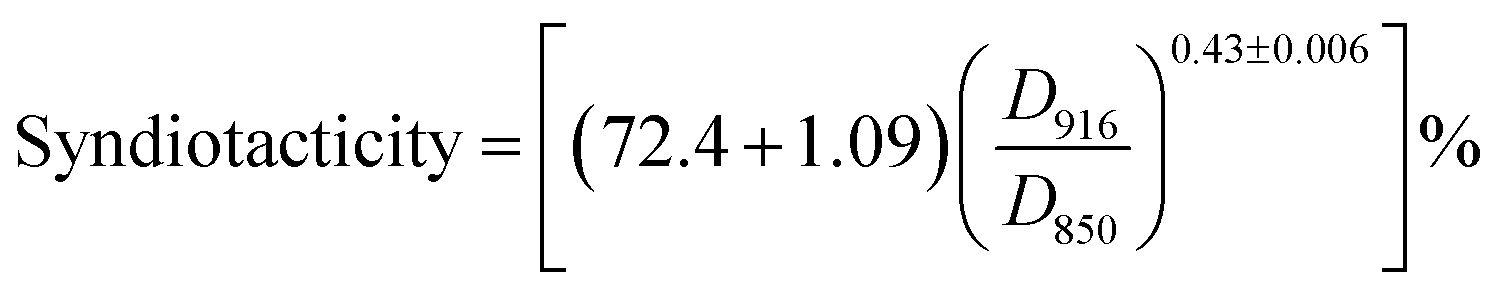DOI:
10.1039/C5RA18437A
(Paper)
RSC Adv., 2015,
5, 86598-86605
Acrylamide modified poly(vinyl alcohol): crystalline and enhanced water solubility
Received
9th September 2015
, Accepted 5th October 2015
First published on 8th October 2015
Abstract
Acrylamide (AAm) modified polyvinyl alcohol (PVA) with enhanced water solubility and tunable tacticity and crystallinity was prepared by alcoholysis of vinyl acetate (VAc) and acrylamide copolymers. The chemical structures and performance of VAc–AAm copolymers and AAm-modified PVA were measured by FTIR, UV-vis, XRD, elemental analysis as well as rheometry. FTIR and XRD analysis reveals that AAm units in PVA chain can reduce the stereoregularity effectively, and suppress the crystallinity. And a decreasing linear relationship between crystallinity and AAm mole fractions is observed. Furthermore, the influence of AAm modification on the water solubility of PVA was studied, and a significant enhancement either at low temperature (30 °C) or at high temperature (70 °C) was achieved through AAm modification. Moreover, the rheological investigation suggested that the relative strength of the hydrogen bonding interactions existing between PVA chains was weakened, while those between PVA chains and water molecules, to some extent, were enhanced after AAm modification. The method presented is an easy but promising way to prepare PVA that can have good water solubility even at low temperature while exhibiting high degrees of alcoholysis.
Introduction
Poly(vinyl alcohol) (PVA), as a well-known water-soluble polymer with high transparency and outstanding mechanical properties, has been utilized in an invaluable component of numerous personal, industrial and electronic products.1–3 More recently, highly alcoholysized PVA has proved attractive for exploitation in the biomedical field due to its water-solubility, low cytotoxicity and biocompatibility.4–6 In these industrial and biomedical applications, the water solubility of PVA is of natural importance.7 However, the water solubility of commercial sourced PVA shows a strong dependence on the degrees of alcoholysis in the PVA chains and highly alcoholysized PVA usually exhibits poor water solubility, particularly at low temperature.8 Worse yet,9,10 aqueous solution of PVA of high degree of alcoholysis usually undergoes viscosity increase with time, and sometimes even gelation, depending on the concentration of polymers and the storage conditions. This increase in viscosity of PVA aqueous solution during storage might lead to a phase separation, as we have revealed previously,11 and critically affect the effectiveness and economic benefits of the industries using aqueous solutions of PVA. To enhance the water solubility of PVA of high degree of alcoholysis, a great number of methods based on copolymerization or post-polymerization modification (esterification occurring on the hydroxyl group or Ce(IV) initiated radical polymerization occurring on the methane/hydroxyl groups in PVA chain) to introduce hydrophilic groups for improving solute–solvent interactions and/or enhance steric hindrances for suppressing crystallinity of PVA chain have been reported.12–14 Recent studies15 found that a noticeable increase of solubility of PVA can be achieved by esterifying PVA with D,L-lactic acid possibly due to the synergism of the increase in steric hindrances and the decrease in alcoholysis degree. However, investigations of the similar PLA modified PVA by Ding et al.16 obtained non-water-soluble polymers in all cases. Moreover, these post-polymerization modifications usually results in the decrease of the degree of alcoholysis and uncontrollable amount of homo-polymerization of modification monomers in the case of Ce(IV) initiated radical polymerization. To circumvent this shortcoming, alternative routes for PVA modification using the strategy of copolymerization with ionic monomers or other hydrophilic monomers were carried out,17,18 while their researches mainly focused on the kinetics studies and the changes of chemical structures and few efforts were devoted to the questions that how the copolymerization will affect the water solubility of PVA and further the rheological properties of the PVA aqueous solution and what kind of monomer for copolymerization will favor the water solubility of PVA with high degrees of alcoholysis, which are of particular interest to our group in the view of the industrial applications of PVA aqueous solution.
