalkynes with a Cu(I)/Cu(III) catalytic cycle
In recent years, haloalkynes have emerged as a type of powerful and versatile building blocks widely used in organic syntheses.8 The alkyne and halide parts, serving as two functional groups, were transformed into various functionalized alkynes and heterocycles through addition, cross coupling or cycloaddition reactions.9 As a continuation of our research interests in Pi acid transition-metal-catalyzed reactions,10 we studied the cyclization reaction of propargylic amide111a. When Ph3PAuNTf2 was used as the catalyst, the desired (Z)-iodoalkene 2a was isolated in 83% yield. However, this product was not stable, probably because it could isomerize into the aromatic oxazole through double bond migration (eqn (1), Scheme 3). To our surprise, when CuI was used as the catalyst, another unknown gem-diiodide product 3a was observed as the major product. Its structure was unambiguously characterized by NMR, Mass and also single crystal X-ray analysis (eqn (2), Scheme 3). Under these conditions, this product was isolated in 45% yield. However, its isolated yield is as high as 90% based on the iodine source. Through a detailed analysis of the reaction mixture, the de-iodine amide 4a was isolated in 42% yield.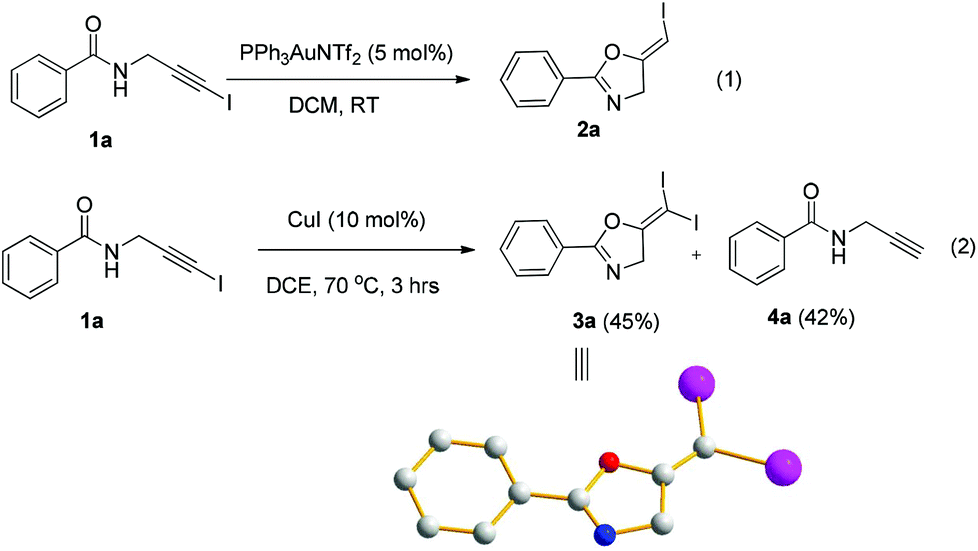 | ||
| Scheme 3 Gold or copper-catalyzed cyclization of propargylic amides. | ||
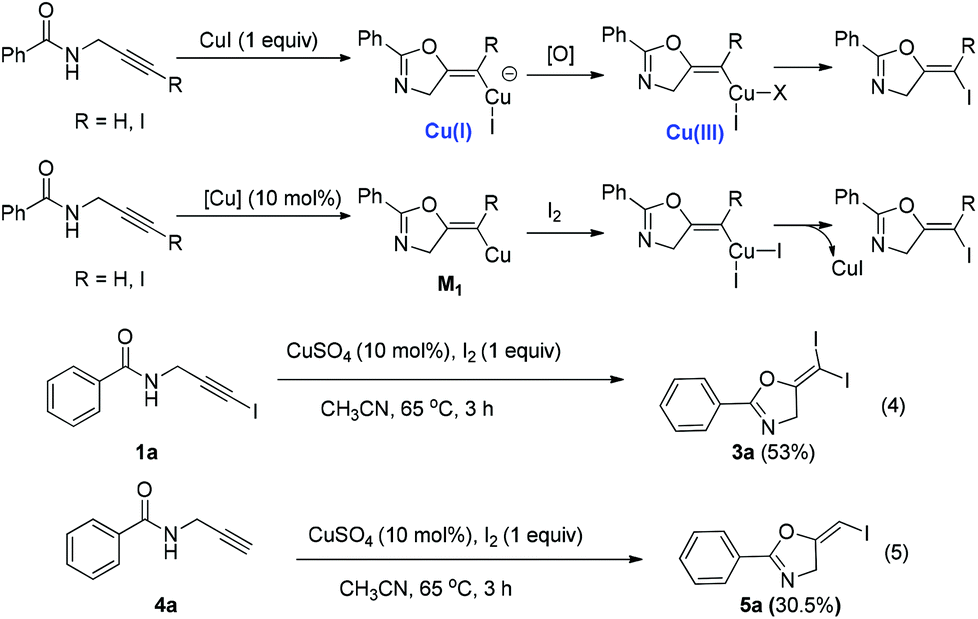
Based on these results, a plausible Cu(I)–Cu(III) catalytic cycle was proposed for this unexpected reaction (Scheme 4).12 Copper(I)-catalyzed intramolecular 5-exo-dig cyclization generated alkene copper intermediate M1. Subsequent oxidative addition with alkyne iodide 1a formed the Cu(III) intermediate M2, which went through reductive elimination generating gem-diiodide alkene 3a and another alkyne copper intermediate. Protonation of this intermediate formed the amide 4a and regenerated the copper(I) catalyst. It should be noted that Hu et al. also observed a similar mechanism in the reaction of AgCF3, benzyne and alkyne iodide.13 Also, Boger and Gevorgyan demonstrated that haloalkynes could serve as an effective source of the corresponding X+ in their earlier reports.14
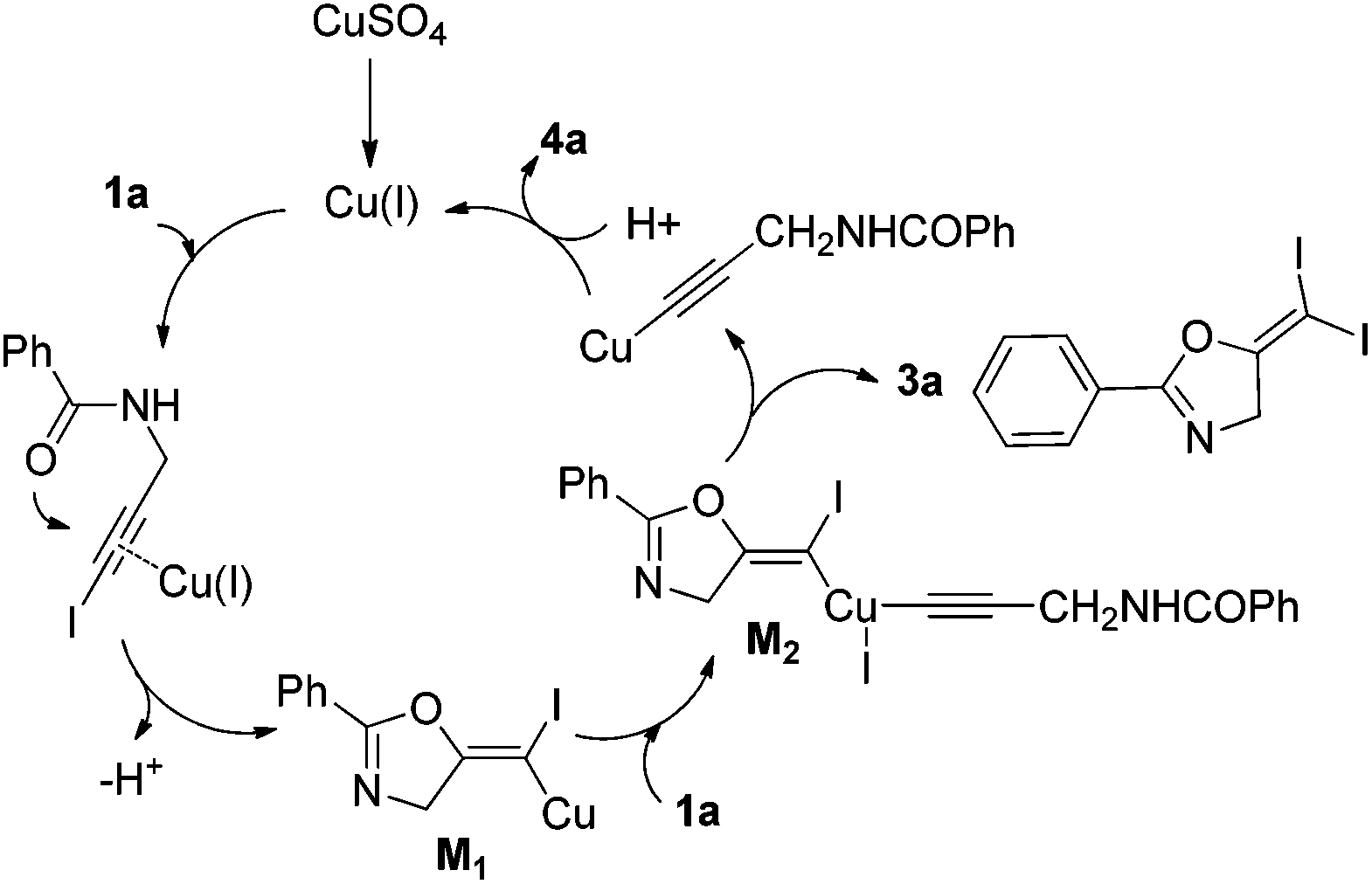 | ||
| Scheme 4 Proposed Cu(I)–Cu(III) catalytic mechanism. | ||
A screening of different copper catalysts indicated that this is a general reaction for all the copper salts tested (Table 1). Not only Cu(I), but also Cu(II) was effective for this transformation. Copper(II) was probably reduced to Cu(I) to catalyze this reaction. Among all the copper catalysts, the very cheap CuSO4 was the optimal choice and the target diiodide 3a was isolated in 95% yield in DCE solution (entry 7). Other solvents led to reduced yields (entries 10–13). A blank test showed that a copper catalyst is necessary for this transformation (entry 14).
