
Hydrothermal synthesis of zinc oxide-reduced graphene oxide nanocomposites for an electrochemical hydrazine sensor
Junwei Dinga,
Shiying Zhua,
Tao Zhua,
Wei Suna,
Qing Lib,
Gang Wei*b and
Zhiqiang Su*a
aState Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, 100029 Beijing, China. E-mail: suzq@mail.buct.edu.cn
bHybrid Materials Interface Group, Faculty of Production Engineering, University of Bremen, D-28359 Bremen, Germany. E-mail: wei@uni-bremen.de
First published on 24th February 2015
Abstract
We report here a facile synthesis of different zinc oxide (ZnO) nanostructures on reduced graphene oxide (RGO) by an in situ hydrothermal reaction. ZnO nanostructures with different morphologies on the surface of RGO were successfully synthesized by adjusting the mass ratio of Zn2+ to RGO in this reaction system. It was found that ZnO nanostructures with nanoparticles, mixed nanoparticles and microspindles, and microspindles were formed on RGO by adjusting the mass ratio of Zn2+ to RGO. The synthesized ZnO–RGO nanocomposites with different structures were immobilized onto glassy carbon electrodes and applied to construct electrochemical hydrazine (N2H4) sensors. The results indicate that the ZnO–RGO nanocomposites created with the mass ratio of 4.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 present the best sensor performance. The fabricated N2H4 sensor exhibited a fast amperometric response to N2H4 with a linear detection range from 1.0 μM to 33.5 mM and a detection limit of 0.8 μM. The superior performance is ascribed to the unique structure of the synthesized ZnO and the excellent conductivity of RGO. In addition, we found that the synthesized ZnO–RGO composites exhibited improved electrochemical stability. Such novel ZnO–RGO hybrid materials represent promising nonenzymatic electrochemical N2H4 sensors with high sensitivity and selectivity, improved stability, and fast amperometric response.
:1 present the best sensor performance. The fabricated N2H4 sensor exhibited a fast amperometric response to N2H4 with a linear detection range from 1.0 μM to 33.5 mM and a detection limit of 0.8 μM. The superior performance is ascribed to the unique structure of the synthesized ZnO and the excellent conductivity of RGO. In addition, we found that the synthesized ZnO–RGO composites exhibited improved electrochemical stability. Such novel ZnO–RGO hybrid materials represent promising nonenzymatic electrochemical N2H4 sensors with high sensitivity and selectivity, improved stability, and fast amperometric response.
1. Introduction
Hydrazine (N2H4) and its derivatives have wide applications in the chemical industry and pharmaceutical fields, including for high-energy propellants, fuel cells, oxygen scavengers, photographic developers, herbicides, and pesticides.1,2 However, N2H4 is also recognized as an environmental pollutant because of its high toxicity and irritant effect. Moreover, as a carcinogenic and mutagenic substance, N2H4 has adverse health effects, especially on the central nervous system.3 Therefore, the accurate and sensitive detection of N2H4 is of great importance. Compared with the methods that have been utilized for the detection of N2H4, such as titrimetry,4 gas chromatography,5 chemiluminescence,6 and spectrophotometry,7,8 the electrochemical method provides clear advantages like high sensitivity, rapid response, wide linear range, low cost, portability, and ease of operating procedure.9–11 It has been reported that the anodic electrochemical oxidation of N2H4 can happen on Au, Ag, Pt, Rh and Pd electrodes, but such metals are too expensive for practical applications and normally relatively high overpotentials were generally required.12 A large overpotential for N2H4 oxidation has also been observed at carbon macroelectrodes.13 One promising method to address the above concern is the modification of electrodes with redox mediators.14,15Graphene (G) is one of the famous materials that has wide applications in the electrochemical field because of its unique properties and potential applications in catalysis,16 sensors,17–19 drug delivery,20 and solar cells.21 Nowadays, reduced graphene oxide (RGO) can be prepared in large quantity by thermal,22 chemical,23,24 and solvothermal reduction25 of graphene oxide (GO). These methods are multipurpose, scalable, and adaptable to a wide variety of applications.
ZnO is biocompatible, non-toxic, thermally stable, and electrochemically active.26–29 ZnO-based nanostructures have been widely used for the fabrication of efficient amperometric sensors, such as N2H4 sensor, and glucose and H2O2 biosensors.30–33 ZnO nanorods and nanowires are particularly promising for electrochemical sensing because of their excellent electron transport path along the length direction. For N2H4 detection, although several ZnO nanowire and rod array electrodes have demonstrated superior performance,34–36 they are not sufficiently stable in PBS solution and alkaline electrolytes, which would affect the stability of the fabricated sensor and limit its potentials for practical applications.
Recently, more attentions have been focused on the fabrication and electrochemical sensor application of transition metal–RGO hybrid nanomaterials owing to the interesting electrochemical and structural properties of RGO and the catalytic characteristics of transition metals.33,37–41 These multifunctional hybrids have been demonstrated to be significantly promising by various outstanding fruits in fields like Li-ion batteries and electrochemical sensors. For instance, Li et al. reported the application of NiAl–RGO nanohybrids for dopamine detection.38 Zhou et al. synthesized a high-rate CuO hollow nanoparticle–RGO composites as an anode material for lithium-ion batteries.39 Kavitha et al. reported a synthesis of ZnO–RGO hybrids through the in situ thermal decomposition of zinc benzoate dihydrazinate complex on the surface of RGO and further used as glucose sensing.41 Palanisamy et al. developed a sensor for detecting H2O2 based on electrochemically prepared ZnO–RGO composites, but the preparation process is very complicated and the morphology of ZnO–RGO is not controllable.33
In this work, we demonstrated a facile hydrothermal synthesis of ZnO–RGO nanocomposites and explored the synthesized materials for fabrication of N2H4 electrochemical sensor. With the hydrothermal reaction, the size and shape of ZnO on RGO surface can be effective controlled by adjusting the mass ratio of Zn2+ to RGO. After the combination of ZnO with RGO, the conductivity of ZnO–RGO composites can be improved, leading to higher sensitivity for N2H4 determination when compared with pristine ZnO sensor. To the best of our knowledge, it is the first time to use ZnO–RGO nanocomposites by electrochemical to detect N2H4.
