DOI:
10.1039/C4RA13083A
(Paper)
RSC Adv., 2015,
5, 3122-3129
Facile synthesis of Ag2O/N-doped helical carbon nanotubes with enhanced visible-light photocatalytic activity
Received
24th October 2014
, Accepted 3rd December 2014
First published on 3rd December 2014
Abstract
Novel Ag2O/N-doped helical carbon nanotubes (Ag2O/N–HCNTs) were successfully synthesized via a simple coprecipitation method and were well characterized by X-ray diffraction (XRD), field emission scanning electron microscopy (FESEM), transmission electron microscopy (TEM), and energy dispersive X-ray spectroscopy (EDS). The photocatalytic activities were evaluated in the degradation of methylene blue (MB) aqueous solution. The results showed that Ag2O nanoparticles sized 3–10 nm were highly anchored on the surface and inner tubes of the N–HCNTs support, and significantly enhanced the visible-light photocatalytic activity compared to bare Ag2O. It was attributed to the combined effects, including highly dispersed smaller Ag2O particles and higher charge separation efficiency. The possible mechanism for the photocatalytic activity of Ag2O/N–HCNTs was also tentatively proposed. In particular, the rate of degradation of the as-prepared Ag2O/N–HCNTs was 3.9 times faster than that of using bare Ag2O nanoparticles under visible light irradiation. Furthermore, the Ag2O/N–HCNTs could be easily recycled in visible photocatalytic activity. In addition, the Ag2O/N–HCNTs could also degrade MB dye in different water sources like Changjiang River water and tap water with high efficiency as well as in deionized water and that will greatly promote their application in the area of environmental remediation.
Introduction
In decades, the environmental problem caused by organic pollutants has increased with the rapid development of industry and become a severe threat to human beings.1–4 With the steady and fast growing field of nanoscience and nanotechnology, semiconductor photocatalysts based on harnessing and converting solar energy into chemical energy are attracting increasing attention because of their potential applications in solving environmental pollution.5–10 Among the semiconductors, titanium dioxide (TiO2), owing to its low cost, ease in preparation, environmental abundance, high photocatalytic activity, good chemical stability, and nontoxicity, is considered to be one of the most important semiconductors being harnessed in photoinduced applications such as photodetection and photocatalysis.11,12 However, due to the large band gap (3.2 eV) of TiO2, it only can be excited under UV light irradiation. Meanwhile, visible-light makes up about 45% of solar energy and UV light only around 4%, the development of visible-light-responsive photocatalysts for environmental remediation has become an active area of research in photocatalysis.13–16 Recently, silver-containing compounds have been demonstrated to be efficient photocatalytic materials under visible-light irradiation.17–21 Among the silver-containing compounds, Ag2O is a well-known visible light photocatalyst which shows excellent photocatalytic performance and good stability.22–24 However, the consumption of a large amount of the noble metal of silver strongly limits their practical environmental applications despite the fact that Ag2O photocatalyst exhibited highly efficient in visible-light photocatalytic performance. On the other hand, the particle size of Ag2O photocatalyst remains relatively large, which hinder their performance in visible photocatalytic processes.23,25 Therefore, it should develop a simple and effective technology to reduce the dosage of silver and to synthesize nanosized Ag2O photocatalyst with high efficient photocatalytic activity for large-scale practical applications.
Carbonaceous materials, such as carbon quantum dots, activated carbon, carbon black, and carbon nanotubes, have been demonstrated to be good supports for semiconductor photocatalysts due to their range of light absorption, cheapness, safety, and corrosion resistance.26–29 Among them, one-dimensional carbon nanotubes (CNTs) have been widely used as ideal electron pathways due to their special one-dimensional (1D) character and good conductivity. And some results have demonstrated that CNTs could efficiently capture and transport of photogenerated electrons through highly conductive long CNTs.30–32 In particular, helical carbon nanotubes (HCNTs), have aroused special attention since they were observed by Amelinckx et al. in 1994 based on their outstanding physical and chemical properties and potential applications.33 It is found that HCNTs exhibit unique electrical and mechanical properties that could be utilized in nanoengineering.34–37 The specific helical geometry (such as diameter and coil pitch, etc.) is determined by the periodic incorporation of pentagon and heptagon pairs in hexagonal carbon network.34 Interestingly, depending on the helical geometry, a HCNT can behave as a semiconductor, a semimetal, a metal, or even a superconductor, which is impossible in the case of straight CNTs.38 Moreover, because doping nitrogen into carbon materials could be a promising way to improve their electronic, mechanical, and optical properties, many efforts have been devoted to study N-doped HCNTs (N–HCNTs).36,37,39–41 In addition, the introduction of nitrogen into carbon materials could create more active sites for anchoring functional metal or metallic oxide nanoparticles.42 Judging from the excellent visible photocatalyst of Ag2O and the efficient electron transfer property of N–HCNTs, combination of Ag2O and N–HCNTs seems to be ideal for hindering the recombination of photogenerated electrons (e−) and holes (h+) and improving the photocatalytic efficiency. Besides, to our best of knowledge, there have been no reports on the synthesis and photocatalytic activity of the Ag2O/N–HCNTs under visible light.
Herein, for the first time, the Ag2O/N–HCNTs sample was prepared via a simple adsorption and coprecipitation method using N–HCNTs as the support material at room temperature. The results indicated that Ag2O was anchored on the surface and inner tubes of N–HCNTs with size about 3–10 nm, causing the composite to display stronger visible absorption and to exhibit enhanced photocatalytic activities for the degradation of methylene blue (MB) under visible light compared with separate N–HCNTs and Ag2O. Moreover, Ag2O/N–HCNTs sample could be easily recycled in visible photocatalytic activity and remove dyes in living water samples such as tap water and Changjiang River water with high efficiency as well as in deionized water.
Experimental section
Materials
Pyrrole, ammonium persulfate (APS), silver nitrate (AgNO3), ethanol (EtOH), methanol (MeOH), sodium hydroxide (NaOH) and methylene blue (MB, C16H18ClN3S) were purchased from Aladdin Chemical Regent Co., Ltd. (Shanghai, China). All the reagents in this experiment are analytically pure and used without further purification.
Synthesis of N–HCNTs
The preparation of N–HCNTs is similar to the method described by Liu et al.43 N–HCNTs were prepared with the template of C14-L-Glu, which was synthesized from myristoyl chloride and L-glutamic acid in a mixture of water and acetone as descried previously.44,45 Typically, 0.3 mmol of C14-L-Glu was dissolved in 65 mL of MeOH at room temperature, then 12 mmol of pyrrole and 300 mL of deionized water were added. The aforementioned mixture was stirred for 30 min. Subsequently, 6 mL of APS solution (2 M) was added into the mixture and stirred for another 60 min at room temperature, yielding a dark suspension. Finally, a dark precipitation was collected and washed thoroughly with deionized water and ethanol for several times and finally dried under vacuum. Then the resulting dark product was calcined at 550 °C under a flow of N2 for 6 h. After slowly cooling to room temperature, N–HCNTs sample was obtained.