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (0.1 mmol), [Cu] (0.01 mmol) in the solvent (1 mL) was stirred at 70 °C for 3 h. b Isolated yields based on the iodine source. | |||
| 1 | CuI | DCE | 90 |
| 2 | CuCl | DCE | 86 |
| 3 | CuBr | DCE | 72 |
| 4 | CuPF6(CH3CN)4 | DCE | 50 |
| 5 | IprCuI | DCE | 69 |
| 6 | Cu(acac)2 | DCE | 75 |
| 7 | CuSO4 | DCE | 95 |
| 8 | CuCl2 | DCE | 79 |
| 9 | Cu(OAc)2 | DCE | 75 |
| 10 | CuSO4 | CH3CN | 26 |
| 11 | CuSO4 | Dioxane | 76 |
| 12 | CuSO4 | THF | 84 |
| 13 | CuSO4 | Toluene | 75 |
| 14 | — | DCE | 0 |
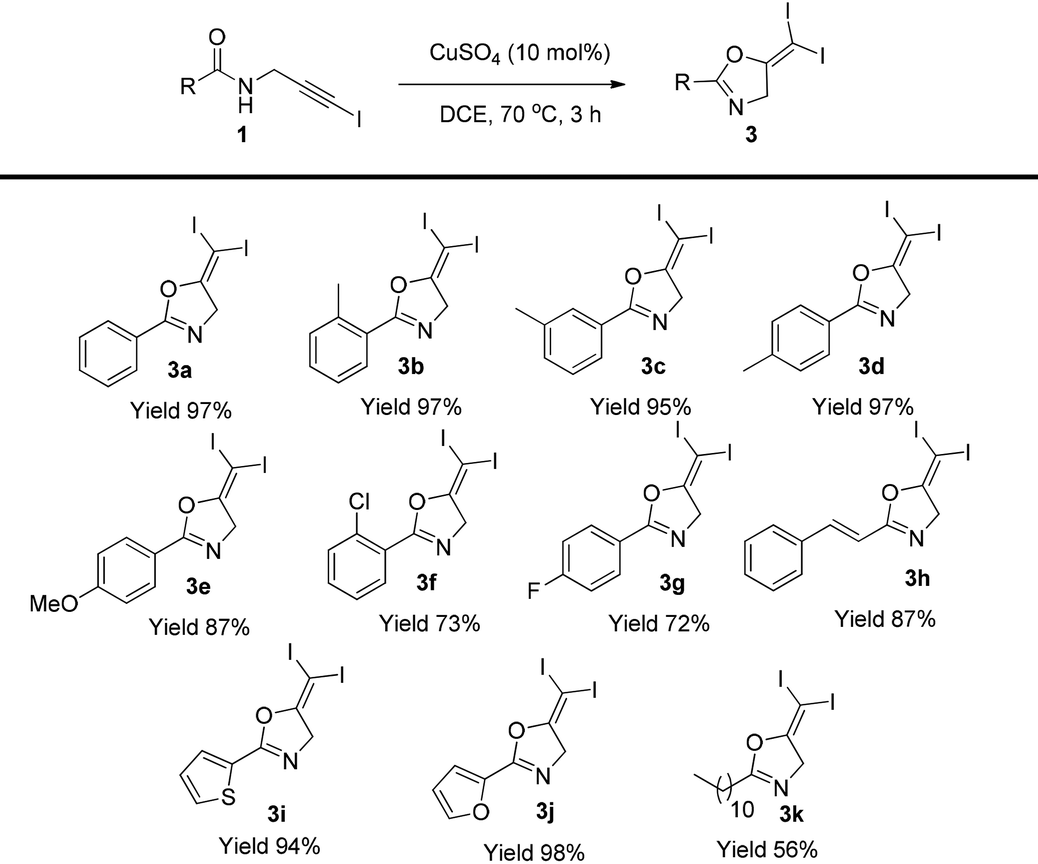
With the optimized conditions established, the substrate scope was next examined (Table 2). Different substituents at the aromatic group, including methyl, methoxyl, and halogen did not affect the reaction (3b–3g). The styryl group and heterocycles are all tolerated in this reaction and the corresponding gem-diiodides were isolated in excellent yields (3h–3j). Aliphatic substrates gave the corresponding product in reduced yield (3k).