2. Experimental
2.1. Reagents
Natural graphite flake (99.8% purity), chloroauric acid (HAuCl4·3H2O, ≥49.0% Au basis), sodium citrate tribasic dehydrate (≥99.0% purity), and cetyltriethylammonium bromide (CTAB) were purchased from Sigma-Aldrich. Silver nitrate (AgNO3) and absolute ethyl alcohol were purchased from Beijing Chemicals Co., Ltd. (Beijing, China). H2O2 (analytical grade, 30% aqueous solution) was supplied by Tianjin Dongfang Chemical Plant (Tianjin, China). Other reagents were purchased from Aladdin. All chemicals used in this work were of analytical reagent grade and obtained from commercial sources and directly used without additional purification. The water used was purified through a Millipore system (∼18.2 MΩ cm).2.2 Preparation of GO and RGO
GO was synthesized directly from graphite by a modified Hummers method.42 In brief, graphite (1 g) was ground with NaCl (50 g) for 10 min. NaCl was then dissolved and removed by filtration with water. The remaining graphite was stirred in 23 mL of 98% H2SO4 for 8 h. KMnO4 (3 g) was gradually added while keeping the temperature less than 20 °C. The mixture was then stirred at 80 °C for 45 min. Next, the redistilled water of 46 mL was added and the mixture was heated at 105 °C for 30 min. The reaction was terminated by addition of redistilled water (140 mL) and 30% H2O2 solution (10 mL). The resulting mixture was washed by repeated centrifugation and filtration, first with 5% HCl aqueous solution and then with distilled water. Finally, the graphite oxide product was obtained after dried in vacuum.The homogeneous GO nanosheet dispersion (0.2 mg mL−1) was prepared by ultrasonication with bath sonicator (100 W, 40 kHz) for 3 h. After adding polyvinylpyrrolidone (PVP, 0.2 mg mL−1, K30, Mw = 30000–40000) and stirring for 2 h, the GO dispersion was mixed with 64 mg glucose and 20 μL ammonia solution (25%, w/w), according to a previous work.43 The mixture was further stirred for 1 h at 95 °C. Finally, the glucose-reduced RGO nanosheets were synthesized and the RGO solution was centrifuged (13 K rpm) and washed with distilled water twice for next use.
2.3. Preparation of ZnO–RGO nanocomposites and ZnO
For the preparation of ZnO–RGO nanocomposites with the mass ratio of Zn2+ to RGO equals 7.4:1, 0.5063 g of Zn(NO3)2·6H2O was added to 30 mL deionized water firstly, then 1 g sodium hydroxide was added under low speed stirring. After that, 1 g CTAB was added to the above solution and stirred until the solution is clear. Finally 0.015 g RGO was added and the resulting mixture was ultrasonicated for 30 minutes. The final mixture was transferred to a Teflon-lined autoclave for hydrothermal synthesis at 120 °C in an oven for 10 h. The precipitate was washed with deionized water and acetone three times each, and dried at 80 °C.
For the preparation of ZnO–RGO nanocomposites with the mass ratio of Zn2+ to G equals 4.4:1 and 2.2:1, 0.025 and 0.05 g RGO were added, respectively, while kept all other experimental parameters as in the typical run.
In the control experiment, ZnO microspindles were prepared with the same synthesis conditions without adding RGO.
2.4. Electrochemical measurements
All electrochemical measurements were performed on a CHI760D electrochemical workstation (CH Instruments, Shanghai, China) at room temperature. A conventional three-electrode cell was used, including a glass carbon electrode (GCE, 3 mm in diameter) as the working electrode, an Ag/AgCl electrode (SCE, saturated KCl) as the reference electrode, and a Pt wire as auxiliary electrode. The test solution was 0.1 M NaOH, which deoxygenated with high-purify nitrogen for 30 minutes before electrochemical experiments.The GCE was polished with 1 and 0.3 μm alumina powder and washed with distilled water, followed by sonication in ethanol solution and distilled water, respectively. Then, the cleaned GCE was dried with a high-purify nitrogen steam for next modification. A total of 5 μL of active materials solution (1.0 mg mL−1) was dropped on the GCE surface and dried at room temperature. Finally, 5 μL Nafion solution (0.1%, diluted with ethanol) was casted onto the electrode to avoid the leakage of modified GCE.
2.5. Characterization techniques
Atomic force microscopy (AFM) images were recorded using a NanoWizard 3 NanoScience atomic force microscope (JPK Instruments AG, Germany) in tapping mode. SEM experiments were performed on JSM-6700F scanning electron microscope (JEOL). Raman spectroscopy (Lab RAM HORIBA JY, Edison, NJ), Fourier transform infrared spectroscopy (FTIR, Nicolet 6700, Thermo-Fisher), and X-ray diffraction (XRD, Rigaku D/max-2500 VB+/PC) were used for the characterization of ZnO–RGO nanocomposites.3. Results and discussion
3.1. Characterization of GO and RGO nanosheets