Formation of Ag2O/N–HCNTs
Typically, 0.03 g of N–HCNTs were dispersed in 300 mL of deionized water by sonication for 60 min to form a uniformly suspension. Subsequently, 0.15 g of AgNO3 was added into the above suspension. After stirring for another 120 min, 15 mL aqueous solution of NaOH (0.5 M) was added to the aforementioned mixture and then stirred for another 30 min. The obtained precipitate was separated by centrifugation and washed thoroughly with deionized water and ethanol for several times to remove any impurities. Finally, the product was dried in vacuum at 45 °C for 12 h. For comparison, bare Ag2O was also synthesized in the same way but in the absence of N–HCNTs.
Characterization
The field emission scanning electron microscopy (FESEM; FEI Inspect F50), transmission electron microscopy (TEM; JEM-1230) and high resolution TEM (HRTEM) were used to characterize the morphologies of the products. Energy dispersive X-ray spectroscopy (EDS) being attached to field emission scanning electron microscopy (FESEM) was used to analyze the composition of samples. X-Ray diffraction (XRD) measurement was carried out using a SmartLab XRD spectrometer (Rigaku) with Cu Kα radiation in the range of 10–70° (2θ). The nitrogen adsorption and desorption isotherms were measured at 77 K on an ASAP 2020 (Micromertics USA). UV-vis diffuse reflectance spectra (DRS) of the samples were recorded on a UV-vis spectrophotometer (UV-3600, Shimadzu) with an integrating sphere attachment.
Evaluation of photocatalytic performance
The photodegradation of MB was chosen as a model reaction to evaluate the photocatalytic activities of the Ag2O/N–HCNTs. A 500 W xenon lamp (Philips) equipped with a UV cut-off filter with λ > 400 nm was used as the light source. The experiments were carried out at room temperature as follows: the photocatalyst (50 mg) was added to a 100 mL of MB aqueous solution (10 mg L−1) in a Pyrex reactor. Before irradiation, the mixed suspension was stirred for 30 min in the dark to reach an adsorption–desorption equilibrium between the photocatalyst and MB dye. Subsequently, the above mixture was irradiated in a photochemical chamber under continuously stirring with reflux water to keep its temperature constant. At intervals of 30 min, 3 mL of the suspension was collected and centrifuged to remove the remnant photocatalyst. The concentration of MB in the supernatant was then performed by measuring its intensity of the absorption peak with a UV-vis spectrophotometer (UV-3600; Shimadzu). For comparison, the photocatalytic activities of bare Ag2O and N–HCNTs were also determined under identical conditions.
Results and discussion
Structure and morphology
X-Ray diffraction (XRD) analysis was employed to investigate the phase structure of the as-synthesized samples. As shown in Fig. 1(a), the N–HCNTs sample shows a broad peak and the diffraction peak at 24.6° corresponds to d spacing of 0.36 nm.46 In the diffraction patterns of the as-prepared Ag2O/N–HCNTs, the major diffraction peaks at 2θ = 32.8°, 38.1°, 54.9°, 65.4° and 68.7° are in accordance with the standard data of well-crystallized cubic Ag2O (JCPDS 41-1104). Meanwhile, the weak and broad peak in the range of 20°–30° (inset of Fig. 1(a)) originates from the N–HCNTs sample. No characteristic peaks for other impurities were observed, which indicated that the product had high purity. And the EDS analysis (Fig. 1(b)) further confirms that the product was only composed of C, N, O and Ag.
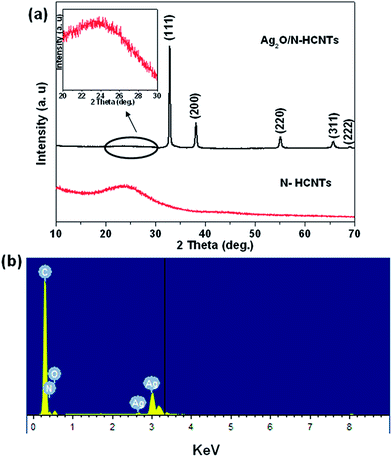 |
| | Fig. 1 XRD patterns of the as-prepared (a) N–HCNTs and Ag2O/N–HCNTs, (b) EDS spectrum of the Ag2O/N–HCNTs. | |
The sizes, morphologies, and microstructures of the as-prepared N–HCNTs, bare Ag2O and Ag2O/N–HCNTs were elucidated by field emission scanning electron microscopy (FESEM), transmission electron microscopy (TEM) and high resolution TEM (HRTEM). Fig. 2(a) and (b) shows representative FESEM images of the synthesized N–HCNTs and bare Ag2O. And Fig. 3 shows the typical TEM and HRTEM images of bare N–HCNTs, individual N–HCNT and Ag2O/N–HCNTs, respectively. As seen from Fig. 2(a) and 3(a), the N–HCNTs sample is composed of exclusively left-handed helical fibers with uniform morphology. The outer diameters of N–HCNTs are in the same range of 70–100 nm and in length of several micrometers. The pitches of outer surface along the rod axis are estimated to be all about 80 nm, as indicated by black arrows in Fig. 3(b). In Fig. 2(b), it can be clearly seen that the Ag2O nanoparticles possess an irregular spherical morphology with an average diameter of approximately 100 nm. In contrast, from Fig. 3(c), the Ag2O nanoparticles are clearly and uniformly anchored on the surface and inner tubes of N–HCNTs to form Ag2O/N–HCNTs, which is propitious to electron transmission between two phases. By measuring the lattice fringes in HRTEM image (Fig. 3(d)), the resolved interplanar distance of 0.235 nm which enlarged inset of Fig. 3(d) agreed well with the lattice spacing of the plane of Ag2O. This is indicative of the formation of Ag2O/N–HCNTs. Compared with the bare Ag2O nanoparticles prepared in the absence of N–HCNTs, the Ag2O particles in the Ag2O/N–HCNTs hybrid are smaller with diameters in the range of 3–10 nm (Fig. 3(d)). The smaller size of the Ag2O nanoparticles may be attributed to the existence of N–HCNTs in the Ag2O/N–HCNTs systems, which probably provide a platform to prevent the agglomeration of Ag2O particles.