| a Reaction conditions: 1 (0.2 mmol), CuSO4 (0.02 mmol) in DCE (2 mL) was stirred at 70 °C for 3 h. Isolated yields were reported. |
|---|
|
2.2. Improved oxidative iodination toward gem-diiodoalkenes
In the above transformation, the yield was almost quantitatively calculated on the iodine element. However, half of the substrates were transformed into another propargylic amide 4a. To improve this atom economy issue, we proposed another reaction pathway to mimic the previous Cu(I)/Cu(III) cycle: CuI catalyzed a similar cyclization of 1a to form M1. This Cu(I) intermediate M1 could be oxidized into a similar Cu(III) intermediate by adding another strong oxidant, which also underwent reductive elimination to form the gem-diiodide 3a. Herein, CuI served as the cyclization catalyst and also the iodine source, so one equivalent of CuI is necessary. Moreover, to inhibit the oxidative addition of alkyne idodides to M1, the choice of oxidant is very crucial for the success of this transformation. Then, a series of oxidants were tested (Table 3). It was found that selectfluor was the best oxidant, leading to the target 3a in 84% yield (entry 1). Other oxidants such as PhI(OAc)2, DDQ, MCPBA, TBHP, H2O2 all led to reduced yields.|
|
||
|---|---|---|
| Entry | Oxidant (1.2 equiv.) | Yieldb (%) |
| a Reaction conditions: 1a (0.1 mmol), CuI (0.1 mmol) , oxidant (0.12 mmol) in the solvent (1 mL) was stirred at 70 °C for 3 h. b Isolated yields were reported. | ||
| 1 | Selectfluor | 84 |
| 2 | PhI(OAc)2 | 43 |
| 3 | DDQ | 28 |
| 4 | K2S2O8 | 51 |
| 5 | 3-Chloroperbenzoic acid | 57 |
| 6 | tert-Butyl hydroperoxide | 37 |
| 7 | H2O2 | 12 |
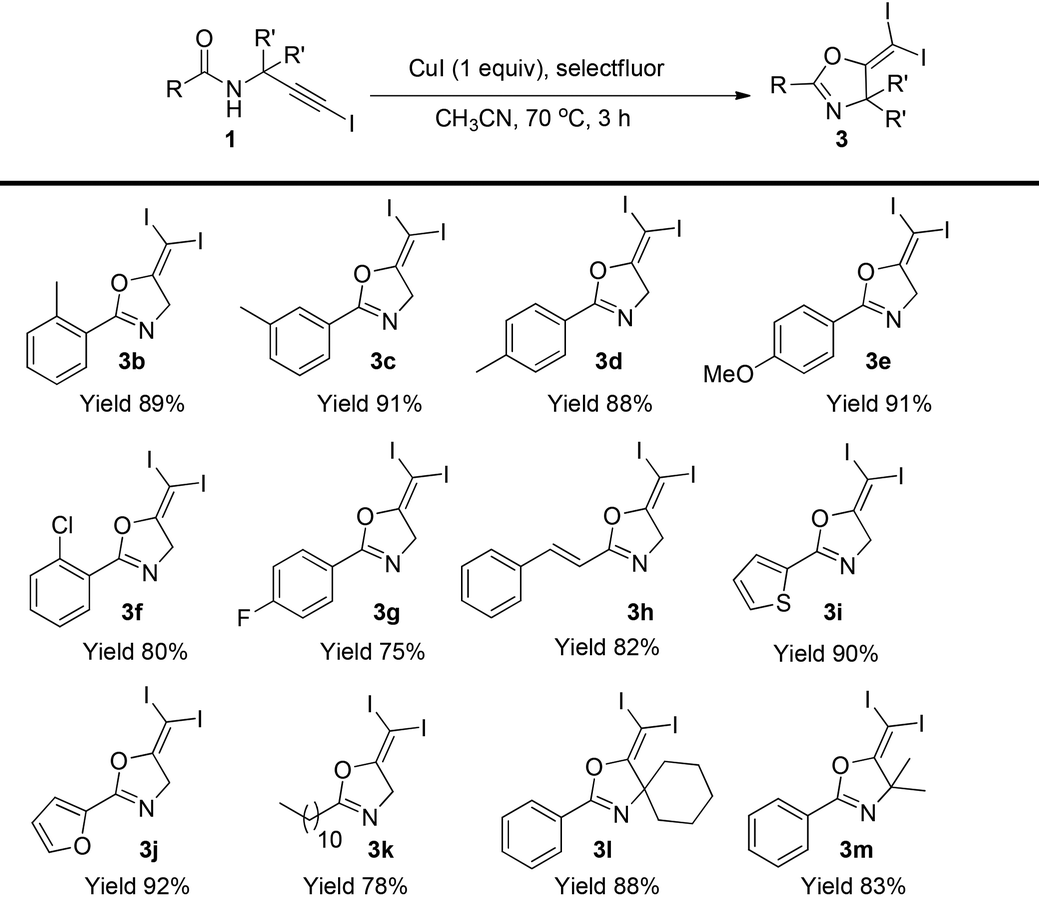
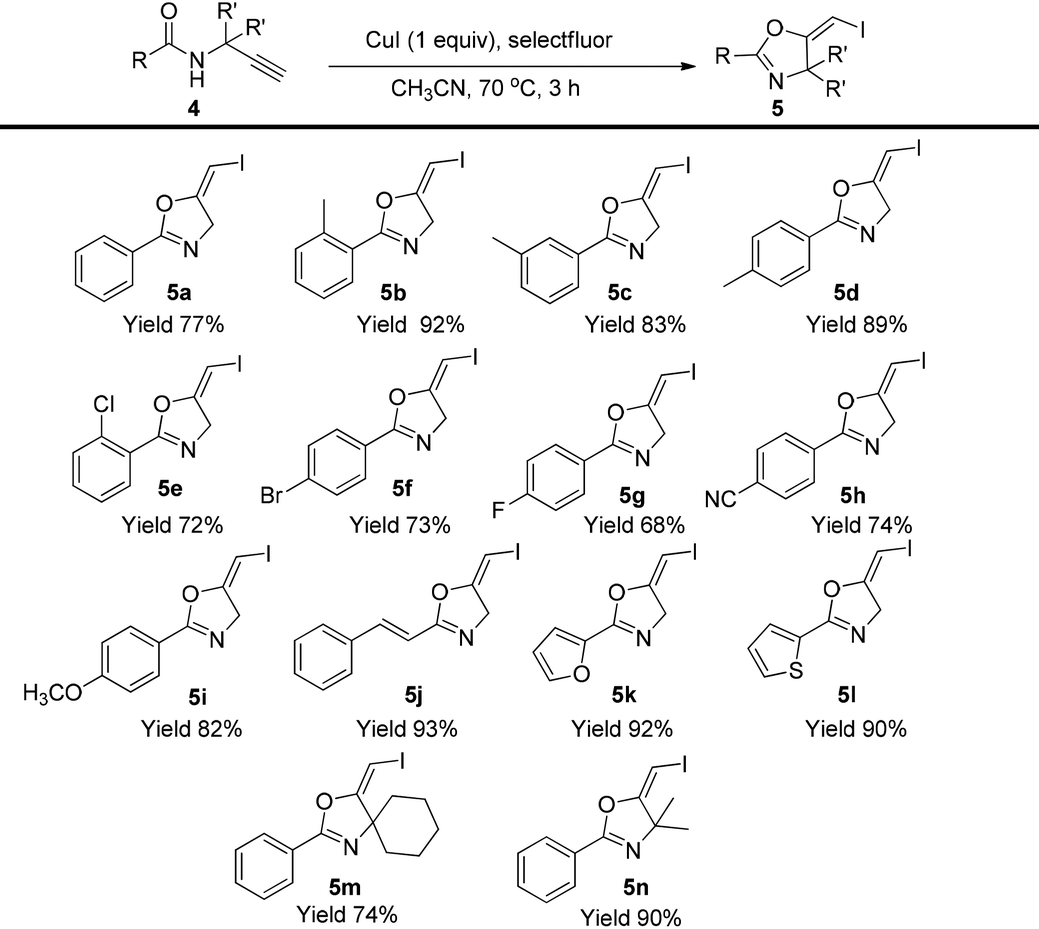
Then, the scope of this protocol was also tested (Table 4). Similarly, this reaction showed a very general scope. Different aromatic substituents, the styryl group, furan or thiophene substituents do not affect the reaction efficiency, and various gem-diiodide alkenes were prepared in good to excellent yields with this very simple procedure (3b–3k). Substrates bearing a quaternary carbon centre also reacted very well, generating the corresponding products in excellent yields (3l–3m). All these reactions did not require an inert atmosphere. Thus, such a simple procedure and mild conditions make this protocol very practical in organic syntheses.
