
Combining Pd(π-allyl)Cp and PPh3 as a unique catalyst for efficient synthesis of alkyliodo indoles via C(sp3)–I reductive elimination†
Wei
Hao
a,
Han
Wang
a,
Patrick J.
Walsh
*b and
Zhenfeng
Xi
*a
aNational Laboratory for Molecular Sciences, Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, College of Chemistry, Peking University, Beijing 100871, China. E-mail: zfxi@pku.edu.cn
bRoy and Diana Vagelos Laboratories, Department of Chemistry, University of Pennsylvania, 231 South 34th Street, Philadelphia, Pennsylvania 19104-6323, USA. E-mail: pwalsh@sas.upenn.edu
First published on 14th July 2015
Abstract
The combination of Pd(π-allyl)Cp and PPh3 was found to generate an efficient catalyst for the formation of indole-containing alkyl iodides from ortho-amino iodobenzenes and both aromatic and aliphatic alkynes. A unique feature of this reaction is a palladium-promoted C(sp3)–I bond formation via reductive elimination. This catalytic process was found to be very sensitive to the size and equivalents of phosphine ligands and the nature of the base.
Introduction
Alkyl iodides are important reagents in synthetic organic chemistry and their synthesis has attracted much attention. Traditionally, they are prepared by the reaction of alcohols with I2/PPh3 or by conversion of alcohols to sulfonates followed by treatment with NaI.1 Despite the widespread use of such reactions, new methods to prepare these classic electrophiles with increasing degrees of functionality remain in demand.Recently it has been shown that alkyl iodides can be prepared using transition metal catalysts via an unusual reductive elimination pathway.2–5 The C–I bond-forming reductive elimination step takes place from an intermediate LnM(alkyl)(I) that can also undergo facial β-hydride elimination,6 limiting the synthetic utility of this approach. As a consequence, the alkyl groups of [LnM(alkyl)(I)] intermediates usually do not contain syn-β-hydrogen atoms to circumvent generation of alkene byproducts.
We recently reported a unique Pd-catalyzed tandem reaction between alkynes and ortho-NR2 iodobenzene derivatives such as 1a to afford indole-containing alkyl iodides (Scheme 1).2 The proposed mechanism (Scheme 2) involves initial oxidative addition of the aryl iodide, alkyne insertion, and a C–N bond cleavage/formation sequence to give intermediate III. The alkyliodide product 2 is formed via a rare reductive elimination of the alkylpalladium iodide intermediate III. This chemistry is particularly noteworthy because it represents the first example of reductive