 |
| | Fig. 2 FESEM images of bare (a) N–HCNTs and (b) Ag2O nanoparticles. | |
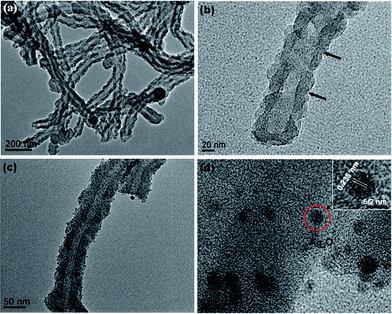 |
| | Fig. 3 Typical TEM images of (a) bare N–HCNTs, (b) an individual N–HCNT and (c) Ag2O/N–HCNTs; (d) HRTEM image of Ag2O/N–HCNTs. Insets in (d) is the enlarged image. | |
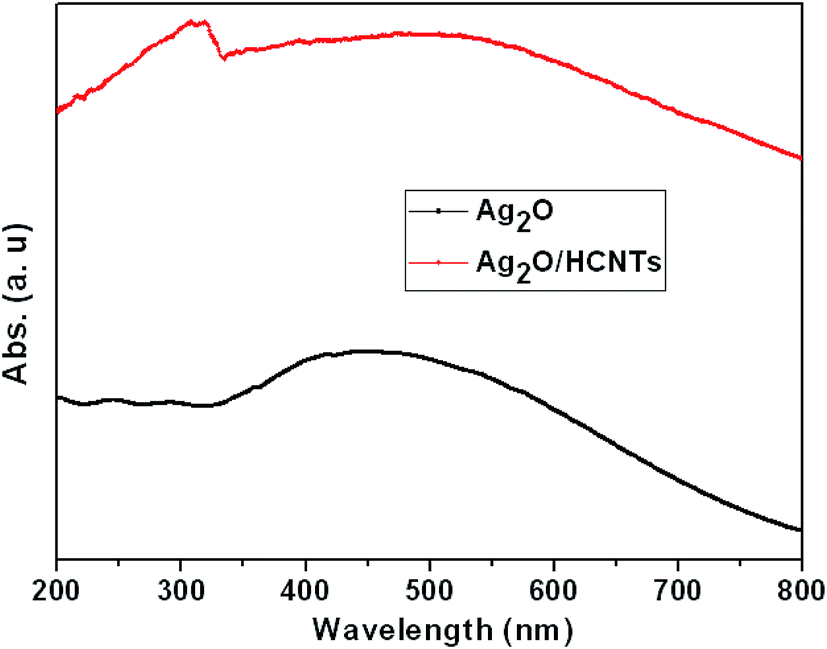
It is well known that optical absorption plays an important role in determining the photocatalytic performance of a catalyst, especially in the visible-light photodegradation of contaminants.47,48 The optical properties of bare Ag2O and Ag2O/N–HCNTs were probed by UV-vis diffuse reflectance spectroscopy (Fig. 4). The UV-vis spectrum of the as-prepared Ag2O clearly exhibits broad and strong absorption intensity in the whole UV and visible-light region of 200–800 nm, which is in agreement with previous reports.22 The Ag2O/N–HCNTs nanocomposites exhibit more intense in the range of 200–800 nm than that of bare Ag2O, which suggests that the Ag2O/N–HCNTs photocatalyst may absorb visible light more efficiently. An analogous phenomenon has also been reported by other researchers.49,50
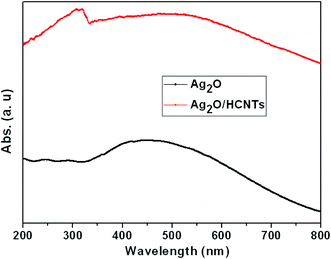 |
| | Fig. 4 UV-vis diffuse reflectance spectra of bare Ag2O and Ag2O/N–HCNTs. | |
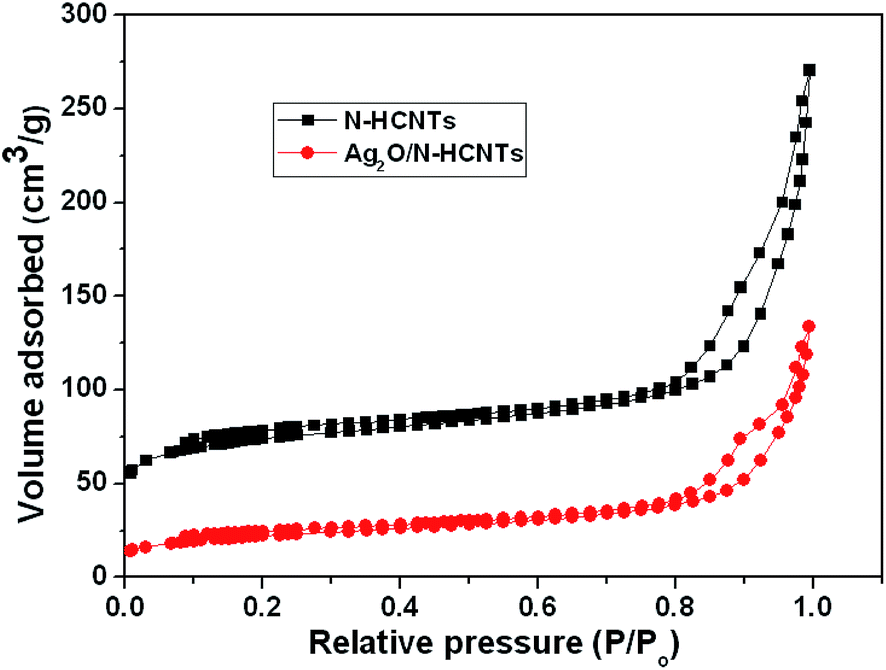
The nitrogen adsorption–desorption isotherms of N–HCNTs and Ag2O/N–HCNTs are shown in Fig. 5. It can be seen that both of the N–HCNTs and Ag2O/N–HCNTs samples show a type IV adsorption isotherm and H3 hysteresis loop, indicating the presence of mesoporous (2–50 nm).51,52 The BET surface area and the single-point total pore volume for N–NCNTs came out to be 272.6 m2 g−1 and 0.42 cm3 g−1, respectively. The average BJH pore diameter calculated from the desorption branch of the isotherms was determined to be 11 nm. However, the values for the BET surface area, total pore volume, and pore diameter of the Ag2O/N–HCNTs sample was calculated at 78 m2 g−1, 0.21 cm3 g−1, and 14.1 nm, respectively. Compared with N–HCNTs, the Ag2O/N–HCNTs sample show a monotonic decrease in BET surface areas and pore volumes and increase in average pore size. This is probably caused by the heavy coating of the N–HCNTs by small crystallite size of Ag2O nanoparticles.53