| a Reaction conditions: 1a (0.2 mmol), CuI (0.2 mmol), selectfluor (0.24 mmol), in CH3CN (2 mL) was stirred at 70 °C for 3 h. Isolated yields were reported. |
|---|
|
2.3. Synthesis of (E)-1-iodo-1-alkenes with the oxidative iodination approach
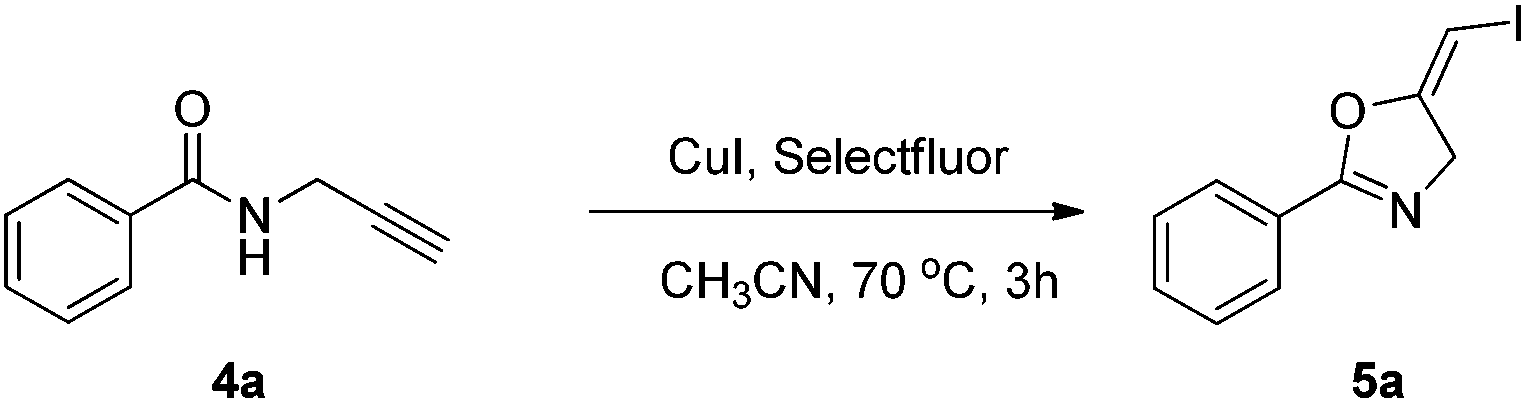
In the gold-catalyzed cyclization reaction of 1a, (Z)-alkene 2a was observed. Similarly iodoalkyne 6 went through 5-exo-dig cyclization in the presence of a Ph3PAuNTf2 catalyst, giving a stable (Z)-alkene 7 in 83% yield (eqn (3)). If the current CuI mediated oxidative iodination reaction is applied to the propargylic amide 4a, the (E)-iodoalkene will be obtained. To our delight, this protocol works equally well on this substrate, generating the (E)-iodoalkene 5a in 77% yield under standard conditions. Other substituted substrates also worked very well and the results are summarized in Table 5. Thus, both configurations of the iodoalkenes could be obtained by gold or copper catalysis, which provided different options for synthetic chemists.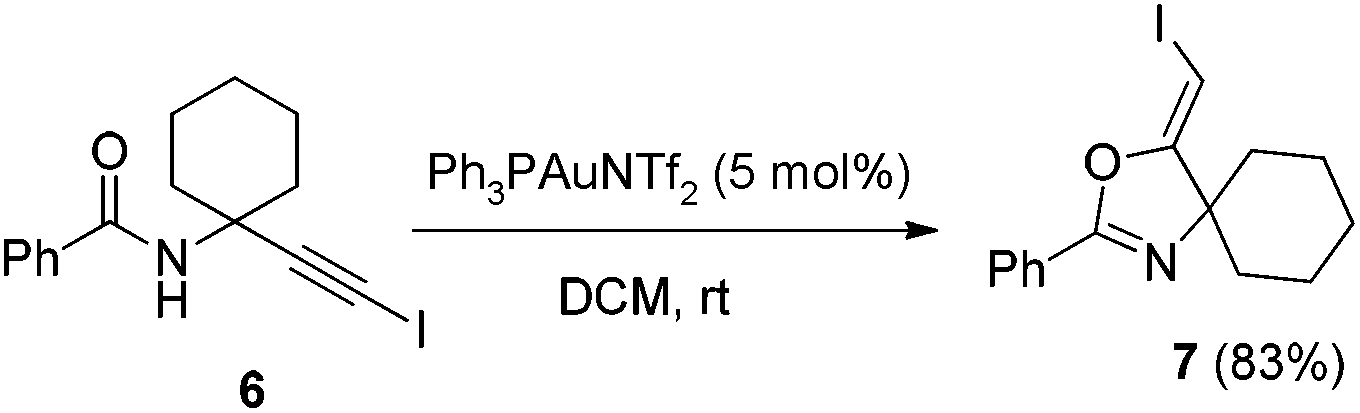 | (3) |
| a Reaction conditions: 1a (0.2 mmol), CuI (0.2 mmol), selectfluor (0.24 mmol), in CH3CN (2 mL) was stirred at 70 °C for 3 h. Isolated yields were reported. |
|---|
|
In the current reaction, CuI catalyzed the first cyclization forming the Cu(I) intermediate, which was oxidized to a Cu(III) intermediate. It went through reductive elimination to form the final products (Scheme 5). When I2 was used as the oxidant and also served as the iodine source, we were able to realize this reaction with only a catalytic amount of the Cu(I) catalyst. We tried the reaction using one equivalent of I2 in the presence of CuSO4 (10 mol%) as the catalyst (eqn (4) and (5)). The expected reactions did happen, however, relatively lower yields were obtained. All these data demonstrated the viability of a Cu(I)/Cu(III) catalytic cycle of this reaction.
 | ||
| Scheme 5 Iodination with a catalytic amount of Cu(I) catalyst. | ||
2.4. Synthesis of trisubstituted and tetrasubstituted alkenes from iodoalkenes
In the next step, we took advantage of the C–I bond to install different functional groups to construct multisubstituted alkenes. Trisubstituted and tetrasubstituted alkenes are important motifs present in bioactive products and also organic functional materials. The regio and stereoselective synthesis of these molecules is still one of the most challenging subjects in organic syntheses.15 The obtained halogen substituted alkenes serve as a perfect template for us to install different functional groups using the palladium-catalyzed cross coupling reactions. Firstly, we chose the Suzuki reaction to install an aromatic group. As shown in Scheme 6, boronic acids coupled with (E)-alkene 5n and (Z)-alkene 7 smoothly and afforded the expected trisubstituted alkenes 8 and 12 in good yields. The Heck reaction of (E)-alkene 5n and (Z)-alkene 7 with methyl acrylate are also successful, giving the corresponding dienes in 84% and 76% yields. The palladium-catalyzed Sonogashira coupling reaction between 5n and a terminal alkyne was also successful and the expected enyne product 10 was isolated in 75% yield.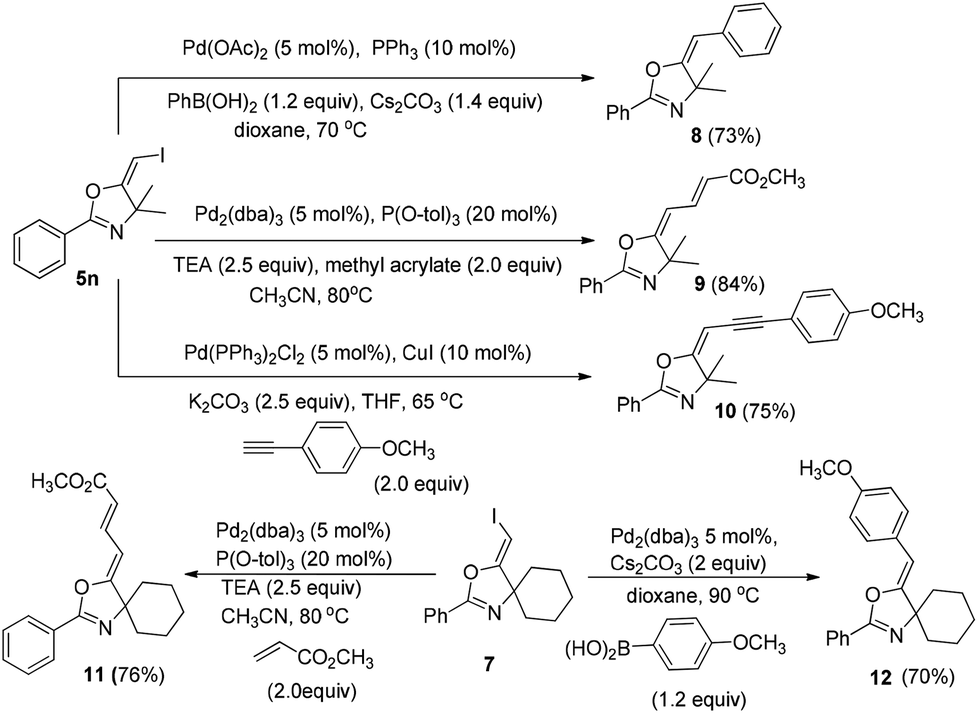 | ||
| Scheme 6 Synthetic transformations of E-iodoalkene and Z-iodoalkene. | ||
Then, we started to investigate whether we could construct tetra-substituted alkenes from gem-diiodoalkenes (Scheme 7). The Suzuki reaction of diiodide 3l and boronic acid in the presence of Pd2(dba)3 and P(o-tol)3 afforded tetrasubstituted alkene 13 in 65% yield. The corresponding Heck reaction and Sonogashira coupling reaction of 3l were also successful, giving the conjugated triene 14 and enyne 15 in good yields. The structures are characterized by NMR and Mass experiments, and triene 14 was further confirmed by single crystal X-ray analysis.16