elimination of alkylpalladium halide complexes possessing syn-β-hydrogens.3–10 A drawback of our initial approach is that different catalysts were required for different alkyne substrates. For aliphatic alkynes such as 3-hexyne, reaction conditions A, employing Pd(PPh3)4 as the palladium source, were found to be effective to generate the alkyl iodide 2a (85% yield). The yield of product 2a was significantly lower (65%) when Pd(OAc)2 and PPh3 were employed (conditions B). On the contrary, for aromatic alkynes such as diphenylacetylene, reaction conditions B were found to afford product 2b in good yield (67%) but reaction conditions A generated only traces of 2b with most of 1a unreacted.
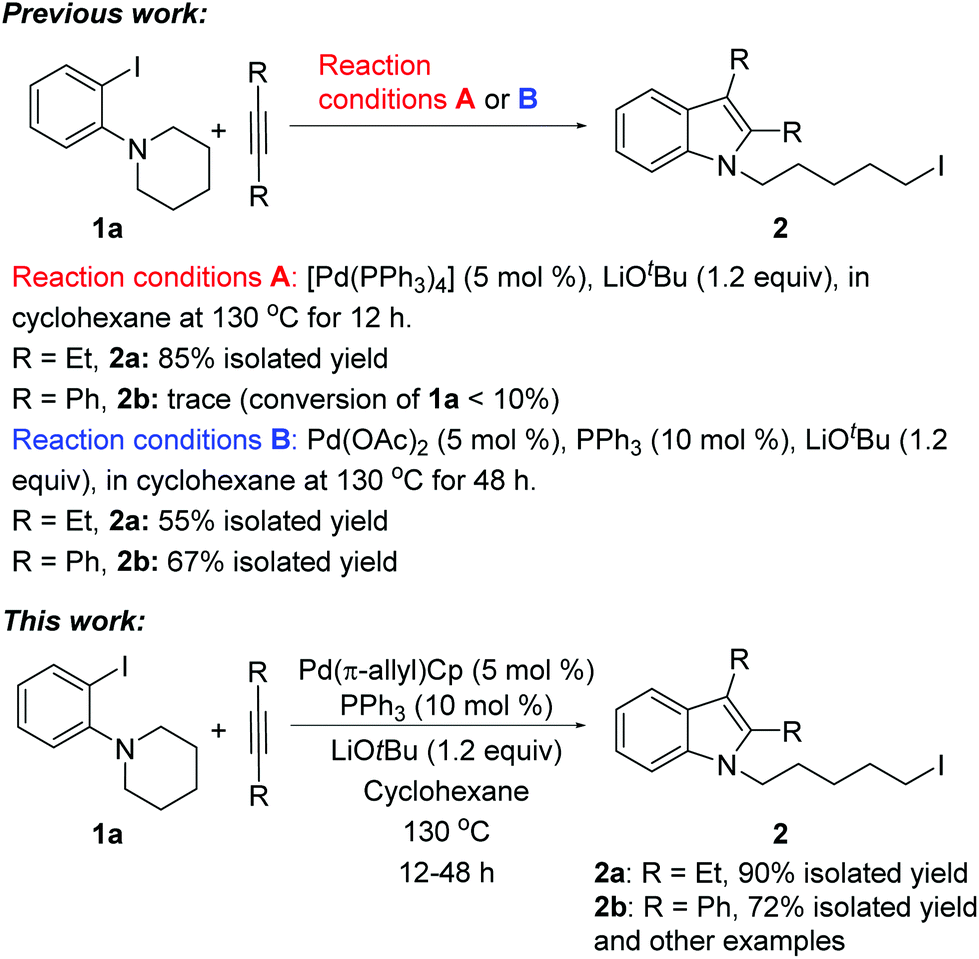 | ||
| Scheme 1 Catalytic formation of alkyliodo indoles from 1a and alkynes. | ||
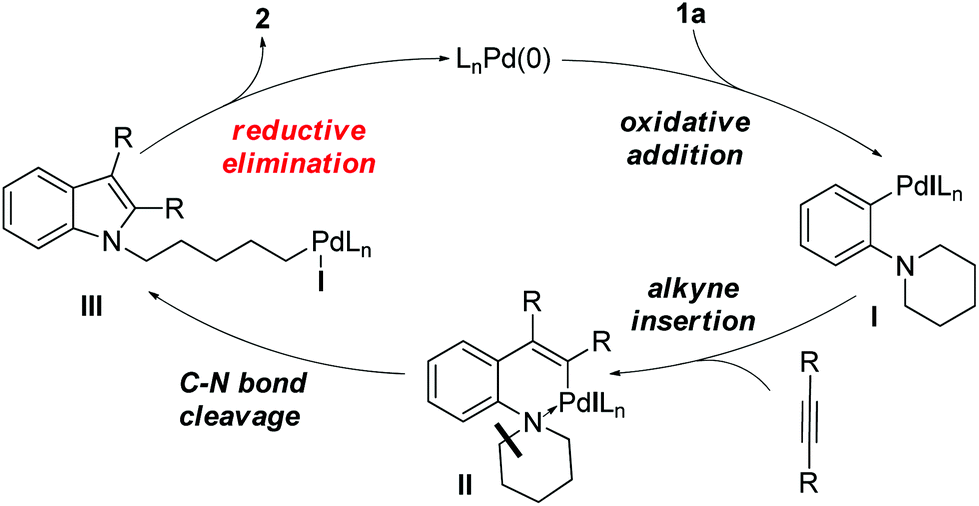 | ||
| Scheme 2 Catalytic cycle for the formation of alkyliodo indoles from 1a and alkynes. | ||
Results and discussion
Based on these results, we set out to find an improved reaction system for this catalytic process that would be effective for both aliphatic and aromatic alkynes. In our preliminary study,2 we realized that the ratio of Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PPh3 was important in optimizing reactivity. Previous theoretical studies also highlight the importance of this ratio in palladium-catalyzed reactions.11 Therefore, we now focus on the use of a precatalyst that will enable better control of this crucial variable.
:PPh3 was important in optimizing reactivity. Previous theoretical studies also highlight the importance of this ratio in palladium-catalyzed reactions.11 Therefore, we now focus on the use of a precatalyst that will enable better control of this crucial variable.
In contrast to other palladium precatalysts,12 a unique feature of the Pd(π-allyl)Cp precatalyst is that it has been recently reported to generate PdL2 reactive species in the presence of 2 equivalents of phosphine (L = phosphine ligand, NHC, etc.) without the assistance of base.13 We anticipated that this property of Pd(π-allyl)Cp, along with the ability to accurately control the Pd:ligand stoichiometry, would be useful for us to investigate the effects of ligands and bases for the above-mentioned Pd-catalyzed alkyl iodide formation. Herein we report that the combination of Pd(π-allyl)Cp and PPh3 (1:2 ratio) is found to lead to an efficient catalyst for the formation of alkyliodides 2 from ortho-NR2 iodobenzene derivatives 1 with both aromatic and aliphatic alkynes.
