
A concise formal synthesis of platencin†
Jie
Wang
a,
Wang-Bin
Sun
ab,
Ying-Zi
Li
a,
Xuan
Wang
a,
Bing-Feng
Sun
*a,
Guo-Qiang
Lin
a and
Jian-Ping
Zou
b
aCAS Key Laboratory of Synthetic Chemistry of Natural Substances, Shanghai Institute of Organic Chemistry, 345 Lingling Road, Shanghai 200032, China. E-mail: bfsun@sioc.ac.cn
bKey Laboratory of Organic Synthesis of Jiangsu Province, College of Chemistry and Chemical Engineering, Soochow University, Suzhou, Jiangsu 215123, China
First published on 13th April 2015
Abstract
A concise formal synthesis of platencin has been realized, featuring an organocatalytic approach to the [2.2.2] bicyclic core, a radical reductive elimination, a Au-catalyzed Meyer–Schuster rearrangement and a Rh-catalyzed chemo- and diastereoselective hydrosilylation.
Platencin (1) and platensimycin (2) have been among the most prominent natural products in recent years (Fig. 1). Identified by a team of Merck scientists from the strains of Streptomyces platensis, these two natural products exhibit broad-spectrum antibacterial activity against Gram-positive pathogens that show resistance to current antibiotics, including methicillin-, macrolide-, and linezolid-resistant S. aureus, vancomycin-resistant enterococci, and Streptococcus pneumoniae.1,2 Platencin shows slightly stronger bioactivities than platensimycin, and blocks both FabF and FabH whereas platensimycin only inhibits the former.2 In addition, platencin even as a racemic form exhibits potent bacteriostatic activities towards Mycobacterium tuberculosis.3 Notably, the poor in vivo efficacy has precluded platencin itself from being a clinical drug.4
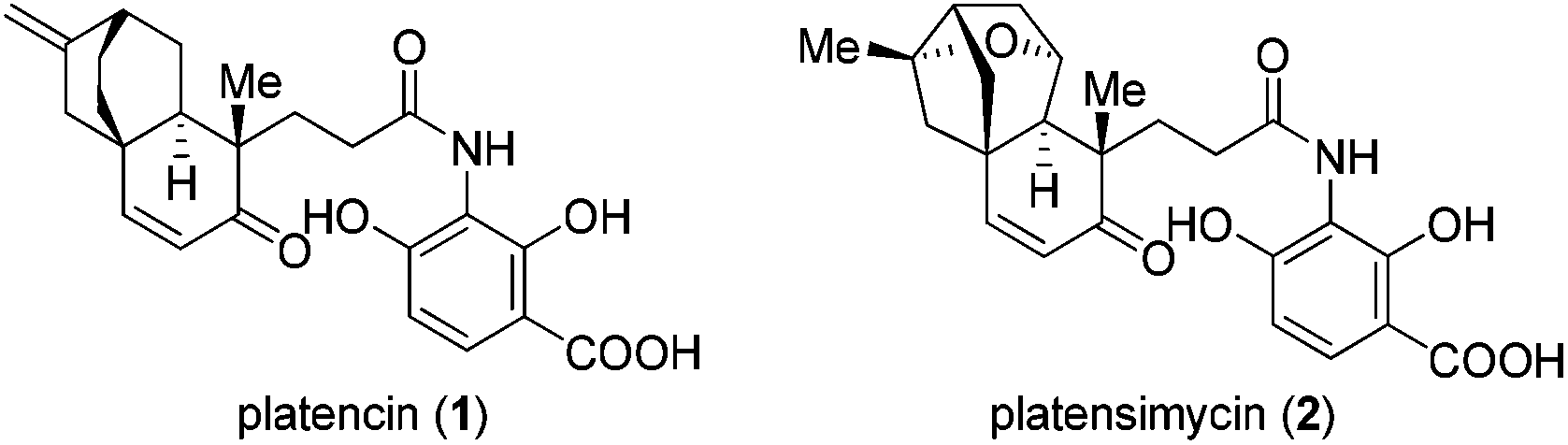 | ||
| Fig. 1 Platencin (1) and platensimycin (2). | ||
Tremendous efforts have been exerted from the synthetic community resulting in numerous syntheses of platencin5a–t and its analogues.5u–x From a perspective of target oriented synthesis, Mulzer and Tiefenbacher's work stands out as the most efficient synthesis of platencin so far.5c Nevertheless, synthetic endeavors based on new strategies are still highly desirable. Recently, we successfully developed a convenient approach to bicyclo[2.2.2]octane-1-carboxylates.6 In this paper, we report a formal synthesis of platencin by virtue of this new method.