To characterize and investigate the topography of GO sheets, AFM was utilized. It is the most direct method to quantify the degree of the GO exfoliation. The typical AFM image was shown in Fig. 1a. GO exfoliated by the ultrasonic treatment at a concentration of 0.1 mg mL−1 in water always has a uniform thickness of about 1.1 nm and a size up to several μm, which agrees well with the data reported for the GO of monolayer.23,44 Previous studies indicate that this increased thickness of GO nanosheets compared to the pristine G (about 0.34 nm) is due to the presence of covalently bound oxygen and the displacement of the sp3 hybridized carbon atoms above and below the original G plane.44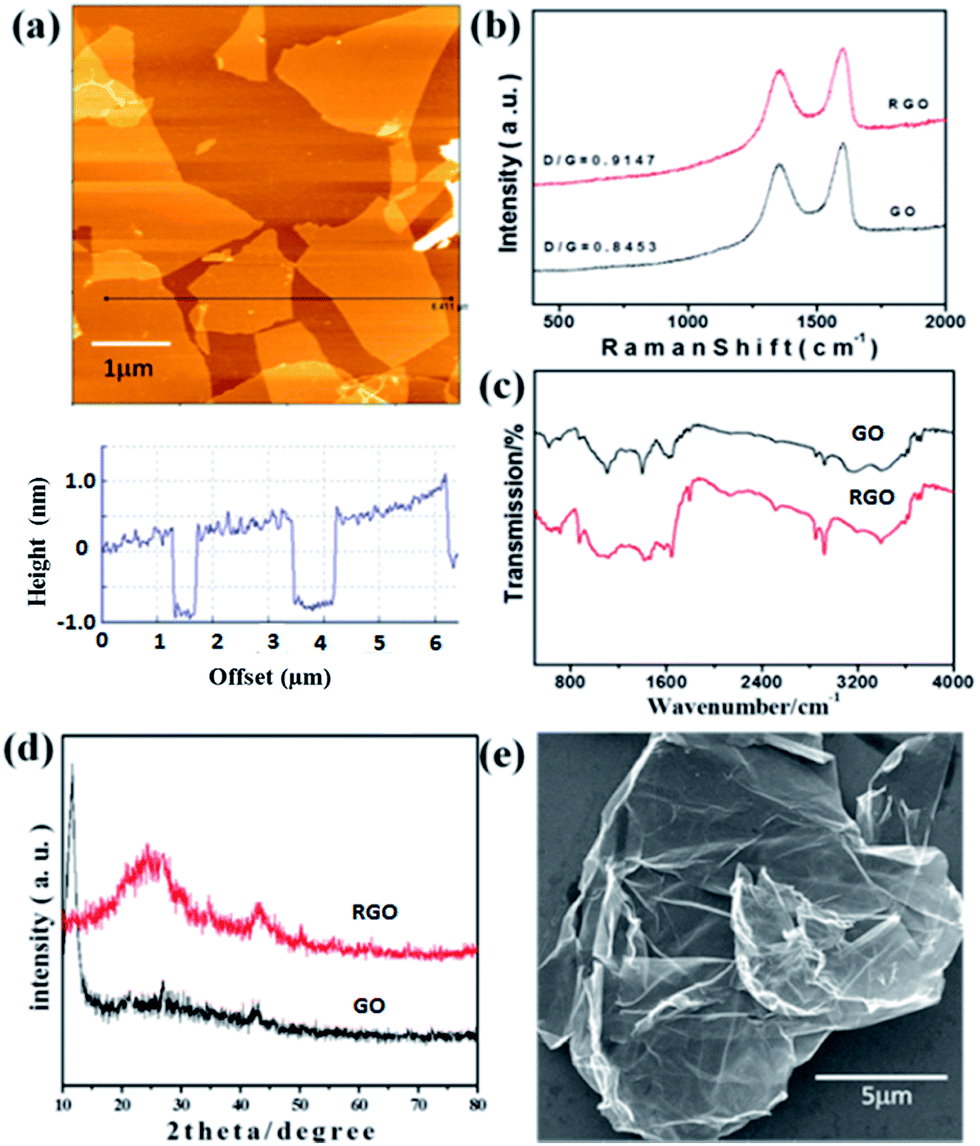 | ||
| Fig. 1 (a) Typical AFM image and section analysis of GO; (b) Raman spectra of GO and RGO; (c) FT-IR spectra of GO and RGO; (d) XRD spectra of GO and RGO; (e) SEM of RGO. | ||
Fig. 1b presents the Raman spectra of GO and RGO, which contain two strong bands at 1589 cm−1 (G band) and 1343 cm−1 (D band). The G and D bands are attributed to the first-order scattering from the E2g phonon of sp2 carbon bonding and structural defects (disorder-induced modes), respectively.45 The Raman spectrum agrees well with the previous reports on GO nanosheets prepared with Hummers method.23,44,45 In Fig. 1b, it was found that ID/IG increases from 0.8453 to 0.9147 after reduction, indicating that most of the oxygenated groups would have been removed during the reduction process.46
The functional groups of GO and RGO were further analyzed by FT-IR spectroscopy and the corresponding spectra is shown in Fig. 1c. The spectrum of GO shows several absorption peaks at 3403 and 1398 cm−1 (OH), 1733 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching), 1645 cm−1 (CC), and 1104 cm−1 (CO).47 For the created RGO, the characteristic absorption peaks of oxide groups (OH, CO, and CO) were found to decrease, indicating that GO has been reduced to RGO.
O stretching), 1645 cm−1 (CC), and 1104 cm−1 (CO).47 For the created RGO, the characteristic absorption peaks of oxide groups (OH, CO, and CO) were found to decrease, indicating that GO has been reduced to RGO.
Power XRD was further used to characterize GO and RGO, and the typical patterns are shown in Fig. 1d. The feature diffraction peak of GO appearing at 11.67° (d spacing ∼8.92 Å) was observed as a result of the introduction of oxygenated functional groups on the carbon sheets.48 For the obtained RGO, the peak located at 11.67° disappears and shows a dramatic shift to higher 2θ angles (24.35°; d spacing ∼3.71 Å), confirming the successful reduction of GO to RGO and the possible exfoliation of the multi-layered RGO.49 The large-scale SEM image of RGO (Fig. 1e) indicates the unique folded morphology of the synthesized RGO nanosheets.
3.2. Synthesis and characterization of ZnO–RGO composites
For the synthesis of ZnO–RGO composites, we utilized a facile one-step hydrothermal method. By this synthesis strategy, three types of ZnO–RGO nanocomposites with different morphologies of ZnO on the RGO surface were obtained by adjusting the mass ratio of Zn2+ to RGO (named as Rm for simplicity). The morphologies of the as-prepared ZnO and ZnO–RGO nanocomposites with different Rm were examined by SEM, and the corresponding images are shown in Fig. 2. | ||
| Fig. 2 Typical SEM images of (a) ZnO, and (b–d) ZnO–RGO nanocomposites with different Rm of (b) 2.2:1, (c) 4.4:1, and (d) 7.4:1. | ||
Fig. 2a shows the typical SEM image of the as-prepared ZnO microspindles without adding RGO. It is found that the as-prepared ZnO has a spindle-like structure. Fig. 2b–d present the typical SEM images of the as-prepared ZnO–RGO nanocomposites with the Rm of 2.2:1, 4.4:1, and 7.4:1, respectively. When the Rm was 2.2:1, the spindle-like ZnO was not seen, which means the introduction of RGO into the reaction system affects the morphology of ZnO. When the ratio was increased to 4.4:1 and 7.4:1, with the increase of the amount of Zn2+, the spindle-like ZnO with smaller size were seen. The above result shows that in the reaction system, only after reaching a certain amount of Zn2+ can form the spindle-like ZnO. Here, by covering on the ZnO nanocrystals during the synthesis, RGO actually served as an additional surfactant, thus resulting in the smaller spindle-like structure ZnO. Compared to the unsupported ZnO, RGO nanosheets wrapping on ZnO can not only reduce the spindle-like structure ZnO size and prevent the particle aggregation but also protect the ZnO effectively and thus improve the electrochemical stability. Moreover, due to the high specific surface area and high electrical conductivity, the wrapped RGO may enhance the electron transfer during the electrochemical reactions.