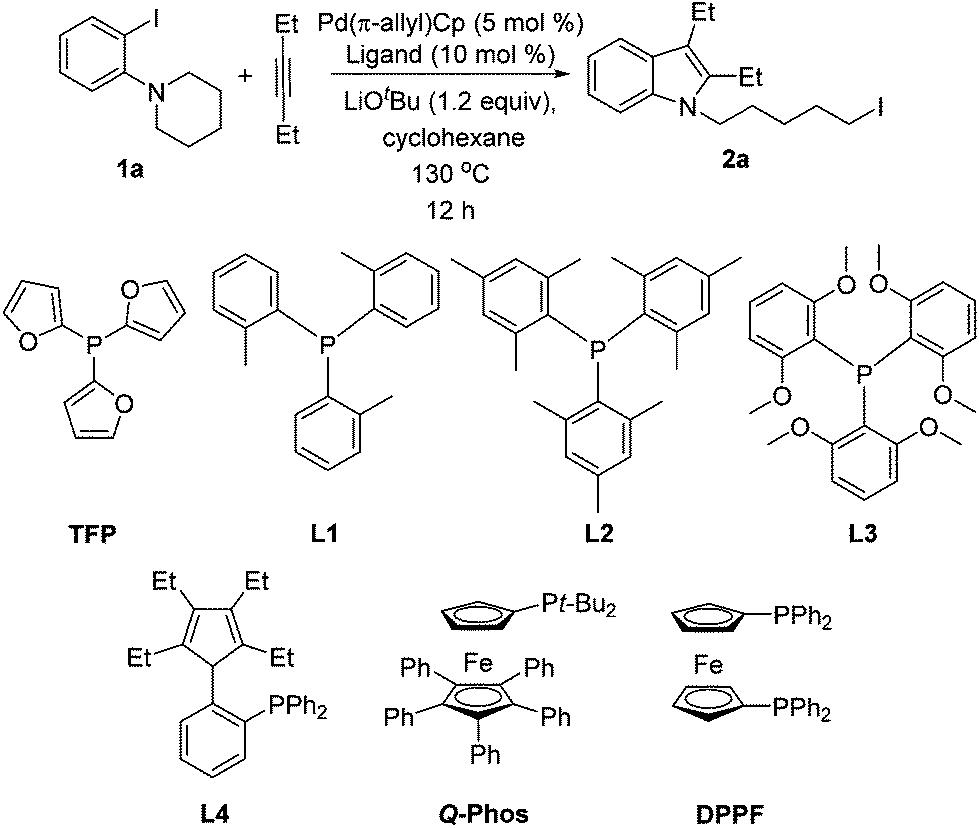
We initially screened various mono- and bidentate phosphine ligands in the reaction of 3-hexyne with orthoamino iodobenzene 1a to give iodo indole 2a (Table 1). When Pd(π-allyl)Cp was substituted for Pd(PPh3)4 (conditions A, but with no added phosphine, Table 1), no reaction took place (entry 1). However, when Pd(π-allyl)Cp (5 mol %) was employed with 10 mol % PPh3, the alkyliodide product 2a was obtained in 92% GC yield (entry 2). Notably, with additional equivalents of PPh3, the yield of 2a decreased dramatically (entries 3, 4). Use of tri(2-fural) phosphine (TFP) in place of triphenyl phosphine resulted in a drop in yield to 78% (entry 5). The yield of 2a was found to be highly dependent on the cone angle of phosphine ligands used. Thus, with increasing size of the ligands (TFP, L1, L2, L3), the yield of 2a went from moderate to traces (entries 5–8), with most of the starting material 1a found unreacted. Q-phos14 has been successfully applied by Lautens's group in palladium-catalyzed generation of alkyl iodides that do not possess accessible β-hydrogens.4a,d–h However, in the present system we observed a maximum of 32% yield of 2a with this ligand (entries 9–10). The cyclopentadiene-phosphine ligand L4, which exhibited high efficiency in our Pd-catalyzed C–N bond cleavage reaction,15 was not effective in the present case, resulting in only 8% yield of 2a (entry 11). Other bulkyl monophosphines and bidentate phosphine ligands such as PCy3, PtBu2Me, PtBu3, DPPF, DPPE and DPPP were also tested. Yields with these ligands ranged from 33% down to trace product (entries 12–17).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | Yield of 2ab (%) | Entry | Ligand | Yield of 2ab (%) |
| a Reaction conditions: 1a (0.3 mmol), 3-hexyne (0.36 mmol), Pd(π-allyl)Cp (5 mol %, 0.015 mmol), ligand (10 mol %, 0.03 mmol), LiOtBu (1.2 equiv., 0.36 mmol), 2 mL cyclohexane, 12 h. b Yields were determined by GC analysis of the crude reaction mixture using dodecane as an internal standard. c PPh3 (30 mol %, 0.09 mmol) was added. d PPh3 (50 mol %, 0.15 mmol) was added. e LiOtBu was not added. | |||||
| 1 | — | 0 | 10e | Q-Phos | <5 |
| 2 | PPh3 | 92 | 11 | L4 | 8 |
| 3c | PPh3 | 65 | 12 | PCy3 | 33 |
| 4d | PPh3 | 15 | 13 | PtBu2Me | 32 |
| 5 | TFP | 78 | 14 | PtBu3 | 28 |
| 6 | L1 | 9 | 15 | DPPF | <5 |
| 7 | L2 | <5 | 16 | DPPE | <5 |
| 8 | L3 | <5 | 17 | DPPP | <5 |
| 9 | Q-Phos | 32 | |||