Our retrosynthesis is depicted in Scheme 1. Nicolaou's intermediate enone 3 was chosen as the direct synthetic target.5a Enone 3 could be reduced to dicarbonyl 4, which in turn could be traced back to 5. Eventually, in light of our methodology, a formal [4 + 2] cycloaddition reaction of nitroethylene (6) with α′-ethoxycarbonyl cyclohexenone (7) was envisaged to access 5.
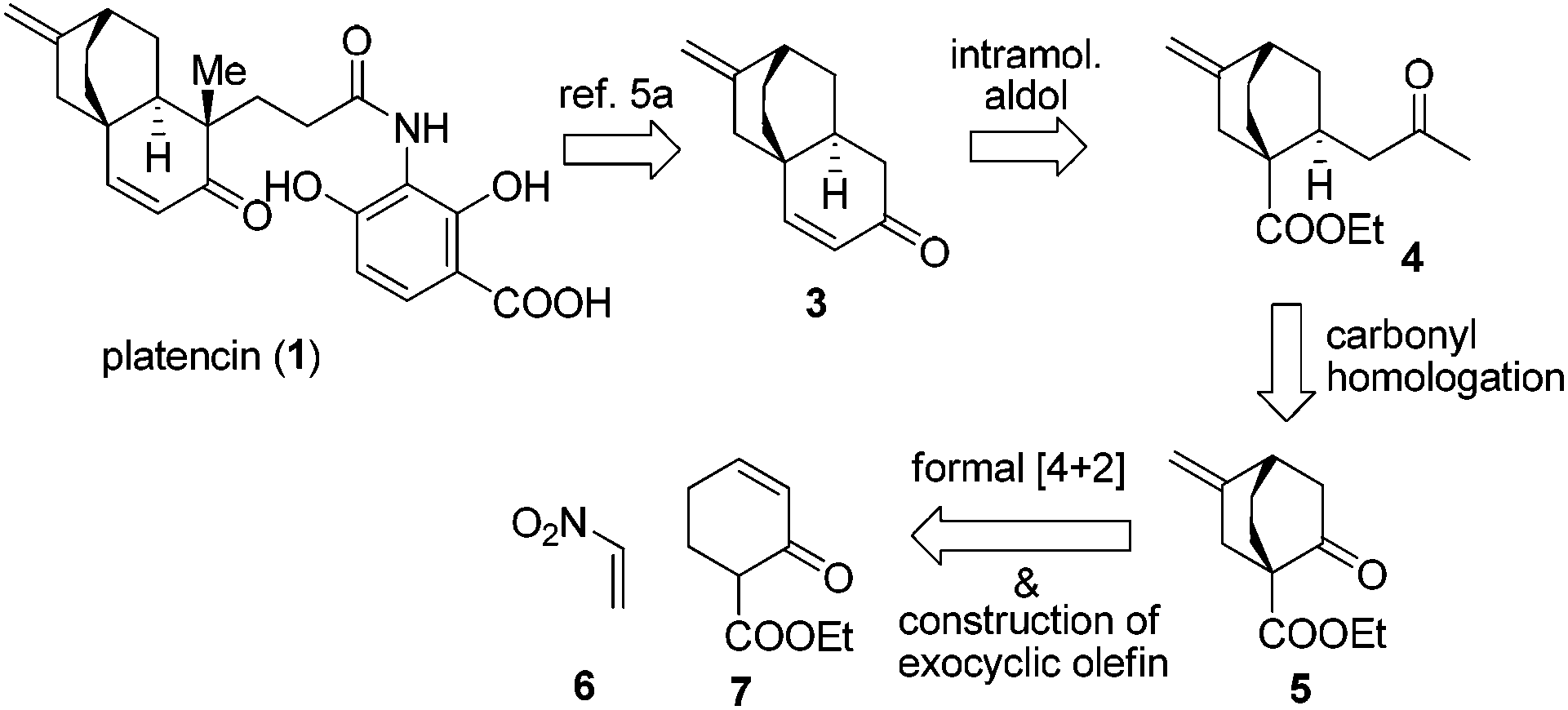 | ||
| Scheme 1 Retrosynthetic analysis of platencin (1). | ||
Our synthesis commenced with the critical [4 + 2] reaction of 6 and 7 (Scheme 2). Surprisingly, by employing the original conditions involving CAT-1 as the catalyst and dichloromethane as the solvent,6 only trace amounts of 8 could be isolated, probably as a result of the high propensity of 6 undergoing polymerization under the reaction conditions. To our delight, when toluene was used as the solvent and 6 was slowly introduced to the reaction mixture, the polymerization of 6 could be significantly suppressed, with 8 being isolated in 74% yield, albeit with an enantioselectivity of only 15% ee which could be ascribed to the absence of any substituent at the β-carbon of 6.