We suggest that the mass ratio of reactants, Rm, is very important for the morphology and size of the synthesized ZnO. When the Rm was 2.2:1, it can be found that there are relatively high content of ZnO nanoparticles but very little ZnO microspindles formed in the product (Fig. 2b). When the ratio was increased to 4.4:1 and 7.4:1, ZnO microspindles can be clearly seen (Fig. 2c and d). Comparing the corresponding SEM images of ZnO–RGO composites, it can be concluded that the ZnO microspindles could become more uniform with high content when the proportion of Zn2+ in the reaction system was increased.
The structure of the ZnO and ZnO–RGO nanocomposites with different Rm was determined by XRD firstly, as shown in Fig. 3a. In comparison, the XRD pattern of ZnO microspindles (Fig. 3a) is also presented. The diffraction peaks of ZnO microspindles appear at 2θ values of 31.9°, 34.5°, 36.4°, 47.6°, 56.6°, 62.9°, 66.5°, 68.1° and 69.2°. All these characteristic diffraction peaks can be indexed to the ZnO hexagonal Wurtzite structure of (100), (002), (101), (102), (110), (103), (200), (112) and (201) planes, which match well with the standard ZnO peaks (JCPDS 36-1451, a = 0.325 nm, c = 0.521 nm).50 The strong diffraction peaks present highly crystallized ZnO. No characteristic peak is observed for other impurities. The RGO does not arise in the pattern due to the small quantity.51 In other reports dealing with the vertically aligned ZnO nanorods, typically the peak (002) in XRD pattern is much more intense than other peaks.52 The intense peak of (002) suggests the preferential growth of ZnO nanorods along c axis. However, due to the reaction system, the ZnO in our sample is polydirectional and the peak (002) is not outstanding comparing to other peaks.
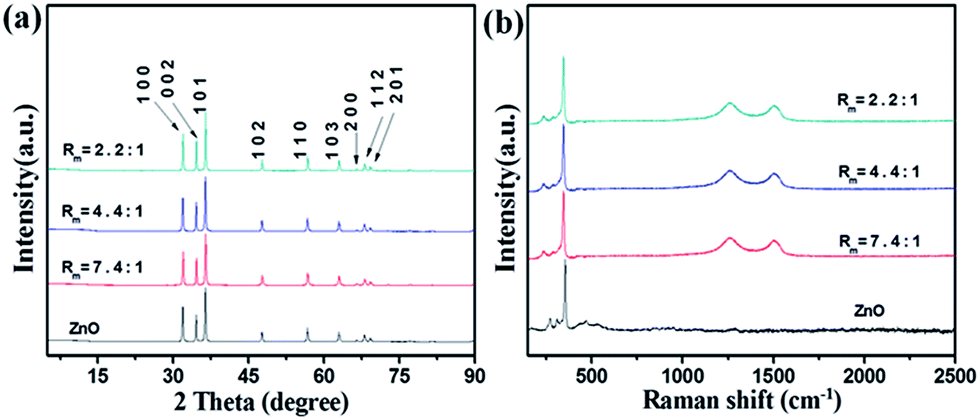 | ||
| Fig. 3 ZnO and ZnO–RGO composites with different Rm of (a) XRD characterization and (b) Raman spectra. | ||
Fig. 3b shows the Raman spectra of the synthesized ZnO microspindles and ZnO–RGO nanocomposites with different Rm. The Raman spectrum of ZnO contains a sharp peak at ∼341 cm−1 corresponding to the vibration mode of E2, and several smaller peaks at ∼270, ∼310 and ∼472 cm−1 resulting from the multiple-phonon scattering process.53 The spectra of ZnO–RGO nanocomposites are essentially the superimposition of that of ZnO and RGO, except the small change of D and G band (likely arising from the defects introduced in the hydrothermal condition54) and the shifts of D and G band (likely due to the doping effects of ZnO55).
3.3. Possible nucleation mechanism
The typical formation process of ZnO nanocrystals in a hydrothermal reaction can be presented as follows.56–58 Firstly, the precursor of zinc nitrate hexahydrate (Zn(NO3)2·6H2O) starts to hydrolysis and induce the formation of zinc hydroxide (Zn(OH)2) hydrosol (formula (1)). During the hydrothermal process, part of the Zn(OH)2 colloids dissolves into Zn2+ and OH− according to reaction (2). When the concentration of Zn2+ and OH− reaches the supersaturation degree of ZnO, ZnO nuclei is formed according to the reaction (3). The growth units of Zn(OH)42− (reaction (4)) have a tetrahedron geometry. Finally, ZnO is formed according to the reaction (5).| Zn(NO3)2·6H2O + 2OH− → Zn(OH)2 + 2NO3− + 6H2O | (1) |
| Zn(OH)2 → Zn2+ + 2OH− | (2) |
| Zn2+ + 2OH− → ZnO + H2O | (3) |
| Zn(OH)2 + 2OH− → Zn(OH)42− | (4) |
| Zn(OH)42− → ZnO + H2O + 2OH− | (5) |
CTAB is a cationic surfactant which ionizes completely in water. CTA+ is positively charged with a tetrahedral head and a long hydrophobic tail. The resulted cation is also a tetrahedron with a long hydrophobic tail (seen in Fig. 4a). Therefore, ion pairs between Zn(OH)42− and CTA+ could form due to the electrostatic interaction. In the crystallization process, CTAB serves not only as an ionic carrier but also as a soft template.59,60
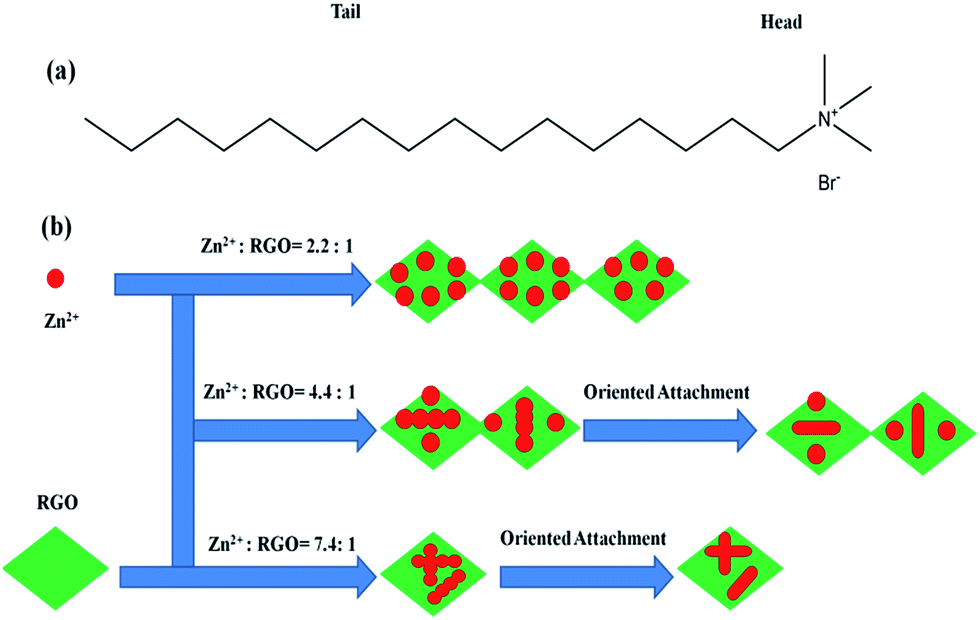 | ||
| Fig. 4 (a) The structure of the hydrophobic tail and hydrophilic head of CTAB; (b) schematic illustration of the formation of ZnO–RGO nanocomposites. | ||
According to our previous report, GO was chemically reduced into RGO by glucose, and the zeta potential of the obtained RGO was −26.05 mV.16 When adding RGO into the reaction system, RGO may affect the electrostatic interaction of Zn(OH)42− and CTA+. Moreover, with the decrease of Rm from 7.4:1, 4.4:1, to 2.2:1, the content of RGO is increasing, and the effect of RGO is also increasing. Finally, the products formed different morphologies.