The effect of bases was next investigated (Table 2).16 Our previous work2 and Lautens's4e–g publications showed that base was necessary to promote the reaction, although the exact role of the base remains elusive. We reported that LiOtBu could promote this reaction efficiently and in its absence, no product was observed (Table 2, entries 1 and 2). Furthermore, we demonstrated that the alkyliodide products were stable to both E2 elimination and SN2 with –OtBu in cyclohexane at 130 °C.2 According to the literature,13 Pd(π-allyl)Cp reacts with 2 equiv. PPh3 to form Pd(PPh3)2 along with the allyl/Cp coupling product C8H10 without base assistance. With the precatalyst Pd(π-allyl)Cp, LiOtBu was found to be the most effective with other bases, including NaOtBu and KOtBu, giving much lower yields of product 2a. The amount of LiOtBu also had a significant influence on the yield of 2a, with 1.2 equiv. giving better yield (92%, entry 2) compared to 0.5 equiv. (65%, entry 5) or 2.0 equiv. (80%, entry 6). We previously demonstrated that crown ethers can be beneficial additives in palladium-catalyzed reactions with strong bases.17 When 12-crown-4 was added to the reaction mixture, however, the yield of 2a decreased to 36% (entry 7). As shown in Table 2, entries 8–17, weaker bases, including both anionic and neutral organic bases, were less effective than LiOtBu.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Yield of 2ab (%) | Entry | Base | Yield of 2ab (%) |
| a Reaction conditions: 1a (0.3 mmol), 3-hexyne (0.36 mmol), [Pd] (5 mol %, 0.015 mmol), PPh3 (10 mol %, 0.03 mmol), base (1.2 equiv., 0.36 mmol), 2 mL cyclohexane, 12 h. b Yields were determined by GC analysis of the crude reaction mixture using dodecane as an internal standard. c LiOtBu (50 mol %, 0.15 mmol) was added. d LiOtBu (200 mol %, 0.6 mmol) was added. e 2.4 equiv. of 12-crown-4 was added. | |||||
| 1 | — | 0 | 10 | Li2CO3 | 0 |
| 2 | LiOtBu | 92 | 11 | LiOEt | 54 |
| 3 | NaOtBu | 52 | 12 | K2CO3 | 54 |
| 4 | KOtBu | 0 | 13 | K3PO4 | 68 |
| 5c | LiOtBu | 65 | 14 | KF | 0 |
| 6d | LiOtBu | 80 | 15 | CsF | 30 |
| 7e | LiOtBu | 36 | 16 | DBU | <5 |
| 8 | LiOAc | 0 | 17 | Et3N | 25 |
| 9 | LiOH | 28 | |||
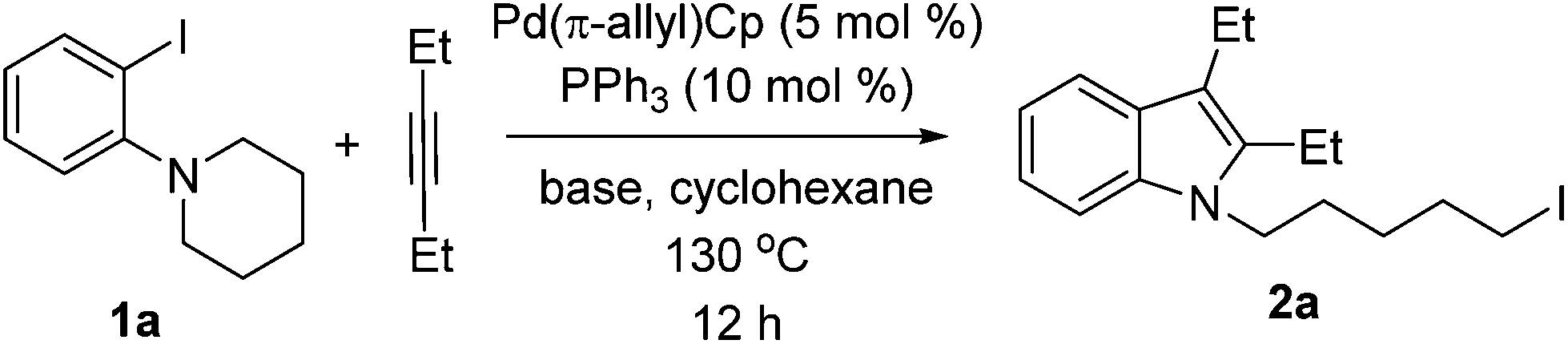
With the results of ligand and base screening in hand, the optimized reaction conditions are 1a (0.3 mmol), alkyne (0.36 mmol), Pd(π-allyl)Cp (5 mol %, 0.015 mmol), PPh3 (10 mol %, 0.03 mmol), LiOtBu (1.2 equiv., 0.36 mmol) in 2 mL of cyclohexane at 130 °C as a homogeneous reaction solution. Fortunately, these conditions were suitable for both aliphatic alkynes and aromatic alkynes, enabling the synthesis of a variety of iodoindoles in 62–90% yields (Scheme 3). For the substrate 1-(2-iodophenyl)piperidine (1a), the yields of corresponding products using Pd(π-allyl)Cp are higher than with our previous conditions. The aryl iodide with a five-membered azacycle could be also used, thereby affording the alkyliodide products with shorter linkers. For aromatic alkynes, the reaction times are longer than those with aliphatic alkynes. This may be a result of their lower reactivity due to conjugation. The yields with aromatic alkynes are 62–75%.