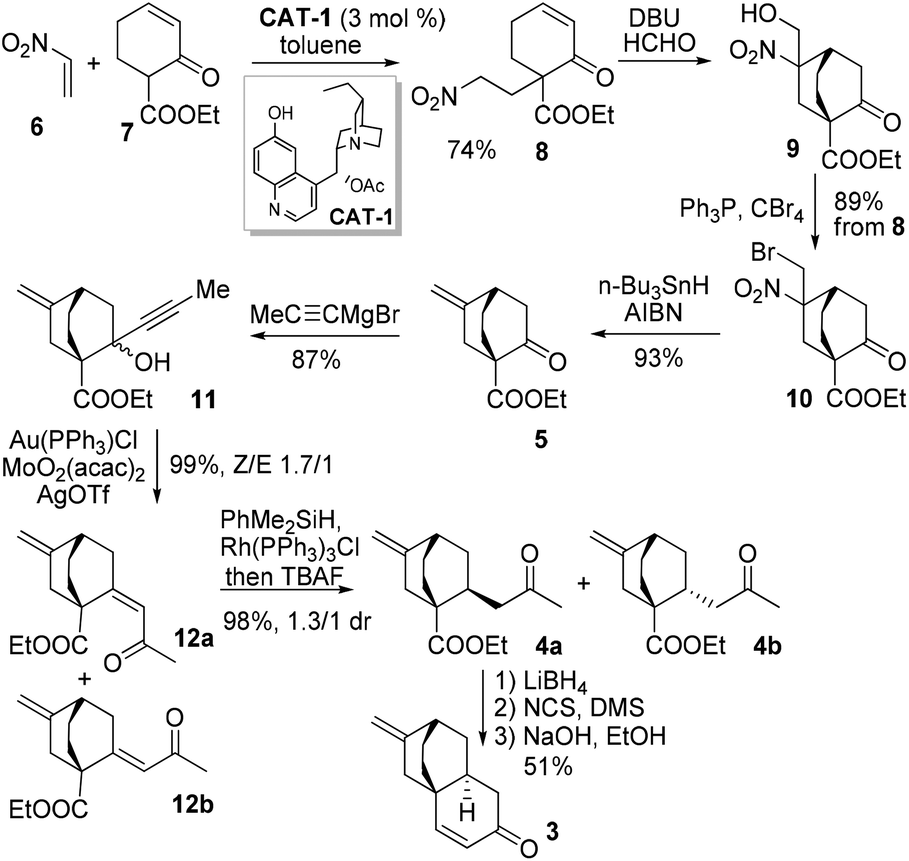 | ||
| Scheme 2 Synthesis of 3. | ||
A tandem Michael–Henry reaction was then realized with DBU and formaldehyde, providing 9 as a 1/1 diastereomeric mixture, which without chromatographic purification was brominated with PPh3/CBr4 to furnish 10 in 89% overall yield spanning three steps from 8. The subsequent unprecedented conversion of 10 to 5 was examined with various reducing agents (Table 1). The optimal set of conditions involving n-Bu3SnH/AIBN in heated toluene provided 5 in 91% yield.