We report herein the acrylic amide (AAm) modification of PVA to tune its tacticity and crystalline structures and to enhance its water solubility. Choosing AAm as the co-monomer is based on the following design rationale. First, since both AAm monomer and poly(acrylic amide) are water soluble, AAm modified PVA is supposed to exhibit better water solubility comparing with the PVA modified by other water insoluble monomers. In addition, the formation of intermolecular hydrogen bonds between water molecule and the amido group, a strong hydrogen bond donor, should to some extent accelerate the dissolving process. Second, as amido group is larger than hydroxyl group, the increase in steric hindrance and the decrease in steric regularity can be expected. And due to the increased steric hindrances after AAm modification, the percentage of tacticity and crystallinity of the modified PVA should be suppressed, may resulting in dramatically enhancement of its water solubility. Thus, through copolymerization vinyl acetate (VAc) with AAm and the following alcoholysis, controlling the chemical structures and crystalline behavior of PVA and further improving its water solubility could be achieved. As discussed below, our characterization results confirmed this design, showing that the modified PVA exhibits the tacticity and crystalline of PVA changing upon AAm mole fraction in PVA chain, while its water solubility, either at low temperature (30 °C) or at high temperature (70 °C) shows a significant enhancement. Moreover, the rheological properties of the AAm modified PVA aqueous solution has been investigated.
Experimental
Synthesis
Materials. Vinyl acetate (VAc) (99%), commercial product from Chengdu Kelong Chemical Reagent Factory (Chengdu, China), was purified by filtration through a basic alumina column to remove inhibitors followed by distillation under reduced pressure and was stored at −18 °C before use. Azobisisobutyronitrile (AIBN) (99%), purchased from Chengdu Kelong Chemical Reagent Factory (Chengdu, China), was recrystallized twice from ethanol. Acrylamide (AAm) (99%) was recrystallized from CHCl3, and then vacuum dried at 25 °C and kept at −18 °C. Methanol (Analytics Reagent) and sodium hydroxide (Analytics Reagent), supplied by Chengdu Kelong Chemical Reagent Factory (Chengdu, China), were used as received.
Alcoholysis of poly(VAc-co-AAm). Acrylamide modified PVA was prepared following the well-established alcoholysis method using sodium hydroxide as catalyst. Typically, into a 50 mL three-necked round-bottomed flask equipped with a mechanical stirrer and a reflux condenser were added P(VAc-co-AAm) (0.9 g) and sodium hydroxide (5 mol% relative to theoretical content of acetate groups) and suspended in methanol (30 mL). The reaction was carried out for 4 hours at 50 °C. After cooling, the supernatant liquid was decanted off and the resultant PVA gel was purified by dialysis against pure methanol until no monomers or sodium hydroxide in dialysis methanol could be detected by FTIR and UV-vis spectrum. The obtained acrylamide modified PVA was then collected by filtration and dried to constant mass under vacuum at 40 °C.
Characterizations
Fourier transform infrared (FTIR) analysis. 10 μm thick films of PVA and AAm-modified PVA were subjected to FTIR spectroscopy in the range of 4000 to 500 cm−1. A FTIR Nicolet 560 FTIR spectrometer was used for the study. The film samples were prepared by casting 5 wt% aqueous solutions of PVA or modified PVA on Teflon substrates, following by slowly evaporating the solvent at 35 °C in an oven for a week, and then drying in vacuum oven at 50 °C to remove residual solvent.
X-ray diffraction (XRD) analysis. X-ray diffraction analysis was performed on an X'Pert Pro MPD (Philips Analytical Co. Ltd., Netherland) X-ray diffractometer with CuKα radiation generated at 50 kV and 35 mA. Samples were scanned at 0.08° s−1 in the range of 2θ = 15–35° using a step size of 0.2°.
Elemental analysis. Nitrogen elemental analysis was performed on a Euro EA 3000 Element analyzer (Euro Vector SPA, Italy).
Polymerization degree. The viscosity-average degrees of polymerization (Pv) of PVA and AAm-modified PVA were calculated from the respective intrinsic viscosities by using the Sakurada equation:19| |
 | (1) |
where [η] is the intrinsic viscosity of polymer. The intrinsic viscosities of PVA and AAm-modified PVA, [η](dL g−1), were measured in deionized H2O for PVA, and in aqueous solution of sodium chloride (1 M) using Ubbelohde capillary viscometer at 30 ± 0.01 °C.17,20
Water-solubility. Water-solubility of samples was evaluated by determining the mass percentage of dissolved polymer based on the total polymer added to solvent. Typically, water-solubility of all samples at 30 °C was measured as follow: 0.5 g PVA or modified PVA films (1 cm × 1 cm, 10 μm thickness) and 10 mL deionized water were mixed together, and the mixture was kept undisturbed for 120 min at 30 °C. Then 2 mL aliquots were sampled and filtrated to remove un-dissolved polymer films or gels. A Shimadzu UV-720 UV-vis spectrophotometer was used to measure absorption spectra of the filtrate. Prior to calculating concentration, a serious of calibration curves (plots of UV-vis absorbance versus solution concentration) for all samples were established following a well-accepted method.21,22 Calibrating the absorbance at maximum absorption band (λ = 190 nm for all samples) against the corresponding absorption–concentration calibration curve, concentration of the filtrate (Cf) could be achieved. Then the mass percentage (D%) of dissolved polymer based on the total polymer added to solvent could be calculated by eqn (2):where Cf is the concentration of filtrate; Vs is the total volume of deionized water using for dissolving samples, which is 10 mL in this case; and Wp is the total mass of samples using for water-solubility measurement, which is 0.5 g in this case.Evaluating water solubility of samples at 70 °C was processed similar to that at 30 °C as mention above except that time for dissolving samples was changed from 120 min to 20 min.
Viscosity of aqueous solution. Viscosity of PVA or AAm-modified PVA aqueous solution (5 wt%, 10 °C) was measured using a Brookfield LVDV-III viscometer (Brookfield, USA). Since viscosity of an aqueous PVA solution might increase gradually during storage, all viscosity measurements of these PVA and AAm modified PVA aqueous solutions were carried out 2 hours storage at 10 °C after preparation of the solution.