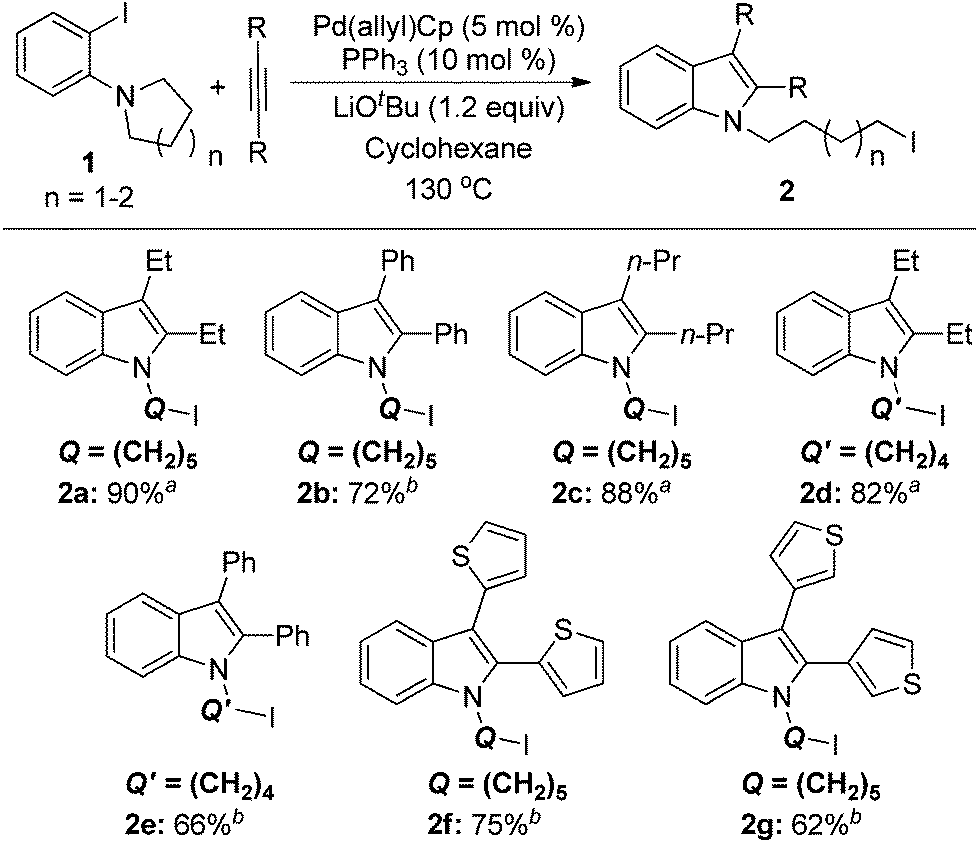 | ||
| Scheme 3 Substrate scope of alkynes and orthoamino iodobenzenes 1 in the synthesis of iodoindoles 2. a 12 h reaction time. b 48 h reaction time. | ||
The results in Scheme 3 led us to reflect on the possibility of reductive elimination to form other C(sp3)–X bonds.9 Previous theoretical and experimental results suggest that reductive elimination of alkyl bromides is more difficult than reductive elimination of alkyl iodides.2,8 These observations are consistent with a recent study by Tong's group on reductive elimination of C–X bonds (X = I, Br, Cl),4i which was published during the preparation of this manuscript. Like previous works, Tong's substrates were limited to those lacking of β-hydrogens to circumvent the β-hydride elimination pathway.
We were encouraged by the high yields in the formation of alkyl iodides described in Scheme 3, so we explored the possibility of reductive elimination of alkyl bromides from our palladium catalyst. As demonstrated in Scheme 4, when the orthoamino bromobenzene 3 was used, the expected reductive elimination product 4 was obtained in 32% isolated yield, with most 3 remaining unreacted. Although the yield is moderate, this result indicates such a reaction is indeed possible and an interesting topic of future study.