Based on the above results, we proposed a possible growth mechanism for the formation of ZnO–RGO nanocomposites, which can be rationally expressed by the Ostwald-ripening assisted oriented attachment mechanism, as shown in Fig. 4b. First, many tiny ZnO particles are formed on RGO according to the above reaction equation with the assistance of CTAB. The generated ZnO has a low solubility in the water solution, and thereby forms nanoparticle-composed aggregates on the surface of RGO. The poorly crystallized spherical intermediate phase arranged on the RGO surface is initially formed owing to the kinetic advantage. With time elapsing, the intermediate is transformed to well-crystallized spheres through Ostwald ripening,61 which involves the growth of larger particles at the expense of the smaller ones driven by the tendency of the solid phase in the systems to adjust it to achieve a minimum total surface free energy. Finally, the formation of ZnO micro-spindles through ZnO nanoparticles oriented attachment and binding with each other along the different crystal face directions.
3.4. Electrochemical N2H4 sensor application
To demonstrate the potential of the synthesized ZnO–RGO composites as sensor materials, we carried out the preliminary investigations on the electrochemical performances of these materials.Fig. 5a shows the CVs of RGO-modified GCE (RGO/GCE) in the absence and presence of N2H4. It is clear that there is no obvious electrochemical oxidation peak observed. Meanwhile, for ZnO/GCE and ZnO–RGO/GCE with different Rm of 2.2:1, 4.4:1, and 7.4:1, when N2H4 was added to the system, a clear increase of the oxidation peak was observed in deoxygenized environment (Fig. 5b and c). However, for ZnO/GCE, the increase of the oxidation peak current is small with the increase of the amount of N2H4 (Fig. 5b). For ZnO/GCE and ZnO–RGO/GCE with different Rm of 2.2:1, 4.4:1, and 7.4:1, when 0.5 mM N2H4 was added to the system, the highest oxidation peak was seen when the Rm of 4.4:1 was used (Fig. 5b and c). Therefore, we expected that the ZnO–RGO (Rm = 4.4:1)/GCE will has the highest performances as N2H4 sensor, and then utilized this modified GCE as sensor application.
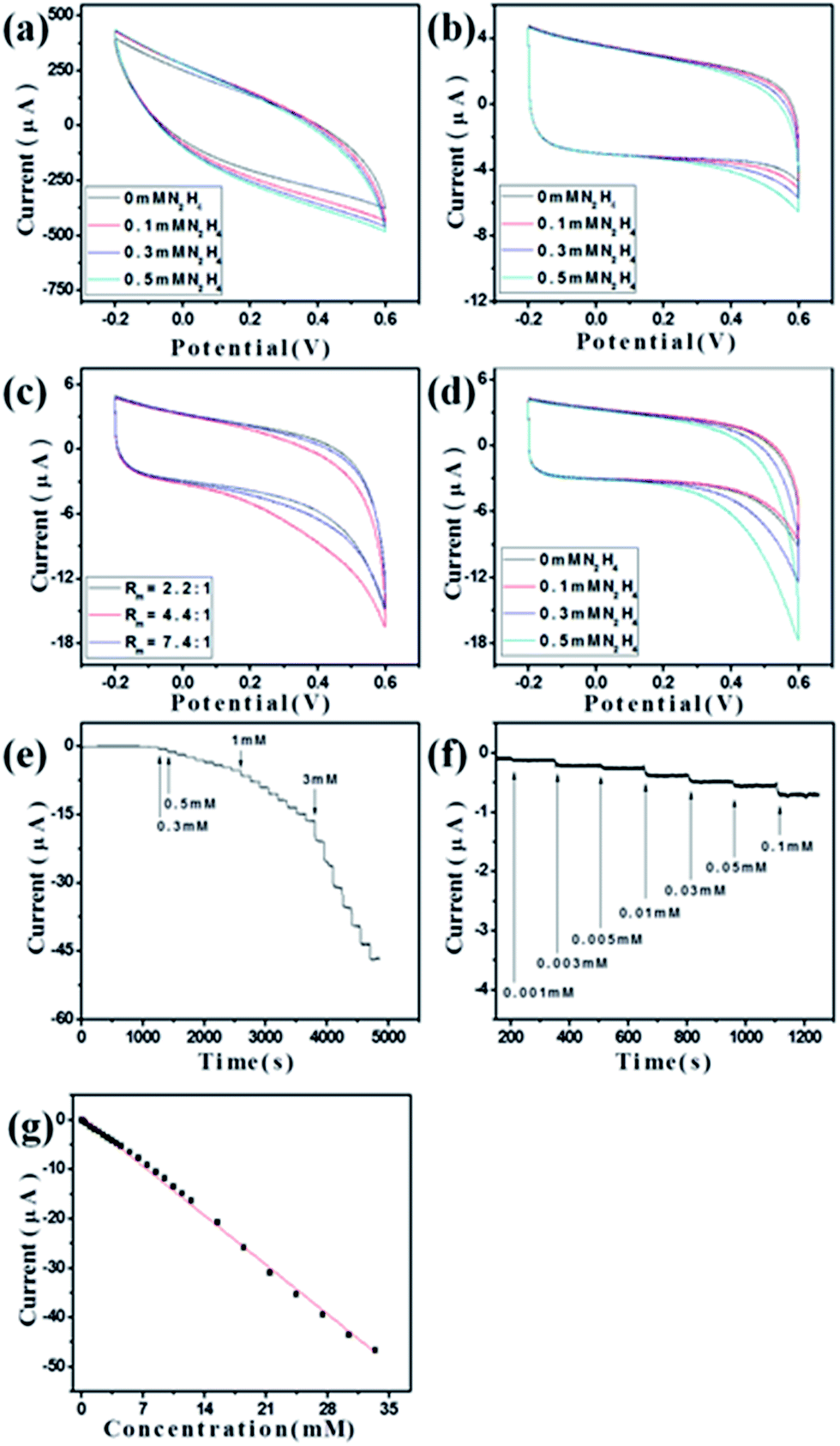 | ||
| Fig. 5 Electrochemical N2H4 sensor application: (a) CVs of RGO/GCE in 0.1 M NaOH in the absence and presence of N2H4 at a scan rate of 0.5 V s−1; (b) CVs of ZnO/GCE in 0.1 M NaOH in the absence and presence of N2H4 at a scan rate of 0.5 V s−1; (c) CVs of ZnO–RGO/GCE composites with different Rm in 0.1 M NaOH with 0.5 mM N2H4 at the scan rate of 0.5 V s−1; (d) CVs of ZnO–RGO (Rm = 4.4:1)/GCE in 0.1 M NaOH in the absence and presence of N2H4 at a scan rate of 0.5 V s−1; (e) and (f) amperometric responses of ZnO–RGO (Rm = 4.4:1)/GCE upon the successive addition of N2H4 at 0.6 V; and (g) calibration line. | ||
Fig. 5c shows the CVs of ZnO–RGO (Rm = 4.4:1)/GCE in the absence and presence of N2H4. When N2H4 was added to the system, an obvious increase of the oxidation peak was observed in deoxygenized environment compared with the system without N2H4. In addition, the increase of the oxidation peaks is greater with the increase of N2H4 concentration from 0.3 to 0.5 mM. The electrochemical response is irreversible, as no cathodic current is observed during the reverse sweep. According to the previous reports,35,36 