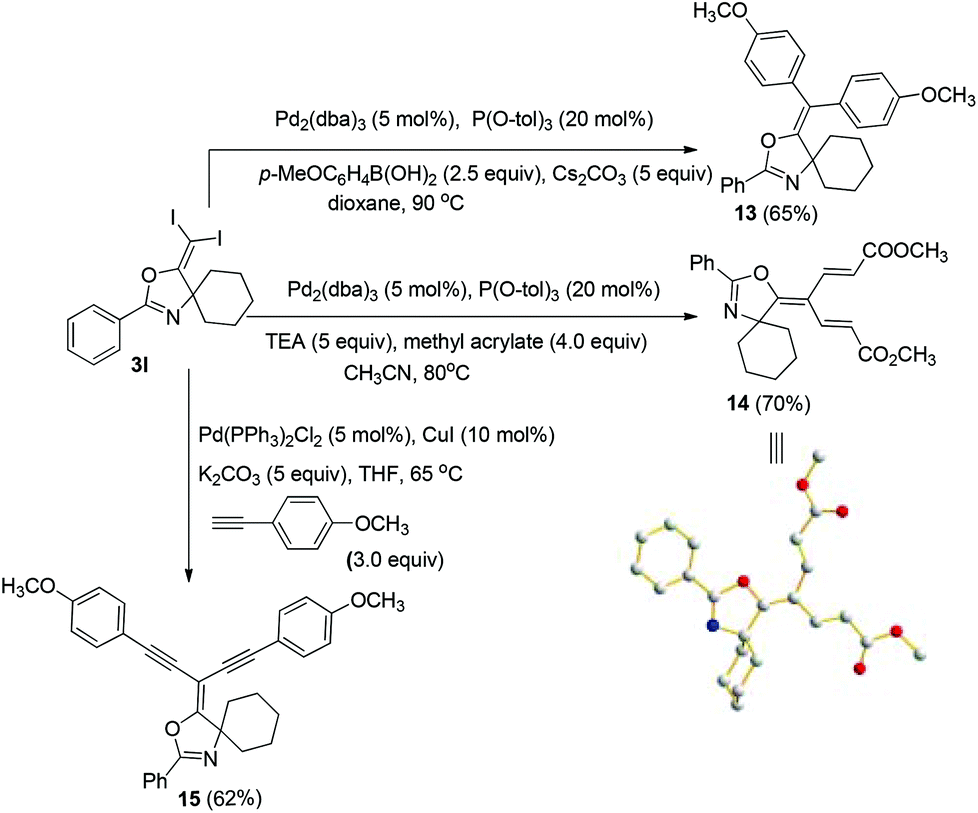 | ||
| Scheme 7 Synthesis of functionalized tetrasubstituted alkenes from gem-diiodoalkene. | ||
3. Conclusions
In summary, starting from an unexpected reaction of propargyl amides, we developed a new approach to the synthesis of gem-diiodoalkenes. This reaction features a very simple procedure, high efficiency and broad scope. Stereoselective synthesis of (E) or (Z)-iodoalkenes could also be achieved with copper catalysis or gold catalysis. A possible Cu(I)/Cu(III) catalytic cycle was proposed. The obtained iodoalkenes were successfully used in the further palladium-catalyzed Suzuki, Heck, and Sonogashira coupling reactions, allowing access to a wide variety of multi-substituted alkenes, dienes, trienes and enynes.4. Experimental section
4.1 General procedure for the synthesis of 1-iodoalkynyl amides 1a–1m
The corresponding propynylamine (10 mmol) was dissolved in MeOH (30 mL). A solution of KOH (40 mmol) in H2O (10 mL) was prepared, cooled to 0 °C, and added to the reaction mixture. I2 (10 mmol) was added in one portion, and the solution was stirred at room temperature overnight. The reaction mixture was extracted with CH2Cl2. The organic phase was washed with saturated Na2S2O3 solution, dried over Na2SO4, filtered, and concentrated in vacuo to yield the crude 1-iodopropynylamine. To a cooled solution of 1-iodopropynylamine in CH2Cl2 (20 mL) were added triethylamine (24 mmol), chloride (8 mmol) and 4-dimethylaminopyridine (0.8 mmol) and the resulting solution was allowed to reach room temperature. The reaction was stirred at room temperature for 3 h. Then it was diluted with water and the aqueous layer extracted with CH2Cl2. The combined organic layers were washed with satd. NaHCO3 followed by water and brine, dried over Na2SO4 and concentrated under reduced pressure to obtain the crude 1-iodoalkynyl amides. Silica gel chromatography gave the desired 1-iodoalkynyl amides 1a–1m in 45%–72% yields respectively.
4.2. General procedure for the synthesis of gem-diiodoalkenes (Table 2)
Compound 1a (0.2 mmol) was dissolved in DCE (1 mL), CuSO4 (0.02 mmol) was added and the system was stirred at 70 °C for 3 h. The resulting mixture was washed with water and extracted with DCM. The organic layer was filtered on Celite and evaporated under reduced pressure. Purification by flash chromatography afforded the desired product 3a (38.95 mg, 95%). 1H NMR (400 MHz, DMSO-d6) δ 7.96 (m, 2H), 7.54 (t, J = 7.4 Hz, 1H), 7.46 (t, J = 7.8 Hz, 2H), 4.61 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 161.39, 159.16, 132.92, 129.51, 127.92, 126.39, 61.64, −14.83. HRMS (ESI, m/z) calcd for C10H7I2NO [M + H]+ 411.8690, found 411.8675.
4.3. General procedure for the synthesis of gem-diiodoalkenes (Table 4)
Compound 1a (0.2 mmol), CuI (0.2 mmol) and selectfluor (0.24 mmol) were dissolved in CH3CN (2 mL). The system was stirred at 70 °C for 3 h. The resulting mixture was washed with water and extracted with DCM. The organic layer was filtered on Celite and evaporated under reduced pressure. Purification by flash chromatography afforded the desired product 3a (69.05 mg, 84%).
4.4. General procedure for the synthesis of (E)-1-iodo-1-alkenes (Table 5)
Compound 4a (0.2 mmol), CuI (0.2 mmol) and selectfluor (0.24 mmol) were dissolved in CH3CN (2 mL). The system was stirred at 70 °C for 3 h. The resulting mixture was washed with water and extracted with DCM. The organic layer was filtered on Celite and evaporated under reduced pressure. Purification by flash chromatography afforded the desired product 5a (43.89 mg, 77%). 1H NMR (400 MHz, CDCl3) δ 7.96 (d, J = 7.5 Hz, 2H), 7.55–7.33 (m, 3H), 5.77 (t, J = 2.8 Hz, 1H), 4.63 (d, J = 3.1 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 162.39, 157.43, 132.27, 128.93, 127.48, 126.08, 60.44, 49.30. HRMS (ESI, m/z) calcd for C10H8INO [M + H]+ 285.9723, found 285.9723.