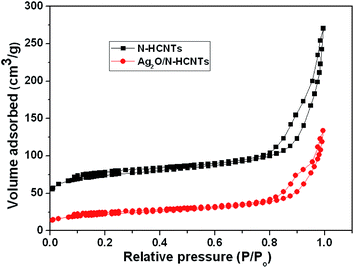 |
| | Fig. 5 N2 adsorption–desorption isotherms of N–HCNTs and Ag2O/N–HCNTs. | |
Formation mechanism of Ag2O/N–HCNTs
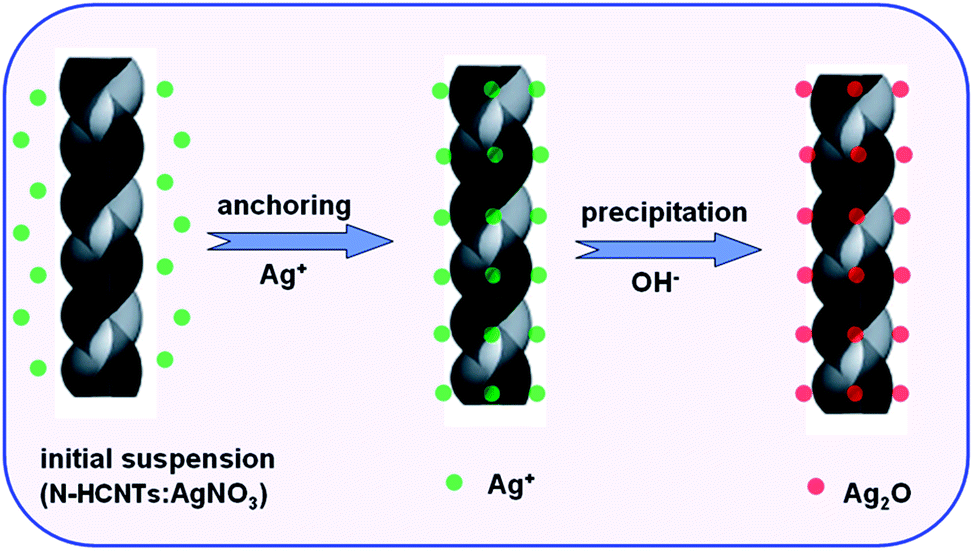
Based on the above results, we speculated a possible formation mechanism for the preparation of Ag2O/N–HCNTs, as illustrated in Scheme 1. In the previous reports, the incorporation of nitrogen atoms into CCNTs could create more active sites for anchoring functional metal nanoparticles, such as Pt and Ag, thus resulting in the use of CCNTs as novel supports of catalysts for direct methanol fuel cells (DMFC), bactericides, and sensors.46,54 Herein, the fabrication of Ag2O/N–HCNTs is based on the anchoring of positively charged Ag+ ions on the surface and inner tubes of N–HCNTs, and followed by subsequent in situ growth of Ag2O.
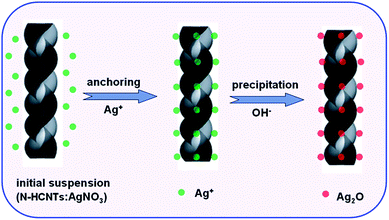 |
| | Scheme 1 Schematic illustration of the fabrication route of Ag2O/N–HCNTs. | |
Photocatalytic activity
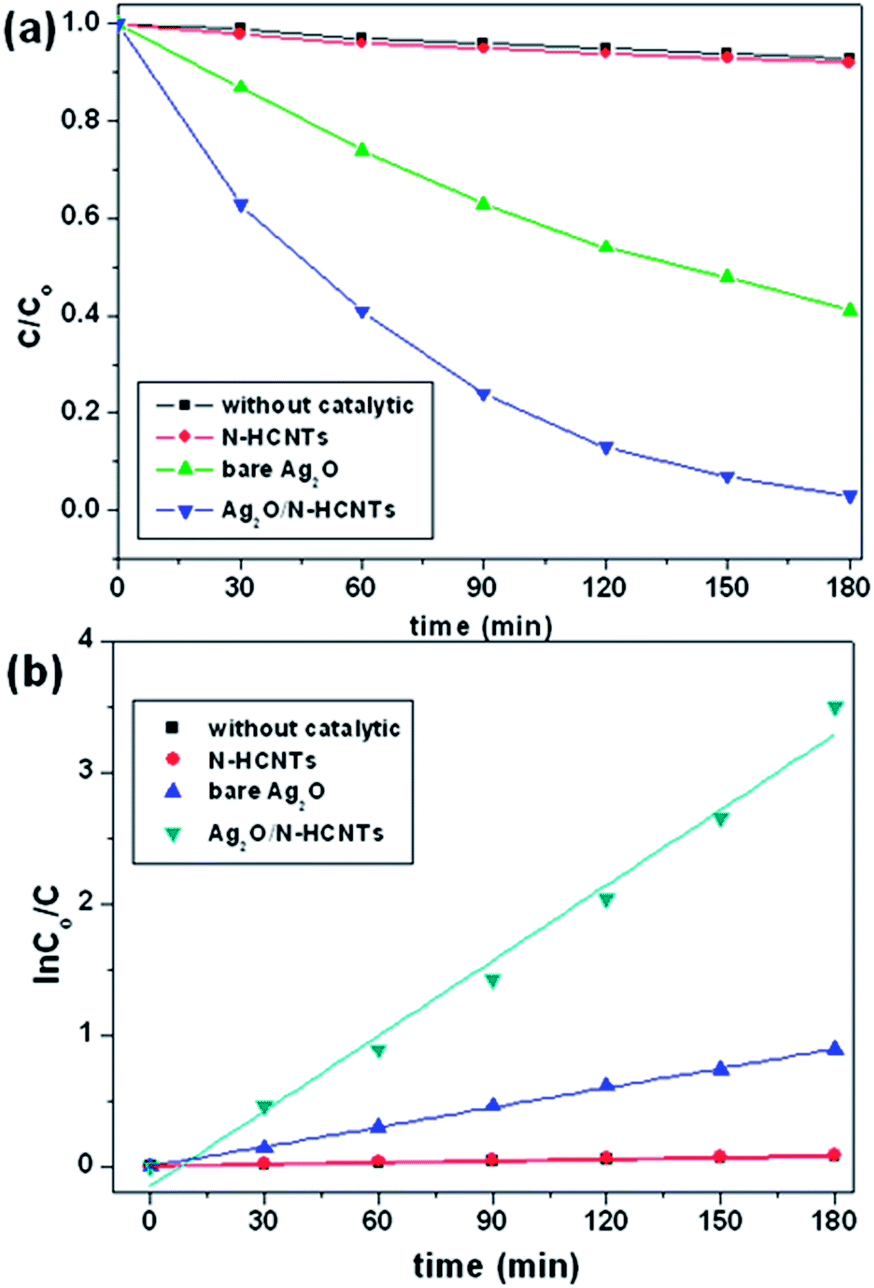
The photcatalytic performances in UV and visible light regions were investigated via the degradation of MB, which is a typical cationic organic pollutant usually discharged by the textile industry after used. According to the Beer–Lambert law, the concentration of MB is linearly proportional to the intensity of the absorption peak at 664 nm, and thus the decomposition efficiency of MB can be calculated using the following expression:| | |
MB decomposition (%) = 100 × (Co − C)/Co
| (1) |
where, Co and C are the equilibrium concentrations of MB before and after UV irradiation, respectively. Fig. 6(a) shows the photocatalytic decomposition of MB monitored according to the concentration change versus time for the various samples. As can be seen, the concentrations of the MB solution are almost unchanged in the absence of any catalyst or with bare N–HCNTs. However, it is noticeable that the