| Entry | Conditions | Yield (%) |
|---|---|---|
| 1 | Mg, THF, 40 °C | 21 |
| 2 | Zn, NaI, MeOH, 50 °C | 25 |
| 3 | Zn, NH4Cl, MeOH, r.t. | — |
| 4 | SmI2, r.t. | — |
| 5 | t-BuLi, −78 °C | — |
| 6 | AIBN, n-Bu3SnH, toluene, 90 °C | 93 |
With a robust protocol established for the synthesis of 5, we proceeded to the next synthetic stage. The Grignard addition of propargylmagnesium bromide to ketone 5 afforded 11 in 87% yield as a 1.5/1 mixture. The subsequent Au-catalyzed Meyer–Schuster rearrangement of 11 proved highly efficient.7 With 2 mol% catalyst in dichloromethane, enone 12 could be obtained in a quantitative yield as a 1.7/1 mixture favouring the (Z)-isomer 12a.8 The chemo- as well as diastereoselective hydrogenation of 12a was investigated (Table 2). A nearly quantitative yield of 4a and its epimer 4b in 1.3/1 ratio could be obtained by rhodium catalyzed hydrosilylation (entry 3).8 Compared to the result obtained with t-BuCu/DIBAL-H (entry 2), the interaction between the exocyclic terminal double bond and the rhodium catalyst probably overrode the adverse inherent steric facial bias, leading to 4a as the major product. Eventually, ketoester 4a was subjected to the reduction–oxidation sequence to provide a ketoaldehyde before undergoing the intramolecular aldol condensation to give enone 3 in 51% yield over three steps.
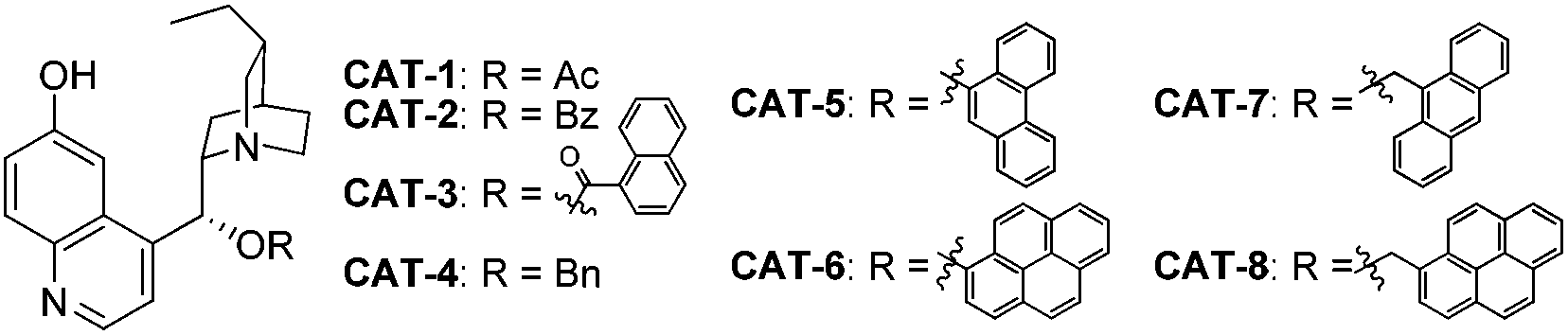
In view of the fact that the enantioselectivity of the initial key reaction delivering 8 was poor, we continued to improve the reaction by screening more catalysts and conditions, as summarized in Table 3. When 6 was dissolved in toluene and introduced slowly in excess, dichloromethane could be employed as the solvent which resulted in a slightly higher ee of 38% with the reversed sense of enantioselectivity as compared to that obtained with toluene (entries 1 and 2). A lower temperature resulted in significantly decreased reactivity (entry 3). Further investigation of the catalysts revealed CAT-5 to be the optimal one (entries 4–10). The best result was obtained with CAT-5 in nitrobenzene, which furnished 8 in 84% yield and 74% ee (entry 12).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Conditionsa | Conv.b (%) | eec (%) |
| a A solution of nitroethylene (6) in toluene (2.0 M) was always employed in excess. b Determined by analysis of the 1H NMR spectra of crude samples. c Determined by chiral HPLC analysis. d 84% isolated yield. | ||||
| 1 | CAT-1 | Toluene, r.t. | 95 | 15 |
| 2 | CAT-1 | DCM, r.t. | 72 | −38 |
| 3 | CAT-1 | DCM, −20 °C | 33 | −42 |
| 4 | CAT-2 | DCM, r.t. | 55 | −42 |
| 5 | CAT-3 | DCM, r.t. | 56 | −41 |
| 6 | CAT-4 | DCM, r.t. | 45 | −52 |
| 7 | CAT-5 | DCM, r.t. | 88 | −72 |
| 8 | CAT-6 | DCM, r.t. | 71 | −63 |
| 9 | CAT-7 | DCM, r.t. | 32 | −68 |