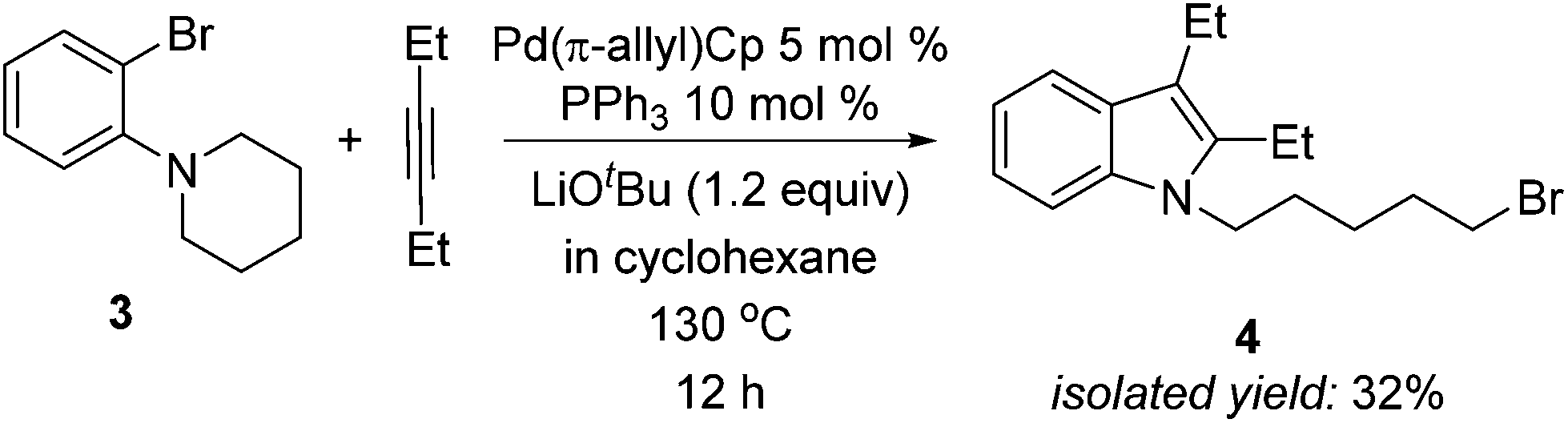 | ||
| Scheme 4 Pd-catalyzed reaction of 1-(2-bromophenyl)piperidine with 3-hexyne. | ||
Conclusions
In summary, we have developed a general catalyst for tandem carboiodination process leading to iodo alkyl indoles. Indoles are among the most important heterocycles in medicinal chemistry and the iodoindoles prepared herein could be used in the synthesis of novel indole-based structures. The synthesis was facilitated by the use of the Pd(π-allyl)Cp precatalyst in combination with PPh3 and LiOtBu as the base. Future work will include the elucidation on the role of base and study of the reaction mechanism to understand the factors that enable these unique C(sp3)–I and C(sp3)–Br reductive elimination processes in the presence of accessible β-hydrogen atoms.Acknowledgements
This work was supported by the 973 Program (2012CB821600) and the National Natural Science Foundation of China (NSFC).Notes and references
- (a) M. V. Pavan, L. Lassiani, F. Berti, G. Stefancich, A. Ciogli, F. Gasparrini, L. Mennuni, F. Ferrari, C. Escrieut, E. Marco, F. Makovec, D. Fourmy and A. Varnavas, J. Med. Chem., 2011, 54, 5769 CrossRef CAS PubMed; (b) S. O. Alapafuja, S. P. Nikas, I. T. Bharathan, V. G. Shukla, M. L. Nasr, A. L. Bowman, N. Zvonok, J. Li, X. Shi, J. R. Engen and A. Makriyannis, J. Med. Chem., 2012, 55, 10074 CrossRef CAS PubMed.
- W. Hao, J. Wei, W.-X. Zhang and Z. Xi, Angew. Chem., Int. Ed., 2014, 53, 14533 CrossRef CAS PubMed.
- For reviews on reductive elimination, see: (a) A. Vigalok, Chem. – Eur. J., 2008, 14, 5102 CrossRef CAS PubMed; (b) V. V. Grushin, Acc. Chem. Res., 2010, 43, 160 CrossRef CAS PubMed; (c) T. Furuya, J. E. M. N. Klein and T. Ritter, Synthesis, 2010, 1804 CAS; (d) K. M. Engle, T.-S. Mei, X. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2011, 50, 1478 CrossRef CAS PubMed; (e) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS PubMed; (f) X. Jiang, H. Liu and Z. Gu, Asian J. Org. Chem., 2012, 1, 16 CrossRef CAS PubMed; (g) C. Chen and X. Tong, Org. Chem. Front., 2014, 1, 439 RSC.