Ag2O/N–HCNTs composite exhibit significantly enhanced activity compared with bare Ag2O and N–HCNTs. After 180 min of visible light irradiation, the photodegradation efficiency of Ag2O/N–HCNTs nearly reach 97%, while the bare Ag2O and N–HCNTs can only approach 59% and 8% for the same irradiation time, respectively. Furthermore, to quantitatively understand the reaction kinetics of MB degradation over different samples in our experiments, we re-plotted the data in Fig. 5(b) according to the pseudo-first-order kinetic model as expressed by eqn (2), which is generally used for photocatalytic degradation process take place at the interface between the catalysts and the organic pollutants with low concentration.where, t is reaction time, k is the rate constant, Co and C are the concentrations of MB solution at time 0 and t, respectively.55 From Fig. 6(b), the rate constants of Ag2O/N–HCNTs, bare Ag2O and N–HCNTs are 0.0195 min−1, 0.005 min−1 and 0.0005 min−1, respectively. The Ag2O/N–HCNTs exhibit the highest rate constant, which is approximately 3.9 times larger than that of pure Ag2O. The enhanced photocatalytic activities were attributed to combined effects, including the highly dispersed smaller Ag2O particles and the higher separation efficiency of photoinduced electron–hole pairs based on the exist of N–HCNTs.
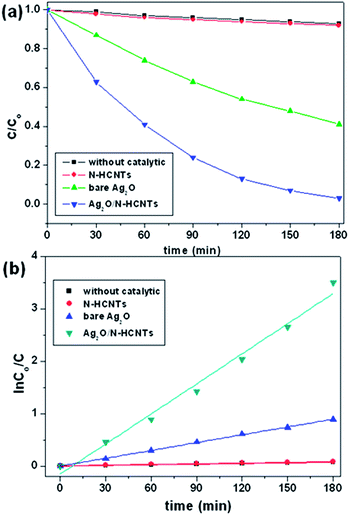 |
| | Fig. 6 Photocatalytic activity (a) and kinetics (b) of the as-prepared Ag2O/N–HCNTs, N–HCNTs, and bare Ag2O for degradation of MB under visible light irradiation. | |
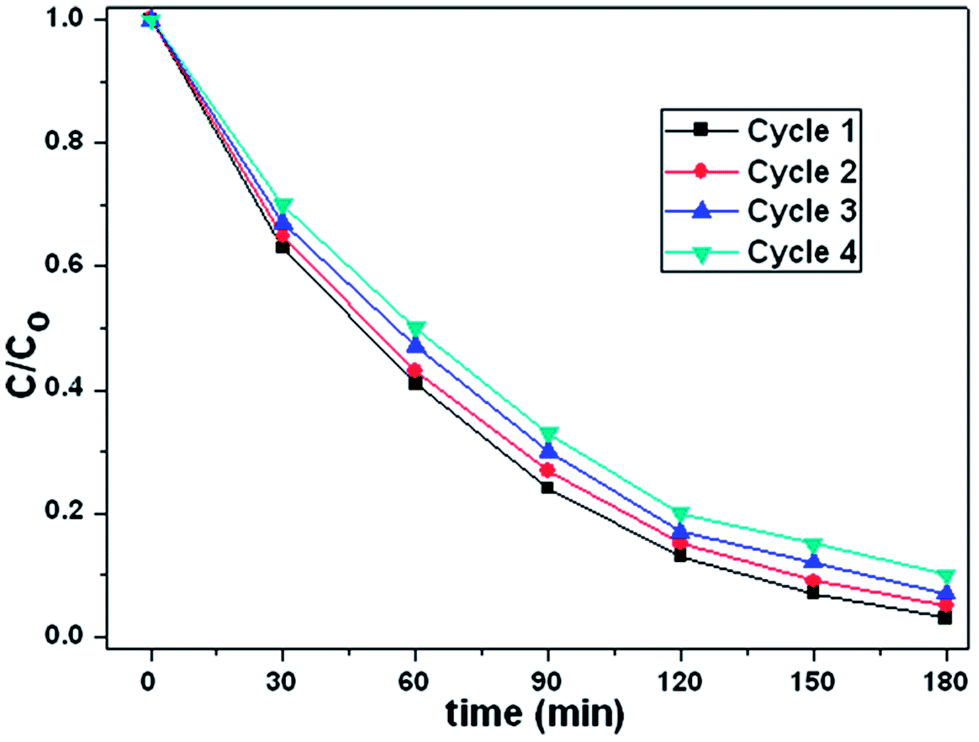
To investigate the stability of photocatalytic performance in visible light region, the as-prepared Ag2O/N–HCNTs were used to degrade MB dye in four repeated cycles, and the results are shown in Fig. 7. It is noteworthy that the photocatalytic performance of the as-prepared Ag2O/N–HCNTs exhibit effective photostability under visible light irradiation, where the photocatalytic efficiency reduces only by 8% after four cycles, indicate that the good stability of the Ag2O/N–HCNTs photocatalyst.