a possible electrochemical reaction for the N2H4 is proposed to be N2H4 + 5/2OH− → 1/2N3− + 1/2NH3 + 5/2H2O + 2e−. As faster electron transfer leads to a sharper and more well-defined peak, the substantial increase in the peak current reflects a faster electron transfer reaction and the increase of reversibility of the electron transfer process. Therefore, N2H4 is effectively detected by oxidation on the modified GCE. The pH of the solution is important to obtain efficient electrocatalytic oxidation of N2H4, and it was reported that the electrocatalytic oxidation of N2H4 can be improved by increasing the pH value of the solution (pH > 7).62 Therefore, we used 0.1 M NaOH for all the electrochemical experiments.
Fig. 5e and f present the current–time (I–T) plot of the fabricated ZnO–RGO (Rm = 4.4:1)/GCE under the optimized experimental conditions with successive adding N2H4. As the N2H4 was injected, the steady-state currents reached another steady-state value (98% of the maximum) in less than 3 s. The linear relationship between the catalytic current and the concentration is shown in Fig. 5g. This nonenzymatic electrochemical N2H4 sensor has a linear response range of 1 μM to 33.5 mM (correlation coefficient: 0.9975) and a detection limit of 0.8 μM at a signal-to-noise ratio of 3. As can be seen from Table 1, our sensor has a larger linear range and lower detection limit compared with the previous ZnO-based N2H4 sensors.
:1) with previous ZnO-based N2H4 sensors
| Electrode materials | Potential (V) | Linear range (mM) | LOD (mM) | Ref. |
|---|---|---|---|---|
| ZnO nanofilm | 0.6 | 5 × 10−4 to 14.2 | 5 × 10−4 | 63 |
| ZnO nanonails | 0.4 | 1 × 10−4 to 1.2 × 10−3 | 2 × 10−4 | 36 |
| ZnO nanowires | 0.06 | 3 × 10−3 to 0.562 | 1.44 × 10−5 | 64 |
| ZnO nanoflowers | 0.4 | 6 × 10−4 to 0.25 | 1.8 × 10−4 | 65 |
| Micro/nano ZnO | 0.1 | 8 × 10−4 to 0.2 | 2.5 × 10−4 | 34 |
| ZnO–RGO (4.4:1) |
0.6 | 1 × 10−3 to 33.5 | 8 × 10−4 | This work |
The selectivity of the fabricated ZnO–RGO (Rm = 4.4:1)/GCE towards N2H4 under the optimum conditions was studied for a number of potential interferents, and the result is shown in Fig. 6a. Here, we defined the tolerance limit as the molar ratio of potential interfering substances/N2H4 that caused the change of peak current less than 5% for the determination of 0.1 mM N2H4. It was found that 8-fold hydroxylamine (NH2OH), 14-fold NH3, 20-fold H2O2, dopamine, and glucose, 25-fold Zn2+, Cu2+, Ca2+, NO3−, Cl−, SO42− have no obvious interferes on the determination of N2H4. The reproducibility of the fabricated ZnO–RGO (Rm = 4.4:1)/GCE was examined by 10 repetitive detection of 0.1 mM N2H4, there was about 2.4% decrease in the response towards 0.1 mM N2H4 after 10 times, demonstrating high antifouling ability of our N2H4 sensor (Fig. 6b). The storage stability measured every 2–3 days over a 21 days period. When not in use, the modified electrode was stored in air at room temperature. There was about 3.44% decrease in the response towards 0.1 mM N2H4 after 21 days, indicating that our ZnO–RGO (Rm = 4.4:1)/GCE maintained its catalytic activity very well and could be used for a long time (Fig. 6c). In addition, we noticed that the amphoteric properties of ZnO nanostructures in NaOH solution have no significant effect on the performances of this ZnO–RGO modified GCE for long-term detecting N2H4.