- For reductive elimination of alkyl halides from alkyl palladium(II) halides, see: (a) S. G. Newman and M. Lautens, J. Am. Chem. Soc., 2011, 133, 1778 CrossRef CAS PubMed; (b) H. Liu, C. Chen, L. Wang and X. Tong, Org. Lett., 2011, 13, 5072 CrossRef CAS PubMed; (c) H. Liu, C. Li, D. Qiu and X. Tong, J. Am. Chem. Soc., 2011, 133, 6187 CrossRef CAS PubMed; (d) S. G. Newman, J. K. Howell, N. Nicolaus and M. Lautens, J. Am. Chem. Soc., 2011, 133, 14916 CrossRef CAS PubMed; (e) D. A. Petrone, H. A. Malik, A. Clemenceau and M. Lautens, Org. Lett., 2012, 14, 4806 CrossRef CAS PubMed; (f) X. Jia, D. A. Petrone and M. Lautens, Angew. Chem., Int. Ed., 2012, 51, 9870 CrossRef CAS PubMed; (g) D. A. Petrone, M. Lischka and M. Lautens, Angew. Chem., Int. Ed., 2013, 52, 10635 CrossRef CAS PubMed; (h) D. A. Petrone, H. Yoon, H. Weinstabl and M. Lautens, Angew. Chem., Int. Ed., 2014, 53, 7908 CrossRef CAS PubMed; (i) C. Chen, L. Hou, M. Cheng, J. Su and X. Tong, Angew. Chem., Int. Ed., 2015, 54, 3092 CrossRef CAS PubMed.
- For reductive elimination of CH3I from Rh complexes, see: (a) C. M. Frech and D. Milstein, J. Am. Chem. Soc., 2006, 128, 12434 CrossRef CAS PubMed; (b) M. Feller, M. A. Iron, L. J. W. Shimon, Y. Diskin-Posner, G. Leitus and D. Milstein, J. Am. Chem. Soc., 2008, 130, 14374 CrossRef CAS PubMed; (c) M. Feller, Y. Diskin-Posner, G. Leitus, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2013, 135, 11040 CrossRef CAS PubMed.
- For β-hydride elimination, see: (a) X. Lu, Top. Catal., 2005, 35, 73 CrossRef; (b) A. C. Bissember, A. Levina and G. C. Fu, J. Am. Chem. Soc., 2012, 134, 14232 CrossRef CAS PubMed; (c) J. Wei, Y. Zhang, W.-X. Zhang and Z. Xi, Org. Chem. Front., 2014, 1, 983 RSC; (d) X. Liu and Z. Gu, Org. Chem. Front., 2015, 2, 778 RSC.
- For reductive elimination of aryl halides from aryl palladium(II) halides: (a) A. H. Roy and J. F. Hartwig, J. Am. Chem. Soc., 2001, 123, 1232 CrossRef CAS; (b) A. H. Roy and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 13944 CrossRef CAS PubMed; (c) A. H. Roy and J. F. Hartwig, Organometallics, 2004, 23, 1533 CrossRef CAS; (d) D. V. Yandulov and N. T. Tran, J. Am. Chem. Soc., 2007, 129, 1342 CrossRef CAS PubMed; (e) V. V. Grushin and W. J. Marshall, Organometallics, 2007, 26, 4997 CrossRef CAS; (f) D. A. Watson, M. Su, G. Teverovskiy, Y. Zhang, J. García-Fortanet, T. Kinzel and S. L. Buchwald, Science, 2009, 325, 1661 CrossRef CAS PubMed; (g) S. G. Newman and M. Lautens, J. Am. Chem. Soc., 2010, 132, 11416 CrossRef CAS PubMed; (h) X. Shen, A. Hyde and S. L. Buchwald, J. Am. Chem. Soc., 2010, 132, 14076 CrossRef CAS PubMed; (i) J. Pan, X. Wang, Y. Zhang and S. L. Buchwald, Org. Lett., 2011, 13, 4974 CrossRef CAS PubMed; (j) T. J. Maimone, P. J. Milner, T. Kinzel, Y. Zhang, M. K. Takase and S. L. Buchwald, J. Am. Chem. Soc., 2011, 133, 18106 CrossRef CAS PubMed; (k) H. G. Lee, P. J. Milner and S. L. Buchwald, Org. Lett., 2013, 15, 5602 CrossRef CAS PubMed.
- For computational study of reductive elimination, see: Y. Lan, P. Liu, S. G. Newman, M. Lautens and K. N. Houk, Chem. Sci., 2012, 3, 1987 RSC.
- For C–Br bond formation through π-allylic-Pd bromide species, see: Z. Gu, X. Wang, W. Shu and S. Ma, J. Am. Chem. Soc., 2007, 129, 10948 CrossRef CAS PubMed.