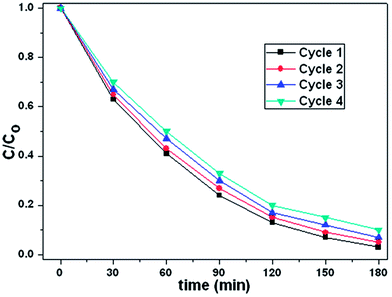 |
| | Fig. 7 Four photocatalytic degradation cycles of MB using Ag2O/N–HCNTs under visible light irradiation. | |
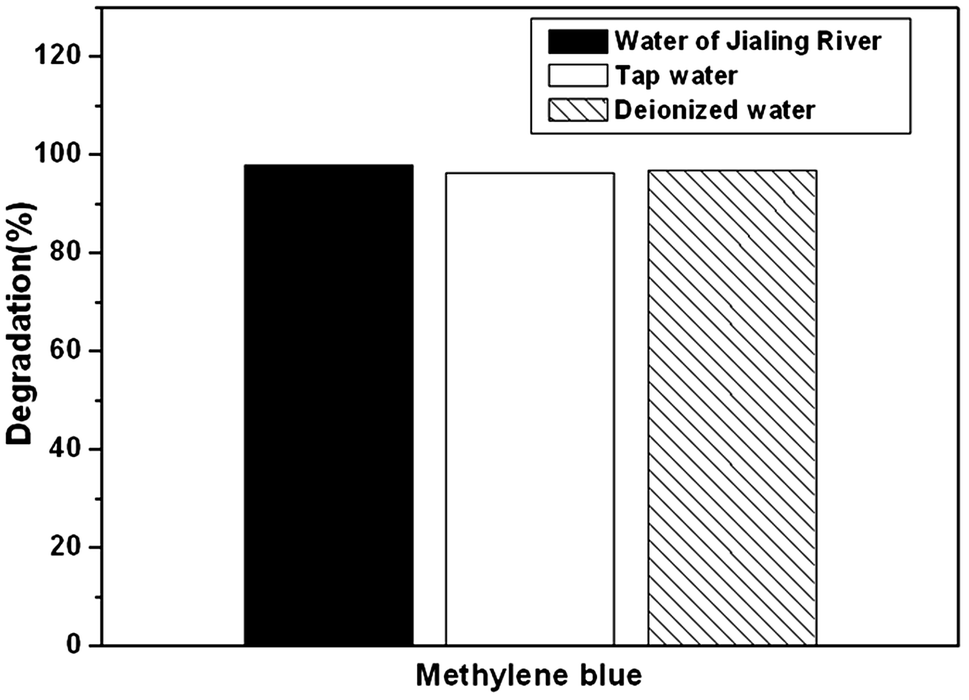
For further practical applications, Fig. 8 shows the photocatalytic activity of the as-prepared Ag2O/N–HCNTs for degradation of MB in different water sources under visible light irradiation. Firstly, MB was added into tap water and Changjiang River water to form different MB solutions with the same concentration in deionized water. Then, the as-prepared Ag2O/N–HCNTs were added into different MB solutions, followed by the same photocatalytic experiment steps mentioned above. As can be seen from Fig. 8, the as-prepared Ag2O/N–HCNTs almost exhibit the same photocatalytic activity in different water sources under visible light irradiation, suggesting that this sample was suitable for removal of dyes from different water sources. It is also indicated that the as-prepared Ag2O/N–HCNTs have great potential applications for pollution control in our environment.
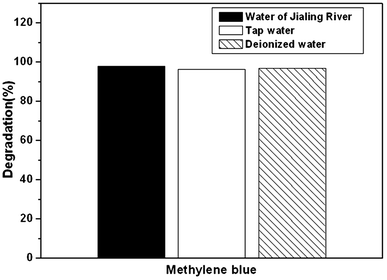 |
| | Fig. 8 Photocatalytic activity of the as-prepared Ag2O/N–HCNTs for degradation of MB in different water sources under visible light irradiation. | |
Mechanisms in enhancing photocatalytic activity
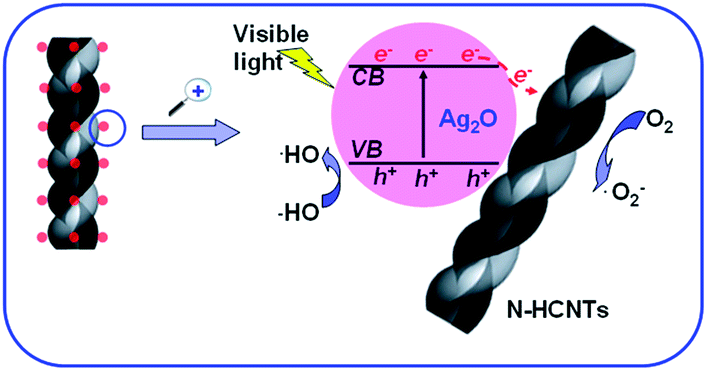
Based on the results above, a possible photocatalytic mechanism of the as-prepared Ag2O/N–HCNTs under visible light irradiation was proposed and illustrated in Fig. 9. The mechanism was that the N–HCNTs supported catalysts were believed to exhibit cooperative or synergetic effects between N–HCNTs and semiconductive metal oxides. The work function of CNTs has been computed to be ranging from 4.3 to 5.1 eV,56 Ag2O semiconductor has the conduction band ca. 0.19 eV and a band gap of about 1.2 eV.57 The conducting band of Ag2O is smaller than work functions of CNTs, such that the photogenerated electrons transfer from Ag2O to CNTs is energetically favorable.58 Here, when the as-prepared Ag2O/N–HCNTs were irradiated with visible light, Ag2O could be excited due to its narrow band gap (1.2 eV) and the photogenerated electrons (e−) were produced in the conduction band (CB) while the photogenerated holes (h+) remained in the valence band (VB).59 Subsequently, photogenerated electrons (e−) in Ag2O conduction band (CB) might move freely toward the surface of the N–HCNTs and excess of valence band holes were left in the Ag2O to migrate to the surface and react with H2O or OH− to produce active species such as OH˙,26 suggesting that the photogenerated electrons and holes were efficiently separated and the lifetime of the excited electrons and holes could be prolonged in the transfer process. Thus, the lifetime of the excited electrons and holes can be prolonged in the transfer process, inducing higher quantum efficiency, and thus the photocatalytic activity of the as-prepared Ag2O/N–HCNTs is enhanced greatly. The mechanism for the photocatalytic degradation of MB in our experiment was proposed as follows:
| Ag2O/N–HCNTs (s) + hν → h+ (Ag2O) + e− (N–HCNTs) |
| OH˙ + MB (dye) → CO2 + H2O |
| h+ + MB (dye) → CO2 + H2O |
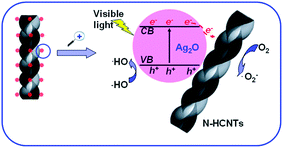 |
| | Fig. 9 Proposed mechanism for the photocatalytic degradation of MB over Ag2O/N–HCNTs under visible-light irradiation. | |
Under visible-light irradiation, photogenerated electrons (e−) in Ag2O moved freely to the surface of the N–HCNTs. Meanwhile, the photogenerated holes (h+) were left in the valence band of Ag2O. Subsequent to various steps, the photogenerated holes (h+) were ultimately trapped by surface hydroxyl groups (or H2O) on the surface of catalyst to yield OH˙ radicals. Simultaneously, the dissolved oxygen molecules react with the surface of the N–HCNTs photogenerated electrons (e−) to yield super oxide radical anions (O2˙−), which on protonation generate the hydroperoxy (HO2˙) radicals, producing hydroxyl radical OH˙, which was a strong oxidizing agent to decompose organic dye.60
Conclusions
In summary, novel visible-light-responsive Ag2O/N–HCNTs have been successfully synthesized by a simple coprecipitation route. The Ag2O nanoparticles were evenly and densely distributed over the surface and inner tubes of N–HCNTs. The incorporation of Ag2O in N–HCNTs can promote the formation of Ag2O nanoparticles with a smaller size and significantly enhance the photocatalytic activity of the composite catalysts in the degradation of the contaminant MB under visible-light irradiation. The enhanced photocatalytic activity of the Ag2O/N–HCNTs can be attributed to the synergic effect of the effective separation of the photogenerated carriers and the smaller particle size of Ag2O when coupled with the N–HCNTs. In addition, the Ag2O/N–HCNTs could be easily recycled in visible photocatalytic activity and degrade MB dye in different water sources such as Changjiang River water and tap water with high efficiency as well as in deionized water. The high and stable catalytic activity makes the Ag2O/N–HCNTs promising photocatalysts for the resolution of energy and environmental issues.