4.5. General procedure for the gold-catalyzed synthesis of (Z)-4-(iodomethylene)-2-phenyl-3-oxa-1-azaspiro[4.5]dec-1-ene (7)
The compound 6 (1 mmol) was dissolved in DCM (10 ml) and PPh3AuNTf2 (0.05 mmol) was added. After stirring for 2 h at room temperature, water was added and the aqueous phase was extracted twice with DCM. After drying of the combined organic phases over Na2SO4, and filtration, the solvent was removed and the crude products were purified by column chromatography affording the pure product 7 (0.29 g, 83%). 1H NMR (400 MHz, CDCl3) δ 8.02 (d, J = 7.1 Hz, 2H), 7.51–7.42 (m, 3H), 5.05 (s, 1H), 1.89–1.52 (m, 10H). 13C NMR (100 MHz, CDCl3) δ 168.38, 158.03, 131.87, 128.51, 128.37, 126.66. HRMS (ESI, m/z) calcd for C15H16INO [M + H]+ 354.0349, found 354.0346.
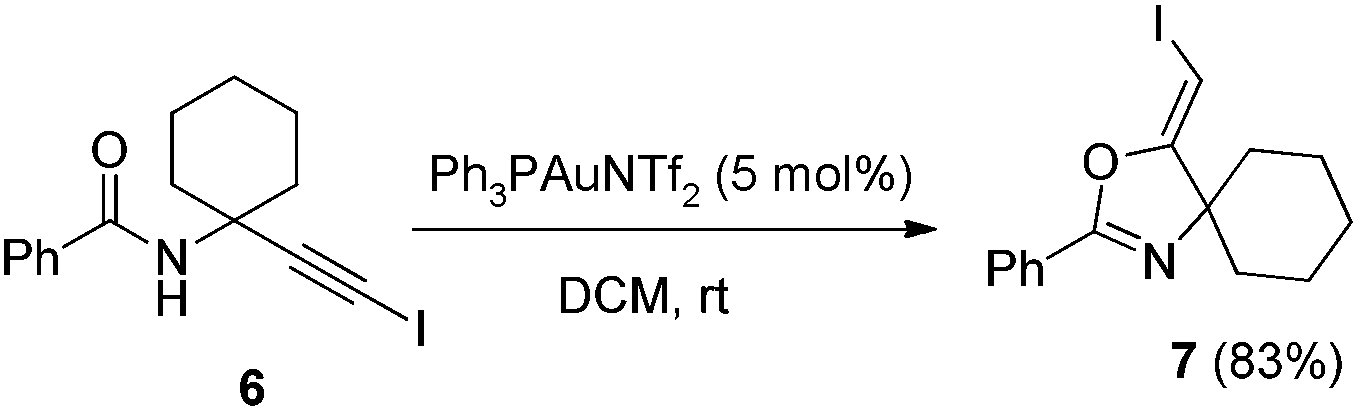
4.6. General procedure for the Suzuki reactions: synthesis of (E)-5-benzylidene-4,4-dimethyl-2-phenyl-4,5-dihydro-oxazole (6)
To a mixture of Pd(OAc)2 (0.01 mmol), PPh3 (0.02 mmol), Cs2CO3 (0.28 mmol), and PhB(OH)2 (0.24 mmol) in dixoane (2 ml) under a N2 atmosphere, compound 5n (0.2 mmol) was added. The system was stirred at 70 °C overnight. The resulting mixture was washed with water and extracted with DCM. The organic layer was filtered on Celite and evaporated under reduced pressure. Purification by flash chromatography afforded the desired product 8 (38.41 mg, 73%).1H NMR (300 MHz, CDCl3) δ 8.10–8.08 (m, 2H), 7.65 (d, J = 7.2 Hz, 2H), 7.55–7.52 (m, 3H), 7.50–7.37 (m, 2H), 7.25–7.20 (m, 1H), 5.55 (s, 1H), 1.54 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 160.15, 159.36, 134.53, 131.36, 128.11, 128.04, 127.75, 127.41, 126.36, 125.66, 98.87, 70.39, 29.17. HRMS (ESI, m/z) calcd for C18H17NO [M + H]+ 264.1383, found 264.1389.
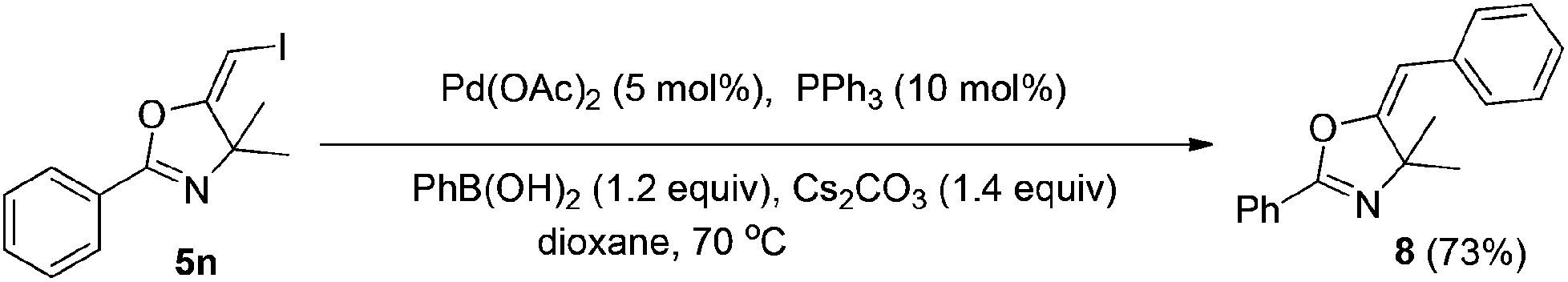
4.7. General procedure for the Heck coupling reactions: synthesis of (2E,4E)-methyl 4-(4,4-dimethyl-2-phenyloxazol-5(4H)-ylidene)but-2-enoate (7)
To a mixture of Pd2(dba)3 (0.01 mmol), P(O-tol)3 (0.04 mmol), and compound 5n (0.2 mmol) in CH3CN (2 ml) under a N2 atmosphere, methyl acrylate (0.4 mmol) and Et3N (0.5 mmol) were added. The system was stirred at 80 °C overnight. The resulting mixture was washed with water and extracted with DCM. The organic layer was filtered on Celite and evaporated under reduced pressure. Purification by flash chromatography afforded the desired product 9 (45.53 mg, 84%).1H NMR (400 MHz, CDCl3) δ 7.96 (d, J = 7.3 Hz, 2H), 7.62–7.50 (m, 2H), 7.46–7.42 (m, 2H), 6.11 (d, J = 12.6 Hz, 1H), 5.83 (d, J = 15.0 Hz, 1H), 3.76 (s, 3H), 1.65 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 169.70, 167.16, 159.17, 138.64, 132.00, 128.58, 128.13, 126.24, 118.47, 101.13, 70.76, 51.46, 51.47, 28.55. HRMS (ESI, m/z) calcd for C16H17NO3 [M + H]+ 272.1281, found 264.1271.