| 10 | CAT-8 | DCM, r.t. | 50 | −65 |
| 11 | CAT-5 | PhCN, r.t. | 94 | −73 |
| 12 | CAT-5 | PhNO2, r.t. | 98d | −74 |
Conclusions
In conclusion, we have accomplished a concise formal synthesis of platencin, featuring an organocatalytic approach to the bicyclo[2.2.2]octane core, a radical reductive elimination, an efficient Au-catalyzed Meyer–Schuster rearrangement, and a Rh-catalyzed chemo- and diastereoselective hydrosilylation. The formal synthesis covers ten steps from simple starting materials in a good overall yield. Importantly, this synthesis demonstrates a new synthetic strategy that may readily lend itself to access to novel platencin analogues valuable for relevant drug discovery. Synthetic endeavours along this line are currently underway in our laboratory and will be reported in due course.Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (grant no. 21172246, 21290180, 21472210), the Youth Innovation Promotion Association of CAS, the Collaborative Innovation Center of Chemical Science and Engineering (Tianjin) and the State Key Laboratory of Bioorganic and Natural Products Chemistry.Notes and references
- (a) J. Wang, S. M. Soisson, K. Young, W. Shoop, S. Kodali, A. Galgoci, R. Painter, G. Parthasarathy, Y. S. Tang, R. Cummings, S. Ha, K. Dorso, M. Motyl, H. Jayasuriya, J. Ondeyka, K. Herath, C. Zhang, L. Hernandez, J. Allocco, A. Basilio, J. R. Tormo, O. Genilloud, F. Vicente, F. Pelaez, L. Colwell, S. H. Lee, B. Michael, T. Felcetto, C. Gill, L. L. Silver, J. D. Hermes, K. Bartizal, J. Barrett, D. Schmatz, J. W. Becker, D. Cully and S. B. Singh, Nature, 2006, 441, 358 CrossRef CAS PubMed; (b) S. B. Singh, H. Jayasuriya, J. G. Ondeyka, K. B. Herath, C. Zhang, D. L. Zink, N. N. Tsou, R. G. Ball, A. Basilio, O. Genilloud, M. T. Diez, F. Vicente, F. Pelaez, K. Young and J. Wang, J. Am. Chem. Soc., 2006, 128, 11916 CrossRef CAS PubMed.
- (a) J. Wang, S. Kodali, S. H. Lee, A. Galgoci, R. Painter, K. Dorso, F. Racine, M. Motyl, L. Hernandez, E. Tinney, S. Colletti, K. Herath, R. Cummings, O. Salazar, I. Gonzalez, A. Basilio, F. Vicente, O. Genilloud, F. Pelaez, H. Jayasuriya, K. Young, D. Cully and S. B. Singh, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7612 CrossRef CAS PubMed; (b) H. Jayasuriya, K. B. Herath, C. Zhang, D. L. Zink, A. Basilio, O. Genilloud, M. T. Diez, F. Vicente, I. Gonzalez, O. Salazar, F. Pelaez, R. Cummings, S. Ha, J. Wang and S. B. Singh, Angew. Chem., Int. Ed., 2007, 46, 4684 CrossRef CAS PubMed.
- G. A. I. Moustafa, S. Nojima, Y. Yamano, A. Aono, M. Arai, S. Mitarai, T. Tanaka and T. Yoshimitsu, MedChemComm, 2013, 4, 720 RSC.
- (a) S. Brinster, G. Lamberet, B. Staels, P. Trieu-Cuot, A. Gruss and C. Poyart, Nature, 2009, 458, 83 CrossRef CAS PubMed; (b) E. Martens and A. L. Demain, J. Antibiot., 2011, 64, 705 CrossRef CAS PubMed.