Acknowledgements
The authors are grateful to National Natural Science Foundation of China (Grant no. 21106017 and 51077013), Fund Project for Transformation of Scientific and Technological Achievements of Jiangsu Province of China (Grant no. BA2011086), Specialized Research Fund for the Doctoral Program of Higher Education of China (Grant no. 20100092120047) and Key Program for the Scientific Research Guiding Found of Basic Scientific Research Operation Expenditure of Southeast University (Grant no. 3207042102).
References
- C. C. Chen, W. H. Ma and J. C. Zhao, Chem. Soc. Rev., 2010, 39, 4206–4219 RSC.
- D. Q. Zhang, M. C. Wen, B. Jiang, G. S. Li and J. C. Yu, J. Hazard. Mater., 2012, 211, 104–111 CrossRef PubMed.
- M. A. Shannon, P. W. Bohn, M. Elimelech, J. G. Georgiadis, B. J. Marinas and A. M. Mayes, Nature, 2008, 452, 301–310 CrossRef CAS PubMed.
- C. J. Vorosmarty, P. B. McIntyre, M. O. Gessner, D. Dudgeon, A. Prusevich, P. Green, S. Glidden, S. E. Bunn, C. A. Sullivan, C. R. Liermann and P. M. Davies, Nature, 2010, 467, 555–561 CrossRef CAS PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- S. A. K. Leghari, S. Sajjad, F. Chen and J. L. Zhang, Chem.–Eng. J., 2011, 166, 906–915 CrossRef CAS PubMed.
- X. Xu, X. P. Shen, H. Zhou, D. Z. Qiu, G. X. Zhu and K. M. Chen, Appl. Catal., A, 2013, 455, 183–192 CrossRef CAS PubMed.
- N. Watanabe, T. Kaneko, Y. Uchimaru, S. Yanagida, A. Yasumori and Y. Sugahara, CrystEngComm, 2013, 15, 10533–10540 RSC.
- H. B. Lu, S. M. Wang, L. Zhao, J. C. Li, B. H. Dong and Z. X. Xu, J. Mater. Chem., 2011, 21, 4228–4234 RSC.
- S. S. Ma, R. Li, C. P. Lv, W. Xu and X. L. Gou, J. Hazard. Mater., 2011, 192, 730–740 CrossRef CAS PubMed.
- X. Zhang, V. Thavasi, S. G. Mhaisalkar and S. Ramakrishna, Nanoscale, 2012, 4, 1707–1716 RSC.
- M. M. Khan, S. A. Ansari, D. Pradhan, M. O. Ansari, D. H. Han, J. Lee and M. H. Cho, J. Mater. Chem. A, 2014, 2, 637–644 CAS.
- J. G. Yu, J. Jin, B. Cheng and M. Jaroniec, J. Mater. Chem. A, 2014, 2, 3407–3416 CAS.
- X. X. Wei, C. M. Chen, S. Q. Guo, F. Guo, X. M. Li, X. X. Wang, H. T. Cui, L. F. Zhao and W. Li, J. Mater. Chem. A, 2014, 2, 4667–4675 CAS.
- X. J. Feng, T. J. LaTempa, J. I. Basham, G. K. Mor, O. K. Varghese and C. A. Grimes, Nano Lett., 2010, 10, 948–952 CrossRef CAS PubMed.
- Z. Y. Liu, D. D. L. Sun, P. Guo and J. O. Leckie, Nano Lett., 2007, 7, 1081–1085 CrossRef CAS PubMed.
- H. Kato, H. Kobayashi and A. Kudo, J. Phys. Chem. B, 2002, 106, 12441–12447 CrossRef CAS.
- R. Konta, H. Kato, H. Kobayashi and A. Kudo, Phys. Chem. Chem. Phys., 2003, 5, 3061–3065 RSC.
- T. Kako, N. Kikugawa and J. Ye, Catal. Today, 2008, 131, 197–202 CrossRef CAS PubMed.
- P. Wang, B. B. Huang, X. Y. Qin, X. Y. Zhang, Y. Dai, J. Y. Wei and M. H. Whangbo, Angew. Chem., Int. Ed., 2008, 47, 7931–7933 CrossRef CAS PubMed.
- D. W. Wang, Y. Li, G. L. Puma, C. Wang, P. F. Wang, W. L. Zhang and Q. Wang, Chem. Commun., 2013, 49, 10367–10369 RSC.
- X. F. Wang, S. F. Li, H. G. Yu, J. G. Yu and S. W. Liu, Chem.–Eur. J., 2011, 17, 7777–7780 CrossRef CAS PubMed.
- Z. Y. Ji, X. P. Shen, J. L. Yang, Y. L. Xu, G. X. Zhu and K. M. Chen, Eur. J. Inorg. Chem., 2013, 2013, 6119–6125 CrossRef CAS.
- G. Wang, X. C. Ma, B. B. Huang, H. F. Cheng, Z. Y. Wang, J. Zhan, X. Y. Qin, X. Y. Zhang and Y. Dai, J. Mater. Chem., 2012, 22, 21189–21194 RSC.
- L. Shi, L. Liang, J. Ma, F. X. Wang and J. M. Sun, Catal. Sci. Technol., 2014, 4, 758–765 CAS.
- K. Woan, G. Pyrgiotakis and W. Sigmund, Adv. Mater., 2009, 21, 2233–2239 CrossRef CAS.