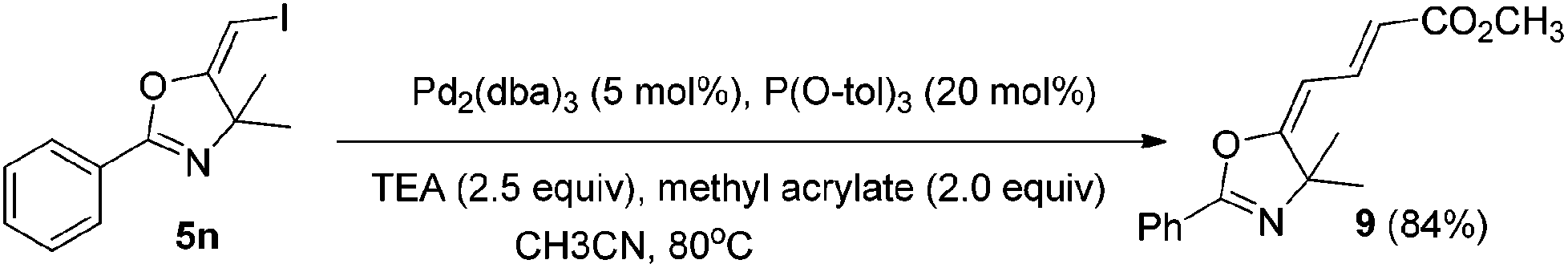
4.8. General procedure for the Sonogashira coupling reactions: synthesis of (E)-5-(3-(4-methoxyphenyl)prop-2-yn-1-ylidene)-4,4-dimethyl-2-phenyl-4,5-dihydrooxazole (8)
To a mixture of Pd(PPh3)2Cl2 (0.01 mmol), CuI (0.02 mmol), K2CO3 (0.5 mmol), and compound 5n (0.2 mmol) in THF (2 mL) under a N2 atmosphere, 4-methoxyphenylacetylene (0.4 mmol) was added. The system was stirred at 65 °C overnight. The resulting mixture was washed with water and extracted with DCM. The organic layer was filtered on Celite and evaporated under reduced pressure. Purification by flash chromatography afforded the desired product 10 (47.55 mg, 75%).1H NMR (400 MHz, CDCl3) δ 7.97 (d, J = 7.4 Hz, 2H), 7.53–7.49 (m, 1H), 7.44 (t, J = 7.6 Hz, 2H), 7.36 (d, J = 8.6 Hz, 2H), 6.85 (d, J = 8.6 Hz, 2H), 5.60 (s, 1H), 3.81 (s, 3H), 1.73 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 170.74, 158.88, 158.69, 131.77, 131.36, 128.03, 127.64, 125.97, 115.47, 113.56, 93.06, 84.99, 81.73, 70.67, 54.82, 25.85. HRMS (ESI, m/z) calcd for C21H19NO2 [M + H]+ 318.1489, found 318.1483.
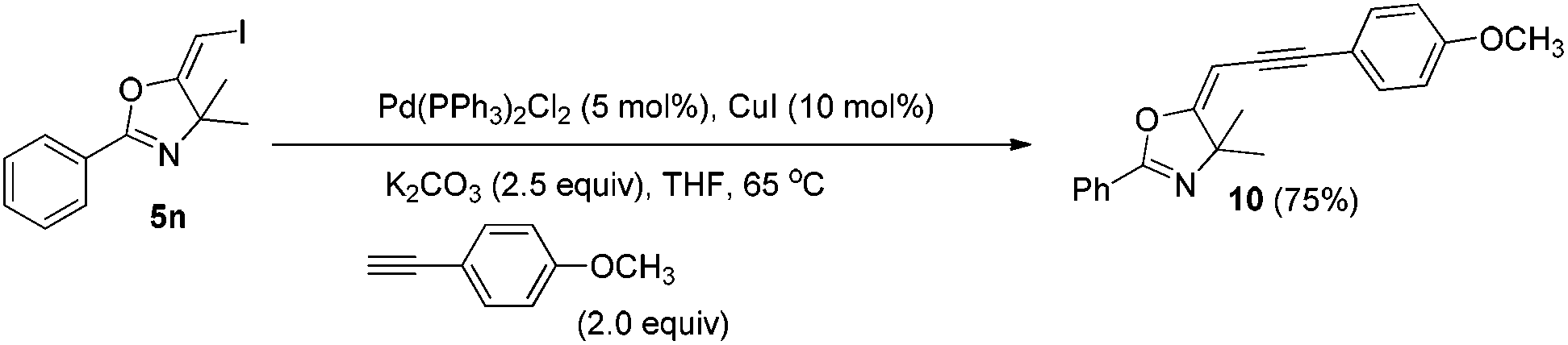
Acknowledgements
We are grateful for financial support from the fundamental research and subject construction funds of Shandong University (no 2014JC008, 104.205.2.5), and the china postdoctoral science foundation. We thank Prof. Dr Di Sun for the analysis of X-ray structure.Notes and references
- (a) D. Arlt, M. Jautelat and R. Lantzsch, Angew. Chem., Int. Ed. Engl., 1981, 20, 703 CrossRef; (b) C. E. Salomon, D. H. Williams and D. J. Faulkner, J. Nat. Prod., 1995, 58, 1463 CrossRef CAS.
- B. Xu and S. Ma, Chin. J. Org. Chem., 2001, 4, 252 Search PubMed.
- (a) E. J. Corey and P. L. Fuchs, Tetrahedron Lett., 1972, 13, 3769 CrossRef; (b) L. F. Tietze, T. Noebel and M. Spescha, J. Am. Chem. Soc., 1998, 120, 8971 CrossRef CAS; (c) M. Braun, Angew. Chem., Int. Ed., 1998, 37, 430 CrossRef; (d) S. Ma and B. Xu, J. Org. Chem., 1998, 63, 9156 CrossRef CAS; (e) S. Ma, B. Xu and B. Ni, J. Org. Chem., 2000, 65, 8532 CrossRef CAS PubMed.
- S. Hirashima, S. Aoyagi and C. Kibayashi, J. Am. Chem. Soc., 1999, 121, 9873 CrossRef CAS.
- (a) Y. Fang and M. Lautens, Org. Lett., 2005, 7, 3549 CrossRef CAS PubMed; (b) M. Nagamochi, Y. Fang and M. Lautens, Org. Lett., 2007, 9, 2955 CrossRef CAS PubMed; (c) C. S. Bryan and M. Lautens, Org. Lett., 2008, 10, 4633 CrossRef CAS PubMed; (d) S. G. Newman, V. Aureggi, C. S. Bryan and M. Lautens, Chem. Commun., 2009, 5236 RSC; (e) C. S. Bryan, J. A. Braunger and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 7064 CrossRef CAS PubMed; (f) W. Chen, Y. Zhang, L. Zhang, M. Wang and L. Wang, Chem. Commun., 2011, 47, 10476 RSC.
- (a) S. Ye, J. Liu and J. Wu, Chem. Commun., 2012, 48, 5028 RSC; (b) S. Ye, X. Yang and J. Wu, Chem. Commun., 2010, 46, 2950 RSC; (c) Z. Wang, J. Yang, F. Yang and W. Bao, Org. Lett., 2010, 12, 3034 CrossRef CAS PubMed; (d) W. Chen, M. Wang, P. Li and L. Wang, Tetrahedron, 2011, 67, 5913 CrossRef CAS PubMed.
- (a) F. Ramirez, N. B. Desai and N. Mckelvie, J. Am. Chem. Soc., 1962, 84, 1745 CrossRef CAS; (b) V. K. Aggarwal and A. Mereu, J. Org. Chem., 2000, 65, 7211 CrossRef CAS; (c) E. Klaps and W. Schmid, J. Org. Chem., 1999, 64, 7537 CrossRef CAS; (d) J. Barluenga, M. A. Rodriguez, P. J. Campos and G. Asensio, J. Am. Chem. Soc., 1988, 110, 5567 CrossRef CAS.
- W. Wu and H. Jiang, Acc. Chem. Res., 2014, 47, 2483 CrossRef CAS PubMed.
- (a) Z. Chen, H. Jiang, Y. Li and C. Qi, Chem. Commun., 2010, 46, 8049 RSC; (b) Y. Li, X. Liu, D. Ma, B. Liu and H. Jiang, Adv. Synth. Catal., 2012, 354, 2683 CrossRef CAS; (c) Y. Li, X. Liu, H. Jiang and Z. Feng, Angew. Chem., Int. Ed., 2010, 49, 3338 CrossRef CAS PubMed; (d) J. E. Hein, J. C. Tripp, L. B. Krasnova, K. B. Sharpless and V. V. Fokin, Angew. Chem., Int. Ed., 2009, 48, 8018 CrossRef CAS PubMed; (e) Y. Gao, M. Yin, W. Wu, H. Huang and H. Jiang, Adv. Synth. Catal., 2013, 355, 2263 CrossRef CAS; (f) K. C. Nicolaou, Y. Tang and J. Wang, Angew. Chem., Int. Ed., 2009, 48, 3449 CrossRef CAS PubMed; (g) Y. Li, X. Liu, H. Jiang, B. Liu, Z. Chen and P. Zhou, Angew. Chem., Int. Ed., 2011, 50, 6341 CrossRef CAS PubMed; (h) K. Villeneuve, N. Riddell, R. W. Jordan, G. C. Tsui and W. Tam, Org. Lett., 2004, 6, 4543 CrossRef CAS PubMed.
- (a) X. Wang, Z. Yao, S. Dong, F. Wei, H. Wang and Z. Xu, Org. Lett., 2013, 15, 2234 CrossRef CAS PubMed; (b) X. Wang, S. Dong, Z. Yao, L. Feng, P. Daka, H. Wang and Z. Xu, Org. Lett., 2014, 16, 22 CrossRef CAS PubMed; (c) S. Zhang, Z. Xu, J. Jia, C.-H. Tung and Z. Xu, Chem. Commun., 2014, 50, 12084 RSC; (d) S. Zhang, F. Wei, C. Song, J. Jia and Z. Xu, Chin. J. Chem., 2014, 32, 937 CrossRef CAS; (e) F. Wei, X. Wang, S. Peng, Q. Jia, C. Jiang, S. Liu and Z. Xu, Synthesis, 2014, 2168 CAS; (f) C. Song, L. Ju, M. Wang, P. Liu, Y. Zhang, J. Wang and Z. Xu, Chem. – Eur. J., 2013, 19, 3584 CrossRef CAS PubMed.
- For reviews on the reaction of propargylic amides, see: (a) Y. Hu, X. Xin and B. Wan, Tetrahedron Lett., 2015, 56, 32 CrossRef CAS PubMed; for recent examples, see: (b) Y. Hu, R. Yi, C. Wang, X. Xin, F. Wu and B. Wan, J. Org. Chem., 2014, 79, 3052 CrossRef CAS PubMed; (c) A. Alhalib and W. J. Moran, Org. Biomol. Chem., 2014, 12, 795 RSC; (d) X. Yu, X. Xin, B. Wan and X. Li, J. Org. Chem., 2013, 78, 4895 CrossRef CAS PubMed; (e) A. S. K. Hashmi, M. C. B. Jaimes, A. M. Schuster and F. Rominger, J. Org. Chem., 2012, 77, 6394 CrossRef CAS PubMed; (f) G. C. Senadi, W.-P. Hu, J.-S. Hsiao, J. K. Vandavasi, C.-Y. Chen and J.-J. Wang, Org. Lett., 2012, 14, 4478 CrossRef CAS PubMed; (g) O. A. Egorova, H. Seo, Y. Kim, D. Moon, Y. M. Rhee and K. H. Ahn, Angew. Chem., Int. Ed., 2011, 50, 11446 CrossRef CAS PubMed; (h) S. Yasuhara, M. Sasa, T. kusakabe, H. Takayama, M. Kimura, T. Mochida and K. Kato, Angew. Chem., Int. Ed., 2011, 50, 3912 CrossRef CAS PubMed; (i) A. S. K. Hashmi, A. M. Schuster, M. Schmuck and F. Rominger, Eur. J. Org. Chem., 2011, 4595 CrossRef CAS; (j) J. P. Weyrauch, A. S. K. Hashmi, A. Schuster, T. Hengst, S. Schetter, A. Littmann, M. Rudolph, M. Hamzic, J. Visus, F. Rominger, W. Frey and J. W. Bats, Chem. – Eur. J., 2010, 16, 956 CrossRef CAS PubMed; (k) A. S. K. Hashmi, A. M. Schuster and F. Rominger, Angew. Chem., Int. Ed., 2009, 48, 8247 CrossRef CAS PubMed; (l) Y.-M. Pan, F.-J. Zheng, H.-X. Lin and Z.-P. Zhan, J. Org. Chem., 2009, 74, 3148 CrossRef CAS PubMed; (m) P.-Y. Coqueron, C. Didier and M. A. Ciufolini, Angew. Chem., Int. Ed., 2003, 42, 1411 CrossRef CAS PubMed.
- For reviews, see: (a) S. E. Allen, R. R. Walvoord, R. Padilla-Salinas and M. C. Kozlowski, Chem. Rev., 2013, 113, 6234 CrossRef CAS PubMed; (b) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062 CrossRef CAS PubMed; (c) A. Casitas and X. Ribas, Chem. Sci., 2013, 4, 2301 RSC; (d) A. J. Hickman and M. S. Sanford, Nature, 2012, 484, 177 CrossRef CAS PubMed; (e) M.-X. Wang, Acc. Chem. Res., 2012, 45, 182 CrossRef CAS PubMed; For recent examples, see: (f) H. Zhang, B. Yao, L. Zhao, D. Wang, B. Xu and M.-X. Wang, J. Am. Chem. Soc., 2014, 136, 6326 CrossRef CAS PubMed; (g) A. M. Suess, M. Z. Ertem, C. J. Cramer and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 9797 CrossRef CAS PubMed; (h) F. Zhou, C. Tan, J. Tang, Y. Zhang, W. Gao, H. Wu, Y. Yu and J. Zhou, J. Am. Chem. Soc., 2013, 135, 10994 CrossRef CAS PubMed; (i) B. Chen, X.-L. Hou, Y.-X. Li and Y.-D. Wu, J. Am. Chem. Soc., 2011, 133, 7668 CrossRef CAS PubMed; (j) M. Shang, H. Wang, S. Sun, H.-X. Dai and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 11590 CrossRef CAS PubMed; (k) Y. Yan, Y. Yuan and N. Jiao, Org. Chem. Front., 2014, 1, 1176 RSC; (l) X. Yin, Y. Li, J. Yuan, W.-J. Gu and B.-F. Shi, Org. Chem. Front., 2015, 2, 119 RSC; (m) C. Tong and N. Jiao, J. Am. Chem. Soc., 2012, 134, 18924 CrossRef PubMed; (n) L. Chu and F.-L. Qing, J. Am. Chem. Soc., 2010, 132, 7262 CrossRef CAS PubMed; (o) Z.-L. Wang, L. Zhao and M.-X. Wang, Org. Lett., 2012, 14, 1472 CrossRef CAS PubMed; (p) B. Yao, D.-X. Wang, Z.-T. Huang and M.-X. Wang, Chem. Commun., 2009, 2899 RSC; (q) X. Jiang, Z. Chen, X. Xu and F.-L. Qing, Org. Chem. Front., 2014, 1, 774 RSC.
- Y. Zeng, L. Zhang, Y. Zhao, C. Ni and J. Hu, J. Am. Chem. Soc., 2013, 135, 2955 CrossRef CAS PubMed.
- (a) D. L. Boger, S. R. Brunette and R. M. Garbaccio, J. Org. Chem., 2001, 66, 5163 CrossRef CAS PubMed; (b) A. Trofimov, N. Chernyak and V. Gevorgyan, J. Am. Chem. Soc., 2008, 130, 13538 CrossRef CAS PubMed.
- (a) A. B. Flynn and W. W. Ogilvie, Chem. Rev., 2007, 107, 4698 CrossRef CAS PubMed; (b) K. Itami, T. Kamei and J. Yoshida, J. Am. Chem. Soc., 2003, 125, 14670 CrossRef CAS PubMed; (c) D. Zhang, G. Xu, D. Ding, C. Zhu, J. Li and J. Sun, Angew. Chem., Int. Ed., 2014, 53, 11070 CrossRef CAS PubMed.
- CCDC 1046596 (3a) and 1046597 (14) contain the supplementary crystallographic data for this paper.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1046596 and 1046597. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00043b |
| This journal is © the Partner Organisations 2015 |