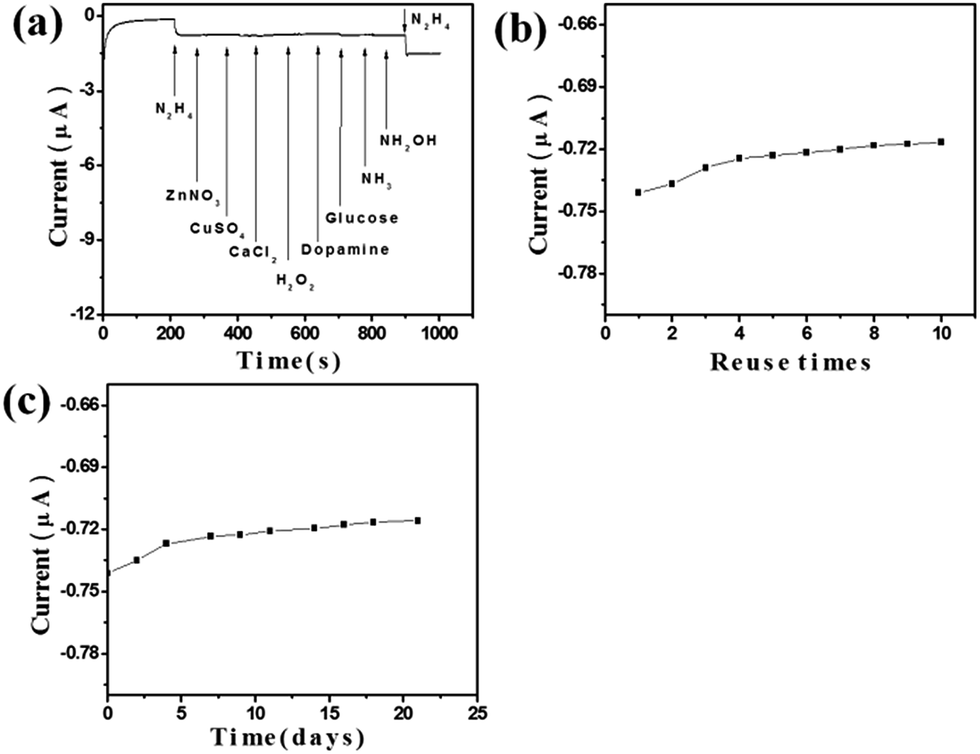 | ||
| Fig. 6 The typical (a) selectivity, (b) reuse ability, and (c) long-term stability of the N2H4 sensor fabricated with ZnO–RGO (Rm = 4.4:1)/GCE. | ||
4. Conclusions
In summary, we demonstrated a simple and effective hydrothermal synthesis for the preparation of ZnO–RGO nanocomposites. We found that the introduction of RGO into the reaction system could affect the morphology of ZnO. Different ZnO nanostructures on the RGO surface can be synthesized by adjusting the mass ratio of Zn2+ to RGO. The synthesized ZnO–RGO nanocomposites showed better electrochemical performances than that with RGO and ZnO. In addition, an optimal mass ratio for the best electrochemical performances was obtained. Such ZnO–RGO nanocomposites represent promising enzyme-free N2H4 sensor with high sensitivity and selectivity, improved stability, and fast amperometric response.Acknowledgements
The authors gratefully acknowledge the financial supports from the Fundamental Research Funds for the Central Universities (project no. ZZ1307). We would like to thank the financial support of the China Scholarship Council (CSC) for a PhD scholarship in University of Bremen.Notes and references
- S. Amlathe and V. K. Gupta, Analyst, 1988, 113, 1481–1483 RSC.
- K. Yamada, K. Yasuda, N. Fujiwara, Z. Siroma, H. Tanaka, Y. Miyazaki and T. Kobayashi, Electrochem. Commun., 2003, 5, 892–896 CrossRef CAS.
- S. Garrod, M. E. Bollard, A. W. Nicholls, S. C. Connor, J. Connelly, J. K. Nicholson and E. Holmes, Chem. Res. Toxicol., 2005, 18, 115–122 CrossRef CAS.
- J. Budkuley, Mikrochim. Acta, 1992, 108, 103–105 CrossRef CAS.
- Y. Y. Liu, I. Schmeltz and D. Hoffmann, Anal. Chem., 1974, 46, 885–889 CrossRef CAS.
- A. Safavi and M. A. Karimi, Talanta, 2002, 58, 785–792 CrossRef CAS.
- S. Wang, L. Du, A. Zhang and D. Liu, Mikrochim. Acta, 2000, 134, 167–170 CrossRef CAS.
- M. George, K. S. Nagaraja and N. Balasubramanian, Talanta, 2008, 75, 27–31 CrossRef CAS.
- H. Zhang, J. Huang, H. Hou and T. You, Electroanalysis, 2009, 21, 1869–1874 CrossRef.
- L. Zheng and J. F. Song, Talanta, 2009, 79, 319–326 CrossRef CAS.
- W. Lu, R. Ning, X. Qin, Y. Zhang, G. Chang, S. Liu, Y. Luo and X. Sun, J. Hazard. Mater., 2011, 197, 320–326 CrossRef CAS PubMed.
- B. K. Jena and C. R. Raj, J. Phys. Chem. C, 2007, 111, 6228–6232 CAS.
- B. Šljukić, C. E. Banks, A. Crossley and R. G. Compton, Electroanalysis, 2006, 18, 1757–1762 CrossRef.
- S. S. Narayanan and F. Scholz, Electroanalysis, 1999, 11, 465–469 CrossRef CAS.
- K. I. Ozoemena and T. Nyokong, Talanta, 2005, 67, 162–168 CrossRef CAS.
- G. Wei, Y. Zhang, S. Steckbeck, Z. Su and Z. Li, J. Mater. Chem., 2012, 22, 17190–17195 RSC.
- J. Wang, X. Zhao, J. Li, X. Kuang, Y. Fan, G. Wei and Z. Su, ACS Macro Lett., 2014, 3, 529–533 CrossRef CAS.
- S. Liu, J. Tian, L. Wang, H. Li, Y. Zhang and X. Sun, Macromolecules, 2010, 43, 10078–10083 CrossRef CAS.
- W. Lu, Y. Luo, G. Chang and X. Sun, Biosens. Bioelectron., 2011, 26, 4791–4797 CrossRef CAS.
- H. Wang, D. Sun, N. Zhao, X. Yang, Y. Shi, J. Li, Z. Su and G. Wei, J. Mater. Chem. B, 2014, 2, 1362–1370 RSC.
- X. Wang, L. Zhi and K. Müllen, Nano Lett., 2007, 8, 323–327 CrossRef.
- W. Chen, L. Yan and P. R. Bangal, Carbon, 2010, 48, 1146–1152 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- J. Tian, S. Liu, Y. Zhang, H. Li, L. Wang, Y. Luo, A. M. Asiri, A. O. Al-Youbi and X. Sun, Inorg. Chem., 2012, 51, 4742–4746 CrossRef CAS.
- C. Nethravathi and M. Rajamathi, Carbon, 2008, 46, 1994–1998 CrossRef CAS.