Results and discussion
Synthesis
To obtain acrylamide (AAm) modified PVA, random copolymerization of VAc and AAm were carried out firstly. Table 1 shows the copolymerization results of typical examples. For homopolymerization of VAc, monomer conversion reached up to 78% within 4 hours, while introducing 1.0 mol% (based on the amount of VAc) of AAm as a co-monomer, the monomer conversion of the copolymerization dropped to 73% under the same reaction conditions. Concomitantly, the monomer conversion slightly decreased with increasing the mole fractions of AAm in the reaction mix (Table 1). Kinetic study of the polymerization of VAc and the copolymerization of VAc with AAm at different mole feed ratio of VAc/AAm (Fig. 1) reveals the same tendency: the rate of copolymerization varies inversely with the AAm content. Considering the reactivity ratios of the monomer pairs, where rVAc = 0.09 and rAAm = 9.28,23 this phenomenon could be explained by the following: as AAm is approximately 10 times as reactive as VAc, AAm monomers hold a strong attraction for both AAm and VAc radicals, leading to the additive reaction of all radicals with AAm monomers, and the radicals will transfer to AAm monomers, and consequently, high reactive VAc radicals and high reactive AAm monomers gradually convert into low reactive AAm radicals. Thus, the concentration of AAm radicals will gradually increase with the polymerization time, while the concentration of AAm monomers will decrease. However, due to the low reactivity, AAm radicals react slowly with VAc monomers, which are also of low reactivity, resulting in the retardant of chain propagation. Similar results are also found in styrene–vinyl acetate and acrylonitrile–vinyl acetate systems as described in other articles.24,25 Furthermore, since both AAm and VAc radicals prefer AAm monomer, mole fraction of AAm units in copolymer chain should be larger than the mole fraction of AAm monomer in the feed until monomers conversion reaches 100%, which was consisted with the results of copolymer components (as listed in Table 1). The copolymer components were calculated from the percent nitrogen of the copolymers. The C, H, and N analyses were done on a Euro EA 3000 Element analyzer.
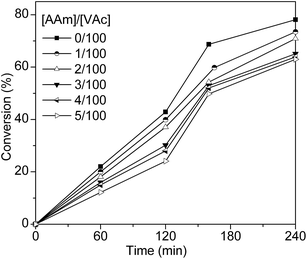 |
| | Fig. 1 Monomer conversion versus polymerization time plots for polymerization of VAc and copolymerization of VAc with AAm at different mole feed ratio of VAc/AAm. See Table 1 for polymerization conditions. | |
Poly(vinyl alcohol) (PVA) and AAM-modified poly(vinyl alcohol) (PVA–AAm) were obtained by alcoholysis of the corresponding precursors. The alcoholysis degree, mole fraction of AAm units and viscosity average molecular weight were given in Table 2. It can be observed that the component of AAm units increased with the increasing of mole fraction of AAm units in corresponding AAm modified PVAc precursor. However, the mole fraction of AAm units in PVA–AAm chain is slightly lower than that in the corresponding PVAc–AAm chain. Taking PVA–AAm-5 for instant, the mole fraction of AAm units in PVAc–AAm-5 is 10.9%, while that in the corresponding PVA–AAm-5 is only 7.1%. The decrease of AAm units may owe to partially hydrolyzing of acrylamide to acrylic acid units during alcoholysis process, resulting in the decrease of nitrogen elemental content. The alcoholysis degrees (AD%) of all samples, measured by back titration method, were also listed in Table 2. The alcoholysis degree information revealed that after 4 hours reaction by using NaOH as catalyst almost all pendent acetate groups were cleaved to end up with another polymers, PVA or acrylamide modified PVA (PVA–AAm). It should be noted that residual acrylic acid groups, generating from the hydrolysis of AAm units, may influence the alcoholysis degree results measured by back titration method. Residual acrylic acid consumes an excess of NaOH, resulting in slightly increase of the measured value of alcoholysis degree. Furthermore, the intrinsic viscosities of obtained PVA–AAm samples were conveniently measured in a Ubbelohde capillary viscometer, and the viscosity average degrees of polymerization were calculated by using the equation presented by Sakurada, Pv = ([η] × 104/8.33)1.61. Listed in the last column of Table 2 are the results. Despite the inhibiting effect of AAm on polymerization of VAc, the degrees of polymerization show no significant difference. In other words, the molecular weights of the as-synthesized PVA and PVA–AAm samples are at the same level.
Table 2 Alcoholysis degree, composition ratio, and Mη for PVA and AAm modified PVA
| Samplea |
Precursor |
ADa (%) |
AAm units ratiob (%) |
Pvc (×103) |
| Alcoholysis degree (AD), measured by back titration method. AAm units ratio in the copolymer composition, calculated from nitrogen elemental analysis. Viscosity-average degree of polymerization (Pv), calculated by using Pv = ([η] × 104/8.33)1.61; [η] was measured at 30 °C in water for PVA and in sodium chloride solution (1 N) for PVA–AAm samples. |
| PVA |
PVAc |
99.2 |
0 |
1.47 |
| PVA–AAm-1 |
PVAc–AAm-1 |
97.8 |
2.3 |
1.45 |
| PVA–AAm-2 |
PVAc–AAm-2 |
97.7 |
3.4 |
1.37 |
| PVA–AAm-3 |
PVAc–AAm-3 |
98.5 |
5.1 |
1.39 |
| PVA–AAm-4 |
PVAc–AAm-4 |
98.5 |
6.0 |
1.46 |
| PVA–AAm-5 |
PVAc–AAm-5 |
97.5 |
7.1 |
1.56 |
Chemical structure and tacticity characterized by FTIR
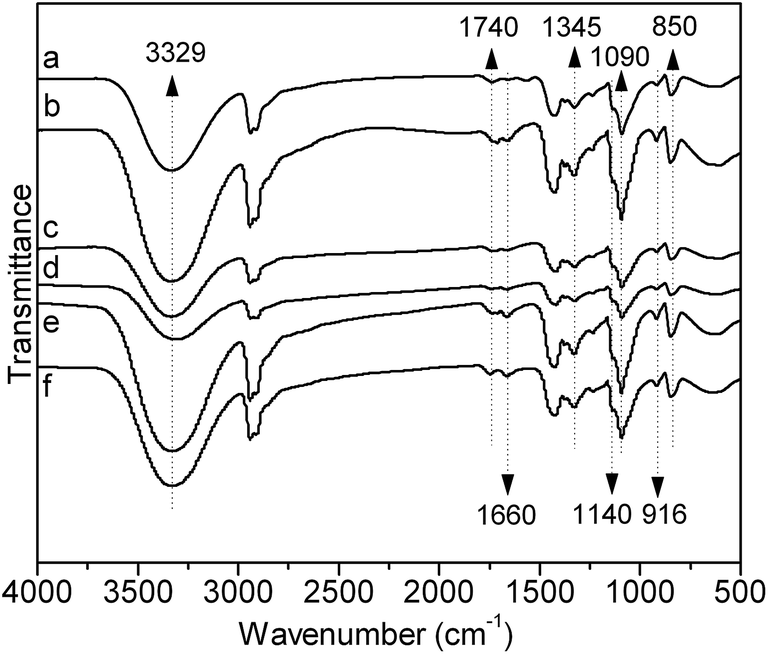
FTIR technique was used to investigate the difference of chemical structures between PVA and AAm modified PVA, PVA–AAm. Prior to recording the FTIR spectra, all samples were firstly purified by precipitation from deionized water solution in cold acetone twice to eliminate the possibility of the existence of homo-polyacrylamide and unreacted monomers. To make sure that all the homo-polyacrylamide and monomers had been extracted, the PVA and PVA–AAm samples were extracted with pure acetone until no impurity in acetone extracts could be detected by FTIR method. Because mixing tough PVA samples with dried KBr powder was not easy to form a homogeneous mixture, thin PVA films (10 μm) were employed instead of KBr pellet in this investigation. Typical infrared spectra of PVA film and PVA–AAm films were presented in Fig. 2.
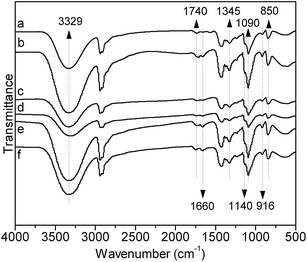 |
| | Fig. 2 FTIR spectra of pure PVA (a) and AAm modified PVA with different mole fraction of AAm units: (b) 2.3%, (c) 3.4%, (d) 5.1%, (e) 6.0%, (f) 7.1%. | |
Fig. 2(a) represents the FTIR spectrum of PVA film, while (b)–(f) show the FTIR spectra of AAm modified PVA films with mole fraction of AAm units of 2.3%, 3.4%, 5.1%, 6.0% and 7.1%, respectively. In Fig. 2(a), the characteristic stretching vibration peaks of –C–O– in PVA at 1090 cm−1 and 1140 cm−1 are observed. The strong absorption between 3000 and 3800 cm−1 should attribute to hydroxyl stretching vibration. Furthermore, weak absorptions at 1740 cm−1 and 1370 cm−1 should be noticed, which represent the stretching vibration of carbonyl (>C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and the stretching vibration of methyl (–COO–CH3) in acetate group, indicating an incomplete alcoholysis. The observation of residual acetate group is coherence with the alcoholysis degree of this sample detected by back titration method. Comparing the FTIR spectra of PVA–AAm, showing in Fig. 2(b)–(f), with that of pure PVA, two major changes could be observed: (1) for all samples (from b to f) at 1660 cm−1 appears a new peak and the peak area increases with the increase of mole fraction of AAm units in PVA chain; (2) the peak area of the band at 1740 cm−1 increases slightly for high AAm content PVA–AAm (samples e and f). As reported by other researchers,26 the characteristic absorption bands of polyacrylamide are at ∼1660 cm−1 corresponding to stretching vibration of carbonyl (>CO) and 1610 cm−1 corresponding to bending vibration of NH group. Considering that reliable purification of samples were performed to remove homo-polyacrylamide and unreacted monomers prior to FTIR measurement, therefore, development of new band at 1660 cm−1 clearly demonstrates the success of introduction of AAm units into PVA chain by copolymerization. The absorption band at 1740 cm−1 should attribute to the residual VAc units, as analyzed above. Therefore, the increase of this band plausibly indicates the decrease of the alcoholysis degree of PVA sample. However, considering the back titration and elemental analysis results, a more reasonably explain for this phenomenon should be the hydrolysis of AAm units, as discussed above. Under base condition, AAm will hydrolyze to acrylic acid, resulting in the blue shift of carbonyl group (>CO) from 1660 to around 1740 cm−1. Duplicating of absorption band of carbonyl group in acrylic acid with that in residual VAc units causes the increase of 1740 cm−1 band area.
O) and the stretching vibration of methyl (–COO–CH3) in acetate group, indicating an incomplete alcoholysis. The observation of residual acetate group is coherence with the alcoholysis degree of this sample detected by back titration method. Comparing the FTIR spectra of PVA–AAm, showing in Fig. 2(b)–(f), with that of pure PVA, two major changes could be observed: (1) for all samples (from b to f) at 1660 cm−1 appears a new peak and the peak area increases with the increase of mole fraction of AAm units in PVA chain; (2) the peak area of the band at 1740 cm−1 increases slightly for high AAm content PVA–AAm (samples e and f). As reported by other researchers,26 the characteristic absorption bands of polyacrylamide are at ∼1660 cm−1 corresponding to stretching vibration of carbonyl (>CO) and 1610 cm−1 corresponding to bending vibration of NH group. Considering that reliable purification of samples were performed to remove homo-polyacrylamide and unreacted monomers prior to FTIR measurement, therefore, development of new band at 1660 cm−1 clearly demonstrates the success of introduction of AAm units into PVA chain by copolymerization. The absorption band at 1740 cm−1 should attribute to the residual VAc units, as analyzed above. Therefore, the increase of this band plausibly indicates the decrease of the alcoholysis degree of PVA sample. However, considering the back titration and elemental analysis results, a more reasonably explain for this phenomenon should be the hydrolysis of AAm units, as discussed above. Under base condition, AAm will hydrolyze to acrylic acid, resulting in the blue shift of carbonyl group (>CO) from 1660 to around 1740 cm−1. Duplicating of absorption band of carbonyl group in acrylic acid with that in residual VAc units causes the increase of 1740 cm−1 band area.
The change in tacticity of PVA due to AAm modification is of special interest by the reason that PVA with higher tacticity, which has higher ability to form intermolecular hydrogen bond and crystals, leads to poor water solubility and eventually insolubility of PVA in hot water.27,28 Though introducing AAm units into PVA chain to decrease the steric regularity of PVA chain, decreases of tacticity of AAm modified PVA samples were expected. To understand the change of tacticity due to AAm modification of PVA, isotactic and syndiotactic contents were determined in respective FT-IR scan using the following formulas:29,30
| |
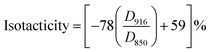 | (3) |
| |
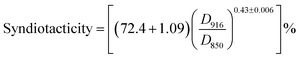 | (4) |
where
D916 is absorbance at 916 cm
−1 and
D850 is the absorbance at 850 cm
−1; calculations are performed on OMNIC 8.1 software after the linear baseline corrections.
Results calculated by eqn (3) and (4) were listed in Table 3. Due to the introduction of AAm units, an obviously decrease of the percentage of isotacticity was spotted as expected. A special notice should be paid to sample PVA–AAm-5. The isotacticity of this sample decreased dramatic from 27.1% (for pure PVA) to 10.3% (with introducing 7.1 mol% AAm units). This indicates that the isotactic regularity of hydroxyl groups of PVA molecule is reduced. However, in case of syndiotacticity, we observed a slight increase in percentage of syndiotacticity after introducing AAm units into PVA chain. Although the reason of this increase has not yet been ascertained, strong intermolecular hydrogen bonding interactions were believed to play an important role in this case as demonstrated by other researchers.31
Table 3 Tacticity of AAm modified PVA calculated by eqn (3) and (4)
| Sample |
AAm units ratio (%) |
Isotacticity (%) |
Syndiotacticity (%) |
| PVA |
0 |
27.1 |
50.0 ± 0.27 |
| PVA–AAm-1 |
2.3 |
25.1 |
51.4 ± 0.26 |
| PVA–AAm-2 |
3.4 |
16.9 |
56.4 ± 0.21 |
| PVA–AAm-3 |
5.1 |
18.9 |
55.2 ± 0.22 |
| PVA–AAm-4 |
6.0 |
22.0 |
53.3 ± 0.24 |
| PVA–AAm-5 |
7.1 |
10.3 |
60.0 ± 0.17 |
Crystallinity of PVA and AAm modified PVA characterized by XRD
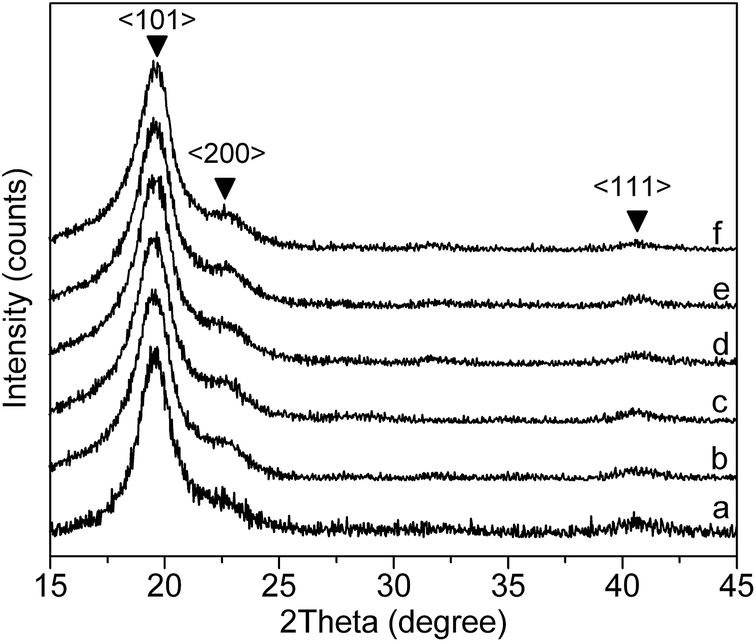
The actual change in the crystallinity of PVA before and after AAm modification was characterized by XRD method and the results were shown in Fig. 3. Fig. 3(a) represents the XRD pattern of the pristine PVA sample, while (b)–(f) show the XRD pattern of AAm modified PVA samples with mole fraction of AAm units of 2.3%, 3.4%, 5.1%, 6.0% and 7.1%, respectively. Typical diffraction peaks at 2θ = 20.12, 23.42 and 41.18 due to 〈101〉, 〈200〉 and 〈111〉 crystalline planes were observed in all cases, indicating that AAm units will not cause observed crystal pattern transition of PVA. By mapping crystalline and amorphous area under the XRD curves, the degrees of crystallinity of PVA and AAm modified PVA samples were estimated. Fig. 4 shows the plot of crystallinity degree against mole fraction of AAm units. This figure illustrated that the degree of crystallinity decreased approximately linearly as mole fraction of AAm units increases, well consisting with the decrease of isotactic regularity of hydroxyl groups as revealed by FTIR measurement.
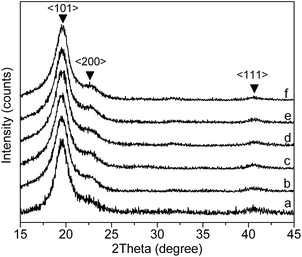 |
| | Fig. 3 XRD spectra of pure PVA (a) and AAm modified PVA with different mole fraction of AAm units: (b) 2.3%, (c) 3.4%, (d) 5.1%, (e) 6.0%, (f) 7.1%. | |
 |
| | Fig. 4 Crystallinity of AAm modified PVA as a function of AAm-content of the copolymers. | |
Effect of copolymerization on water solubility
It is well documented that the water solubility of PVA is closely related to the crystallinity, as well as the tacticity. Previous work by Fuji and coworkers27 reveals that PVA with less crystalline has higher water solubility than more crystalline samples. Meanwhile, the tacticity configuration favours interchain hydrogen bonding, leading to high crystalline and consequently to poor water solubility.28 Triggered by these researches, we carried out the investigation of the influence of AAm units on tacticity and crystallinity of PVA, hoping that we can consequently enhance the water solubility of PVA. As previously discussed, tacticity and crystallinity have been successfully controlled by introducing AAm units, hence, the effects of the AAm units on water solubility (D%) of PVA is of particular interest in this section. The plots of water solubility versus mole fraction of AAm at 30 °C and 70 °C are shown in Fig. 5(a), and the influence of crystalline on the water solubility is shown in Fig. 5(b). The water solubility of PVA and AAm-modified PVA was measured at the same conditions, and polymer film samples with the same surface area (1 cm × 1 cm) and thickness (10 μm thickness) were used in all cases to eliminate errors that might result from experimental design. From Fig. 5 it can be seen that water solubility of the pristine PVA was only 3.9 wt% for 20 hours at 30 °C, and an approximately linearly increase of the water solubility of the materials with mole fraction of AAm units was observed. By introducing 7.1% mole fraction of AAm units into PVA chain, the water solubility increased to 30 wt%, more than 4 times as high as that of pristine PVA. It seems that the improved water solubility of the AAm modified PVA can be most probably attributed to the dramatic decrease of the crystallinity of the materials, as shown in Fig. 5(b). It is also worth mentioning that the water solubility of the materials will also be affected by the competition of hydrogen bonding between AAm units and vinyl alcohol units with that between AAm units and solvent. If the AAm units prefer to form inter molecular hydrogen bonds with water, formation of hydrogen bonds will be good for enhancing the water solubility. Otherwise, if the interactions are dominated by AAm–PVA hydrogen bonding, hydrogen bonding networks forms, resulting in the decrease of water solubility of the materials. From the water solubility results, it seems that AAm–H2O hydrogen bonding interaction might be in domination. And the hydrogen bonding effect will be further discussed in the next section.
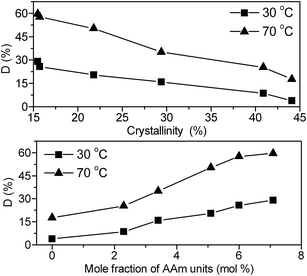 |
| | Fig. 5 Plots of water solubility (D%) versus crystallinity (upper), and AAm-content (lower) of AAm modified PVA. | |
The water solubility measured at 70 °C also indicates the dependence of water solubility on mole fraction of AAm units in PVA chain, confirming that copolymerization with AAm is a promising way to enhance the water solubility of PVA with high alcoholysis degree, in particular in the range of near 97%.
Effect of copolymerization on viscosity
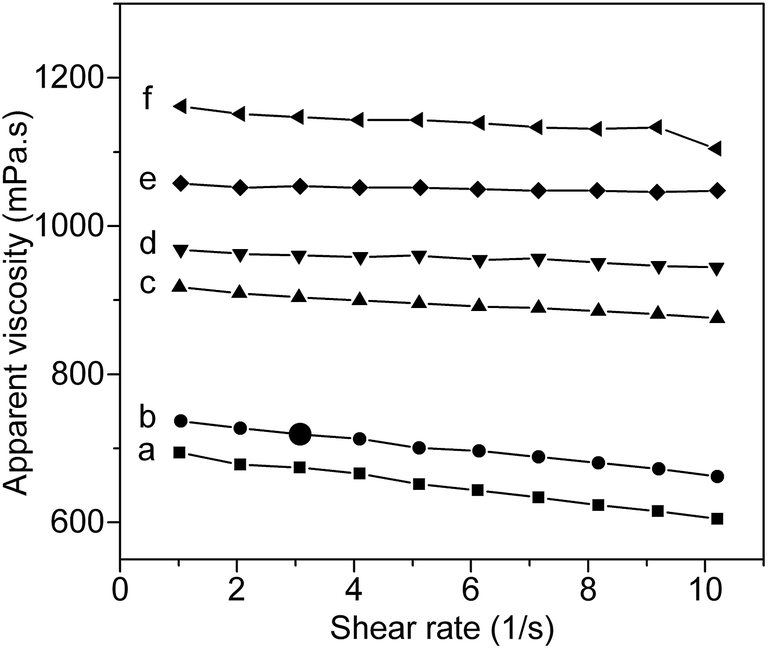
For PVA, it is believed that inter and intra hydrogen bonding in the PVA chains is disrupted by thermal energy during dissolving. However, upon cooling, the inter and intra hydrogen bonding can reform, leading to gradually increase of the viscosity of PVA solution. After introducing AAm units to PVA chain, the viscosity behaviors of PVA–AAm aqueous solution will be more complex due to additional AAm–PVA, AAm–H2O and AAm–AAm hydrogen bonding interactions. To understand the role of AAm units in controlling the inter- and intra-molecular hydrogen bonding of PVA–AAm aqueous solutions, rheological studies of aqueous solution of PVA–AAm with different mole fraction ratio of AAm units were carried out at 10 °C. Fig. 6 shows the interrelationship between apparent viscosity and shear rate for 5 wt% aqueous solutions of PVA–AAm with 6 different mole fraction ratios of AAm units. It is seen from the figure that the apparent viscosity increases significantly when the AAm mole fraction increases from 0% (pristine PVA) to 7.1% (copolymerization with AAm with a feed ratio of VAc![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AAm = 100:5). This increase of viscosity, for PVA–AAm with high AAm mole fraction, is most certainly due to either the strong hydrogen bonding interactions between polymer chains and solvent (water) or that inters polymer chains. If strong polymer chain associations are formed in those aqueous solutions, a pronounced shear thinning behavior will be anticipated. From Fig. 6 it should be special noticed that the shear thinning behavior does have been affected by copolymerization. However, compared with the pristine PVA aqueous solution, no pronounced shear thinning behavior is observed for those PVA–AAm aqueous solutions with high AAm mole fraction (>5.1%). This result indicates that polymer chain associations in the AAm modified PVA are lower than that in the pristine PVA. Considering the experimental results, the hydrogen bonding interactions between PVA–AAm and solvent seems to be in domination here, which is in coherence with the water solubility results as previously discussed. Thus, this study reveals that copolymerization with AAm could be a useful method for enhancing the water solubility of PVA and restraining the formation of hydrogen bonding networks in PVA aqueous solution.
:AAm = 100:5). This increase of viscosity, for PVA–AAm with high AAm mole fraction, is most certainly due to either the strong hydrogen bonding interactions between polymer chains and solvent (water) or that inters polymer chains. If strong polymer chain associations are formed in those aqueous solutions, a pronounced shear thinning behavior will be anticipated. From Fig. 6 it should be special noticed that the shear thinning behavior does have been affected by copolymerization. However, compared with the pristine PVA aqueous solution, no pronounced shear thinning behavior is observed for those PVA–AAm aqueous solutions with high AAm mole fraction (>5.1%). This result indicates that polymer chain associations in the AAm modified PVA are lower than that in the pristine PVA. Considering the experimental results, the hydrogen bonding interactions between PVA–AAm and solvent seems to be in domination here, which is in coherence with the water solubility results as previously discussed. Thus, this study reveals that copolymerization with AAm could be a useful method for enhancing the water solubility of PVA and restraining the formation of hydrogen bonding networks in PVA aqueous solution.
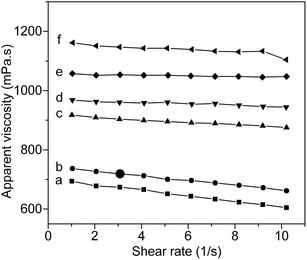 |
| | Fig. 6 Apparent viscosity as a function of shear rate for the AAm modified PVA aqueous solution (5 wt%) at 10 °C. | |
Conclusions
AAm modified PVA with high degree of alcoholysis, 97% or greater, was successfully synthesized by copolymerization of VAc and AAm and following alcoholysis. The introduction of AAm units into PVA chain significantly decreased isotacticity and suppressed crystallinity, resulting in an obvious enhancement of water solubility of AAm modified PVA at either low temperature (30 °C) or high temperature (70 °C) with regard to pristine PVA. Further rheological researches on the PVA and AAm modified PVA aqueous solutions indicated that stronger intermolecular hydrogen bonding interactions with water were found in AAm modified PVA aqueous solution comparing with pristine PVA. Also, strong polymer chain associations through hydrogen bonds in PVA aqueous solution seemed to be weakened by the AAm modification, indicated by rheological investigation. Hence, this research provides a promising way for tuning the microstructure and crystallinity, and improving the water solubility of PVA with high degree of alcoholysis through copolymerization with AAm.
Acknowledgements
The authors are grateful to the National Natural Science Foundation of China (No. 51403140 and 51173116) for support of this research.
Notes and references
- S. Bose, T. Kuila, T. X. H. Nguyen, N. H. Kim, K. T. Lau and J. H. Lee, Polymer membranes for high temperature proton exchange membrane fuel cell: recent advances and challenges, Prog. Polym. Sci., 2011, 36, 813–843 CrossRef CAS PubMed.
- J. M. Yang, N. C. Wang and H. C. Chiu, Preparation and characterization of poly(vinyl alcohol)/sodium alginate blended membrane for alkaline solid polymer electrolytes membrane, J. Membr. Sci., 2014, 457, 139–148 CrossRef CAS PubMed.
- Y. Pan, K. Shi, C. Peng, W. Wang, Z. Liu and X. Ji, Evaluation of hydrophobic polyvinyl-alcohol formaldehyde sponges as absorbents for oil spill, ACS Appl. Mater. Interfaces, 2014, 6, 8651–8659 CAS.
- Y. Zhang, B. Gao and Z. Xu, Adsorption properties of polyvinyl alcohol-grafted particles toward genistein driven by hydrogen-bond interaction, J. Phys. Chem. B, 2013, 117, 5730–5736 CrossRef CAS PubMed.
- A. Sh. Asran, K. Razghandi, N. Aggarwal, G. H. Michler and T. Groth, Nanofibers from blends of polyvinyl alcohol and polyhydroxy butyrate as potential scaffold material for tissue engineering of skin, Biomacromolecules, 2010, 11, 3413–3421 CrossRef CAS PubMed.
- N. D. Thorat, K. P. Shinde, S. H. Pawar, K. C. Barick, C. A. Betty and R. S. Ningthoujam, Polyvinyl alcohol: an efficient fuel for synthesis of superparamagnetic LSMO nanoparticles for biomedical application, Dalton Trans., 2012, 41, 3060–3071 RSC.
- J. L. Manasco, C. Tang, N. A. Burns, C. D. Saquing and S. A. Khan, Rapidly dissolving poly(vinyl alcohol)/cyclodextrin electrospun nanofibrous membranes, RSC Adv., 2014, 4, 13274–13279 RSC.
- Polyvinyl alcohol: properties and applications, ed. C. A. Finch, John Wiley, New York, 1991, ch. 2, pp. 17–66 Search PubMed.
- Y. Satokawa and T. Shikata, Hydration structure and dynamic behavior of poly(vinyl alcohol)s in aqueous solution, Macromolecules, 2008, 41, 2908–2913 CrossRef CAS.
- R. Ricciardi, F. Auriemma, C. de Rosa and F. Laupretre, X-ray diffraction analysis of poly(vinyl alcohol) hydrogels, obtained by freezing and thawing techniques, Macromolecules, 2004, 37, 1921–1927 CrossRef CAS.
- L. Peng, T. Zhou, Y. Huang, L. Jiang and Y. Dan, Microdynamics mechanism of thermal-induced hydrogel network destruction of poly(vinyl alcohol) in D2O studied by two-dimensional infrared correlation spectroscopy, J. Phys. Chem. B, 2014, 118, 9496–9506 CrossRef CAS PubMed.
- Y. Lu, R. Jing, Q. M. Kong and R. X. Zhu, Solid state grafting copolymerization of acrylamide onto poly(vinyl alcohol) initiated by redox system, J. Appl. Polym. Sci., 2014, 131 DOI:10.1002/app.39938.
- S. J. Carlotti and O. Giani-Beaune, Characterization and mechanical properties of water-soluble poly(vinyl alcohol) grafted with lactic acid and glycolic acid, J. Appl. Polym. Sci., 2001, 80, 142–147 CrossRef CAS.
- A. Debuigne, J. Warnant, R. Jerome, I. Voets, A. de Keizer, M. A. C. Stuart and C. Detrembleur, Synthesis of novel well-defined poly(vinyl acetate)-b-poly(acrylonitrile) and derivatized water-soluble poly(vinyl alcohol)-b-poly(acrylic acid) block copolymers by cobalt-mediated radical polymerization, Macromolecules, 2008, 41, 2353–2360 CrossRef CAS.
- A. Lejardi, A. Etxeberria, E. Meaurio and J. R. Sarasua, Novel poly(vinyl alcohol)-g-poly(hydroxy acid) copolymers: synthesis and characterization, Polymer, 2012, 53, 50–59 CrossRef CAS PubMed.
- J. Ding, S. C. Chen, X. L. Wang and Y. Z. Wang, Synthesis and properties of thermoplastic poly(vinyl alcohol)-graft-lactic acid copolymers, Ind. Eng. Chem. Res., 2009, 48, 788–793 CrossRef CAS.
- T. Moritani and K. Kajitani, Functional modification of poly(vinyl alcohol) by copolymerization: 1. Modification with carboxylic monomers, Polymer, 1997, 38, 2933–2945 CrossRef CAS.
- T. Moritani and J. Yamauchi, Functional modification of poly(vinyl alcohol) by copolymerization: III. Modification with cationic monomers, Polymer, 1998, 39, 559–572 CrossRef CAS.
- Polyvinyl Alcohol: Properties and Applications, ed. C. A. Finch, Wiley, New York, 1973, Appendix 3, pp. 561–572 Search PubMed.
- B. M. Budhlall, K. Landfester, E. D. Sudol, V. L. Dimonie, A. Klein and M. S. El-Aasser, Characterization of partially hydrolyzed poly(vinyl alcohol). Effect of poly(vinyl alcohol) molecular architecture on aqueous phase conformation, Macromolecules, 2003, 36, 9477–9484 CrossRef CAS.
- T. Hirai, A. Okazaki and S. Hayashi, Characterization of poly(vinyl alcohol-co-vinyl acetate) by the iodine complexation method, J. Appl. Polym. Sci., 1987, 34, 527–536 CrossRef CAS PubMed.
- H. H. Perkampus, H. C. Grinter and T. L. Threlfall, In UV-vis spectroscopy and its application, Springer, New York, 1992, ch. 4, pp. 26–80 Search PubMed.
- M. Mukherjee, S. K. Chatterjee, A. S. Brar and K. Dutta, Compositional and stereochemical analysis of acrylamide/vinyl acetate copolymers by one- and two-dimensional NMR spectroscopy, Macromolecules, 1998, 31, 8455–8462 CrossRef CAS.
- F. R. Mayo, C. Walling, F. M. Lewis and W. F. Hulse, Copolymerization. V. Some copolymerizations of vinyl acetate, J. Am. Chem. Soc., 1948, 70, 1523–1525 CrossRef CAS.
- P. J. Flory, in Principles of Polymer Chemistry, Cornell University Press, New York, 1953, ch. 5, pp. 178–230 Search PubMed.
- X. Lu and Y. Mi, Characterization of the interfacial interaction between polyacrylamide and silicon substrate by Fourier Transform Infrared Spectroscopy, Macromolecules, 2005, 38, 839–843 CrossRef CAS.
- K. Fuji, S. Imoto, J. Ukida and M. Matsumoto, On the stereoregulation in polymerization of vinyl formate, J. Polym. Sci., Part B: Polym. Lett., 1963, 1, 497–499 CrossRef PubMed.
- H. Ohgi, T. Sato, H. Watanabe and F. Horii, Highly isotactic poly(vinyl alcohol) derived from tert-butyl vinyl ether V. Viscoelastic behavior of highly isotactic poly(vinyl alcohol) films, Polym. J., 2006, 38, 1055–1060 CrossRef CAS.
- Polyvinyl Alcohol Properties and Applications, ed. C. A. Finch, Wiley: New York, 1973, ch 10, pp. 203–232 Search PubMed.
- S. Murahashi, S. Nozakur, M. Sumi, H. Yuki and K. Hatada, NMR study of the tacticity of poly(vinyl acetate), J. Polym. Sci., Part B: Polym. Lett., 1966, 4, 65–69 CrossRef CAS PubMed.
- D. J. Upadhyay and N. V. Bhat, Separation of azeotropic mixture using modified PVA membrane, J. Membr. Sci., 2005, 255, 181–186 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.