- (a) K. C. Nicolaou, G. S. Tria and D. J. Edmonds, Angew. Chem., Int. Ed., 2008, 47, 1780 CrossRef CAS PubMed; (b) J. Hayashida and V. H. Rawal, Angew. Chem., Int. Ed., 2008, 47, 4373 CrossRef CAS PubMed; (c) K. Tiefenbacher and J. Mulzer, Angew. Chem., Int. Ed., 2008, 47, 6199 CrossRef CAS PubMed; (d) S. Y. Yun, J.-C. Zheng and D. Lee, Angew. Chem., Int. Ed., 2008, 47, 6201 CrossRef CAS PubMed; (e) D. C. J. Waalboer, M. C. Schaapman, F. L. van Delft and F. P. J. T. Rutjes, Angew. Chem., Int. Ed., 2008, 47, 6576 CrossRef CAS PubMed; (f) K. C. Nicolaou, Q.-Y. Toh and D. Y.-K. Chen, J. Am. Chem. Soc., 2008, 130, 11292 CrossRef CAS PubMed; (g) K. A. B. Austin, M. G. Banwell and A. C. Willis, Org. Lett., 2008, 10, 4465 CrossRef CAS PubMed; (h) K. Tiefenbacher and J. Mulzer, J. Org. Chem., 2009, 74, 2937 CrossRef CAS PubMed; (i) G. N. Varseev and M. E. Maier, Angew. Chem., Int. Ed., 2009, 48, 3685 CrossRef CAS PubMed; (j) A. K. Ghosh and K. Xi, Angew. Chem., Int. Ed., 2009, 48, 5372 CrossRef CAS PubMed; (k) K. C. Nicolaou, G. S. Tria, D. J. Edmonds and M. Kar, J. Am. Chem. Soc., 2009, 131, 15909 CrossRef CAS PubMed; (l) P. Li and H. Yamamoto, Chem. Commun., 2010, 46, 6294 RSC; (m) V. Singh, B. C. Sahu, V. Bansal and S. M. Mobin, Org. Biomol. Chem., 2010, 8, 4472 RSC; (n) S. Hirai and M. Nakada, Tetrahedron Lett., 2010, 51, 5076 CrossRef CAS PubMed; (o) K. Palanichamy, A. V. Subrahmanyam and K. P. Kaliappan, Org. Biomol. Chem., 2011, 9, 7877 RSC; (p) T. Yoshimitsu, S. Nojima, M. Hashimoto and T. Tanaka, Org. Lett., 2011, 13, 3698 CrossRef CAS PubMed; (q) S. Hirai and M. Nakada, Tetrahedron, 2011, 67, 518 CrossRef CAS PubMed; (r) L. Zhu, C. Zhou, W. Yang, S. He, G.-J. Cheng, X. Zhang and C.-S. Lee, J. Org. Chem., 2013, 78, 7912 CrossRef CAS PubMed; (s) J. S. Yadav, R. Goreti, S. Pabbaraja and B. Sridhar, Org. Lett., 2013, 15, 3782 CrossRef CAS PubMed; (t) E. L. Chang, B. D. Schwartz, A. G. Draffan, M. G. Banwell and A. C. Willis, Chem. – Asian J., 2015, 10, 427 CrossRef CAS PubMed; (u) O. V. Barykina, K. Rossi, L. M. J. Rybak and B. B. Snider, Org. Lett., 2009, 11, 5334 CrossRef CAS PubMed; (v) K. Tiefenbacher, A. Gollner and J. Mulzer, Chem. – Eur. J., 2010, 16, 9616 CrossRef CAS PubMed; (w) D. C. J. Waalboer, S. H. A. M. Leenders, T. Schuelin-Casonato, F. L. van Delft and F. P. J. T. Rutjes, Chem. – Eur. J., 2010, 16, 11233 CrossRef CAS PubMed; (x) G. Y. C. Leung, H. Li, Q.-Y. Toh, A. M.-Y. Ng, R. J. Sum, J. E. Bandow and D. Y.-K. Chen, Eur. J. Org. Chem., 2011, 183 CrossRef CAS PubMed. For a review, see: (y) K. Palanichamy and K. P. Kaliappan, Chem. – Asian J., 2010, 5, 668 CrossRef CAS PubMed.
- Y.-Z. Li, J. Wang, W.-B. Sun, Y.-F. Shan, B.-F. Sun, G.-Q. Lin and J.-P. Zou, Org. Chem. Front., 2015, 2, 274 RSC.
- M. Egi, Y. Yamaguchi, N. Fujiwara and S. Akai, Org. Lett., 2008, 9, 1867 CrossRef PubMed.
- See the ESI† for details.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopy data, and copies of 1H, 13C and 2D NMR spectra. See DOI: 10.1039/c5qo00065c |
| This journal is © the Partner Organisations 2015 |