- For reductive elimination from Pd(IV) and Pd(III) complexes, see: (a) S. Ma and X. Lu, J. Org. Chem., 1993, 58, 1245 CrossRef CAS; (b) R. van Belzen, H. Hoffmann and C. J. Elsevier, Angew. Chem., Int. Ed. Engl., 1997, 36, 1743 CrossRef CAS PubMed; (c) R. van Belzen, C. J. Elsevier, A. Dedieu, N. Veldman and A. L. Spek, Organometallics, 2003, 22, 722 CrossRef CAS; (d) G. Yin and G. Liu, Angew. Chem., Int. Ed., 2008, 47, 5442 CrossRef CAS PubMed; (e) T. Furuya and T. Ritter, J. Am. Chem. Soc., 2008, 130, 10060 CrossRef CAS PubMed; (f) D. C. Powers and T. Ritter, Nat. Chem., 2009, 1, 302 CrossRef CAS PubMed; (g) N. D. Ball and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 3796 CrossRef CAS PubMed; (h) T. Wu, G. Yin and G. Liu, J. Am. Chem. Soc., 2009, 131, 16354 CrossRef CAS PubMed; (i) Y. Li, X. Liu, H. Jiang and Z. Feng, Angew. Chem., Int. Ed., 2010, 49, 3338 CrossRef CAS PubMed; (j) T. Furuya, D. Benitez, E. Tkatchouk, A. E. Strom, P. Tang, W. A. Goddard III and T. Ritter, J. Am. Chem. Soc., 2010, 132, 3793 CrossRef CAS PubMed; (k) J. Vicente, A. Arcas, F. Juliá-Hernández and D. Bautista, Chem. Commun., 2010, 46, 7253 RSC; (l) D. C. Powers, D. Benitez, E. Tkatchouk, W. A. Goddard III and T. Ritter, J. Am. Chem. Soc., 2010, 132, 14092 CrossRef CAS PubMed; (m) W. Oloo, P. Y. Zavalij, J. Zhang, E. Khaskin and A. N. Vedernikov, J. Am. Chem. Soc., 2010, 132, 14400 CrossRef CAS PubMed; (n) D. C. Powers, D. Y. Xiao, M. A. L. Geibel and T. Ritter, J. Am. Chem. Soc., 2010, 132, 14530 CrossRef CAS PubMed; (o) Y. Li, X. Liu, H. Jiang, B. Liu, Z. Chen and P. Zhou, Angew. Chem., Int. Ed., 2011, 50, 6341 CrossRef CAS PubMed; (p) K. S. L. Chan, M. Wasa, X. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2011, 50, 9081 CrossRef CAS PubMed; (q) J. M. Racowski, J. B. Gary and M. S. Sanford, Angew. Chem., Int. Ed., 2012, 51, 3414 CrossRef CAS PubMed; (r) M. H. Pérez-Temprano, J. M. Racowski, J. W. Kampf and M. S. Sanford, J. Am. Chem. Soc., 2014, 136, 4097 CrossRef PubMed; (s) E. P. A. Talbot, T. de A. Fernandes, J. M. McKenna and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 4101 CrossRef CAS PubMed.
- J. F. Hartwig, Organotransition metal chemistry: from bonding to catalysis, University of Science Books, Sausalito, CA, 2010 Search PubMed.
- (a) N. C. Bruno, M. T. Tudge and S. L. Buchwald, Chem. Sci., 2013, 4, 916 RSC; (b) N. C. Bruno and S. L. Buchwald, Org. Lett., 2013, 15, 2876 CrossRef CAS PubMed.
- (a) E. A. Mitchell and M. C. Baird, Organometallics, 2007, 26, 5230 CrossRef CAS; (b) D. M. Norton, E. A. Mitchell, N. R. Botros, P. G. Jessop and M. C. Baird, J. Org. Chem., 2009, 74, 6674 CrossRef CAS PubMed; (c) L. Xue and Z. Lin, Chem. Soc. Rev., 2010, 39, 1692 RSC; (d) B. E. Jaksic, J. Jiang, A. W. Fraser and M. C. Baird, Organometallics, 2013, 32, 4192 CrossRef CAS.
- N. Kataoka, Q. Shelby, J. P. Stambuli and J. F. Hartwig, J. Org. Chem., 2002, 67, 5553 CrossRef CAS PubMed.
- (a) W. Geng, W.-X. Zhang, W. Hao and Z. Xi, J. Am. Chem. Soc., 2012, 134, 20230 CrossRef CAS PubMed; (b) W. Hao, W. Geng, W.-X. Zhang and Z. Xi, Chem. – Eur. J., 2014, 20, 2605 CrossRef CAS PubMed; (c) For our “Organometallic intermediate-based organic synthesis” research strategy, see: W.-X. Zhang and Z. Xi, Org. Chem. Front., 2014, 1, 1132 RSC.
- (a) F. Ozawa, A. Kubo and T. Hayashi, Chem. Lett., 1992, 2177 CrossRef CAS; (b) C. Amatore and A. Jutand, Acc. Chem. Res., 2000, 33, 314 CrossRef CAS PubMed; (c) I. D. Hills and G. C. Fu, J. Am. Chem. Soc., 2004, 126, 13178 CrossRef CAS PubMed; (d) K. Ouyang and Z. Xi, Acta Chim. Sinica, 2013, 71, 13 CrossRef CAS; (e) Y. Sunesson, E. Limé, S. O. N. Lill, R. E. Meadows and P.-O. Norrby, J. Org. Chem., 2014, 79, 11961 CrossRef CAS PubMed.
- A. Bellomo, J. Zhang, N. Trongsiriwat and P. J. Walsh, Chem. Sci., 2013, 4, 849 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qo00197h |
| This journal is © the Partner Organisations 2015 |