- Q. H. Liang, Y. Shi, W. J. Ma, Z. Li and X. M. Yang, Phys. Chem. Chem. Phys., 2012, 14, 15657–15665 RSC.
- B. Y. Yu and S. Y. Kwak, J. Mater. Chem., 2012, 22, 8345–8353 RSC.
- J. B. Mu, C. L. Shao, Z. C. Guo, Z. Y. Zhang, M. Y. Zhang, P. Zhang, B. Chen and Y. C. Liu, ACS Appl. Mater. Interfaces, 2011, 3, 590–596 CAS.
- H. E. Unalan, D. Wei, K. Suzuki, S. Dalal, P. Hiralal, H. Matsumoto, S. Imaizumi, M. Minagawa, A. Tanioka, A. J. Flewitt, W. I. Milne and G. A. J. Amaratunga, Appl. Phys. Lett., 2008, 93, 133116 CrossRef PubMed.
- S. Da Dalt, A. K. Alves and C. P. Bergmann, Mater. Res. Bull., 2013, 48, 1845–1850 CrossRef CAS PubMed.
- S. H. Wang and S. Q. Zhou, J. Hazard. Mater., 2011, 185, 77–85 CrossRef CAS PubMed.
- S. Amelinckx, X. B. Zhang, D. Bernaerts, X. F. Zhang, V. Ivanov and J. B. Nagy, Science, 1994, 265, 635–639 CAS.
- K. Akagi, R. Tamura, M. Tsukada, S. Itoh and S. Ihara, Phys. Rev. Lett., 1995, 74, 2307–2310 CrossRef CAS.
- A. Volodin, D. Buntinx, M. Ahlskog, A. Fonseca, J. B. Nagy and C. Van Haesendonck, Nano Lett., 2004, 4, 1775–1779 CrossRef CAS.
- J. F. Wen, Y. Zhang, N. J. Tang, X. G. Wan, Z. H. Xiong, W. Zhong, Z. L. Wang, X. L. Wu and Y. W. Du, J. Phys. Chem. C, 2011, 115, 12329–12334 CAS.
- Y. Liu, N. J. Tang, W. Kuo, C. W. Jiang, J. F. Wen and Y. W. Du, J. Phys. Chem. C, 2012, 116, 14584–14590 CAS.
- D. M. Chen, K. W. Wang, D. G. Xiang, R. L. Zong, W. Q. Yao and Y. F. Zhu, Appl. Catal., B, 2014, 147, 554–561 CrossRef CAS PubMed.
- Y. Ganesan, C. Peng, Y. Lu, L. Ci, A. Srivastava, P. M. Ajayan and J. Lou, ACS Nano, 2010, 4, 7637–7643 CrossRef CAS PubMed.
- P. Ayala, R. Arenal, M. Rummeli, A. Rubio and T. Pichler, Carbon, 2010, 48, 575–586 CrossRef CAS PubMed.
- K. Xiao, Y. Fu, Y. Q. Liu, G. Yu, J. Zhai, L. Jiang, W. P. Hu, Z. G. Shuai, Y. Luo and D. B. Zhu, Adv. Funct. Mater., 2007, 17, 2842–2846 CrossRef CAS.
- L. L. Wang, L. Shen, L. P. Zhu, H. Y. Jin, N. C. Bing and L. J. Wang, J. Nanomater., 2012, 794625 Search PubMed.
- S. H. Liu, Y. Y. Duan, X. J. Feng, J. Yang and S. A. Che, Angew. Chem., Int. Ed., 2013, 52, 6858–6862 CrossRef CAS PubMed.
- Y. J. Zhang, Y. L. Song, Y. Y. Zhao, T. J. Li, L. Jiang and D. B. Zhu, Langmuir, 2001, 17, 1317–1320 CrossRef CAS.
- M. Takehara, K. Takizawa, I. Yoshimur and R. Yoshida, J. Am. Oil Chem. Soc., 1972, 49, 157–161 CrossRef CAS.
- Y. W. Ma, S. J. Jiang, G. Q. Jian, H. S. Tao, L. S. Yu, X. B. Wang, X. Z. Wang, J. M. Zhu, Z. Hu and Y. Chen, Energy Environ. Sci., 2009, 2, 224–229 CAS.
- N. Zhang, Y. H. Zhang and Y. J. Xu, Nanoscale, 2012, 4, 5792–5813 RSC.
- Y. H. Zhang, Z. R. Tang, X. Z. Fu and Y. J. Xu, ACS Nano, 2010, 4, 7303–7314 CrossRef CAS PubMed.
- G. Z. Liao, S. Chen, X. Quan, H. T. Yu and H. M. Zhao, J. Mater. Chem., 2012, 22, 2721–2726 RSC.
- J. C. Liu, H. W. Bai, Y. J. Wang, Z. Y. Liu, X. W. Zhang and D. D. Sun, Adv. Funct. Mater., 2010, 20, 4175–4181 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- A. Hebrard, D. Oulahna, L. Galet, B. Cuq, J. Abecassis and J. Fages, Powder Technol., 2003, 130, 211–218 CrossRef CAS.
- J. G. Yu and J. Zhang, Dalton Trans., 2010, 39, 5860–5867 RSC.
- A. Zamudio, A. L. Elias, J. A. Rodriguez-Manzo, F. Lopez-Urias, G. Rodriguez-Gattorno, F. Lupo, M. Ruhle, D. J. Smith, H. Terrones, D. Diaz and M. Terrones, Small, 2006, 2, 346–350 CrossRef CAS PubMed.
- M. S. Lee, S. S. Park, G. D. Lee, C. S. Ju and S. S. Hong, Catal. Today, 2005, 101, 283–290 CrossRef CAS PubMed.
- H. Ago, T. Kugler, F. Cacialli, W. R. Salaneck, M. S. P. Shaffer, A. H. Windle and R. H. Friend, J. Phys. Chem. B, 1999, 103, 8116–8121 CrossRef CAS.
- Y. Xu and M. A. A. Schoonen, Am. Mineral., 2000, 85, 543–556 CAS.
- C. X. Guo, H. B. Yang, Z. M. Sheng, Z. S. Lu, Q. L. Song and C. M. Li, Angew. Chem., Int. Ed., 2010, 49, 3014–3017 CrossRef CAS PubMed.
- S. S. Ma, J. J. Xue, Y. M. Zhou and Z. W. Zhang, J. Mater. Chem. A, 2014, 2, 7272–7280 CAS.
- T. Aarthi and G. Madras, Ind. Eng. Chem. Res., 2007, 46, 7–14 CrossRef CAS.
Footnote |
| † J. J. Xue and S. S. Ma contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.