- H. Zeng, X. Xu, Y. Bando, U. K. Gautam, T. Zhai, X. Fang, B. Liu and D. Golberg, Adv. Funct. Mater., 2009, 19, 3165–3172 CrossRef CAS.
- H. J. Fan, Y. Yang and M. Zacharias, J. Mater. Chem., 2009, 19, 885–900 RSC.
- J. Liu, C. Guo, C. M. Li, Y. Li, Q. Chi, X. Huang, L. Liao and T. Yu, Electrochem. Commun., 2009, 11, 202–205 CrossRef CAS.
- J. Tian, S. Liu, H. Li, L. Wang, Y. Zhang, Y. Luo, A. M. Asiri, A. O. Al-Youbi and X. Sun, RSC Adv., 2012, 2, 1318–1321 RSC.
- J. X. Wang, X. W. Sun, A. Wei, Y. Lei, X. P. Cai, C. M. Li and Z. L. Dong, Appl. Phys. Lett., 2006, 88, 233106 CrossRef.
- A. Wei, X. W. Sun, J. X. Wang, Y. Lei, X. P. Cai, C. M. Li, Z. L. Dong and W. Huang, Appl. Phys. Lett., 2006, 89, 123902 CrossRef.
- S. Ameen, M. Shaheer Akhtar and H. S. Shin, Talanta, 2012, 100, 377–383 CrossRef CAS.
- S. Palanisamy, S. M. Chen and R. Sarawathi, Sens. Actuators, B, 2012, 166–167, 372–377 CrossRef CAS.
- Y. Ni, J. Zhu, L. Zhang and J. Hong, CrystEngComm, 2010, 12, 2213–2218 RSC.
- A. Umar, M. M. Rahman and Y. B. Hahn, Talanta, 2009, 77, 1376–1380 CrossRef CAS.
- A. Umar, M. M. Rahman, S. H. Kim and Y. B. Hahn, Chem. Commun., 2008, 166–168, 10.1039/b711215g.
- Y. Xu, X. Huang, Z. Lin, X. Zhong, Y. Huang and X. Duan, Nano Res., 2013, 6, 65–76 CrossRef CAS.
- M. Li, J. E. Zhu, L. Zhang, X. Chen, H. Zhang, F. Zhang, S. Xu and D. G. Evans, Nanoscale, 2011, 3, 4240–4246 RSC.
- J. Zhou, L. Ma, H. Song, B. Wu and X. Chen, Electrochem. Commun., 2011, 13, 1357–1360 CrossRef CAS.
- M. Sathish, T. Tomai and I. Honma, J. Power Sources, 2012, 217, 85–91 CrossRef CAS.
- T. Kavitha, A. I. Gopalan, K. P. Lee and S. Y. Park, Carbon, 2012, 50, 2994–3000 CrossRef CAS PubMed.
- Y. Li and Y. Wu, J. Am. Chem. Soc., 2009, 131, 5851–5857 CrossRef CAS.
- Y. Guo, S. Guo, J. Ren, Y. Zhai, S. Dong and E. Wang, ACS Nano, 2010, 4, 4001–4010 CrossRef CAS.
- F. Xu, Y. Sun, Y. Zhang, Y. Shi, Z. Wen and Z. Li, Electrochem. Commun., 2011, 13, 1131–1134 CrossRef CAS.
- J. Gao, F. Liu, Y. Liu, N. Ma, Z. Wang and X. Zhang, Chem. Mater., 2010, 22, 2213–2218 CrossRef CAS.
- X. Zhong, J. Jin, S. Li, Z. Niu, W. Hu, R. Li and J. Ma, Chem. Commun., 2010, 46, 7340–7342 RSC.
- I. K. Moon, J. Lee, R. S. Ruoff and H. Lee, Nat. Commun., 2010, 1, 73 Search PubMed.
- X. Gao, J. Jang and S. Nagase, J. Phys. Chem. C, 2009, 114, 832–842 Search PubMed.
- H. M. A. Hassan, V. Abdelsayed, A. E. R. S. Khder, K. M. AbouZeid, J. Terner, M. S. El-Shall, S. I. Al-Resayes and A. A. El-Azhary, J. Mater. Chem., 2009, 19, 3832–3837 RSC.
- J. Wang, Z. Gao, Z. Li, B. Wang, Y. Yan, Q. Liu, T. Mann, M. Zhang and Z. Jiang, J. Solid State Chem., 2011, 184, 1421–1427 CrossRef CAS.
- H.-M. Yu, Q. H. Zhang, L. J. Qi, C. W. Lu, T. G. Xi and L. Luo, Thermochim. Acta, 2006, 440, 195–199 CrossRef CAS.
- J. J. Wu and S. C. Liu, J. Phys. Chem. B, 2002, 106, 9546–9551 CrossRef CAS.
- J. Scott, Phys. Rev. B: Solid State, 1970, 2, 1209–1211 CrossRef.
- A. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- X. Dong, D. Fu, W. Fang, Y. Shi, P. Chen and L. J. Li, Small, 2009, 5, 1422–1426 CrossRef CAS.
- L. N. Demianets, D. V. Kostomarov, I. P. Kuźmina and S. V. Pushko, Crystallogr. Rep., 2002, 47, S86–S98 CrossRef CAS.
- C. L. Kuo, T. J. Kuo and M. H. Huang, J. Phys. Chem. B, 2005, 109, 20115–20121 CrossRef CAS.
- S. C. Zhang and X. G. Li, Colloids Surf., A, 2003, 226, 35–44 CrossRef CAS.
- X. M. Sun, X. Chen, Z. X. Deng and Y. D. Li, Mater. Chem. Phys., 2003, 78, 99–104 CrossRef.
- Y. X. Wang, J. Sun, X. Fan and X. Yu, Ceram. Int., 2011, 37, 3431–3436 CrossRef CAS.
- Y. Zhao and L. Jiang, Adv. Mater., 2009, 21, 3621–3638 CrossRef CAS.
- S. V. Guerra, C. R. Xavier, S. Nakagaki and L. T. Kubota, Electroanalysis, 1998, 10, 462–466 CrossRef CAS.
- X. Zhang, W. Ma, H. Nan and G. Wang, Electrochim. Acta, 2014, 144, 186–193 CrossRef CAS.
- Z. Zhao, Y. Sun, P. Li, S. Sang, W. Zhang, J. Hu and K. Lian, J. Electrochem. Soc., 2014, 161, B157–B162 CrossRef CAS.
- B. Fang, C. Zhang, W. Zhang and G. Wang, Electrochim. Acta, 2009, 55, 178–182 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |