
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diazo compounds for the bioreversible esterification of proteins†
Nicholas A.
McGrath‡§
a,
Kristen A.
Andersen‡
b,
Amy K. F.
Davis
c,
Jo E.
Lomax¶
d and
Ronald T.
Raines
*ac
aDepartment of Chemistry, University of Wisconsin–Madison, 1101 University Avenue, Madison, WI 53706, USA. E-mail: rtraines@wisc.edu
bMolecular & Cellular Pharmacology Graduate Training Program, University of Wisconsin–Madison, 1300 University Avenue, Madison, WI 53706, USA
cDepartment of Biochemistry, University of Wisconsin–Madison, 433 Babcock Drive, Madison, WI 53706, USA
dGraduate Program in Cellular and Molecular Biology, University of Wisconsin–Madison, 1525 Linden Drive, Madison, WI 53706, USA
First published on 1st October 2014
Abstract
A diazo compound is shown to convert carboxylic acids to esters efficiently in an aqueous environment. The basicity of the diazo compound is critical: low basicity does not lead to a reaction but high basicity leads to hydrolysis. This reactivity extends to carboxylic acid groups in a protein. The ensuing esters are hydrolyzed by human cellular esterases to regenerate protein carboxyl groups. This new mode of chemical modification could enable the key advantages of prodrugs to be translated from small-molecules to proteins.
Introduction
Esters are nearly absent from human cells, aside from acylglycerols and amino-acyl tRNAs. One reason is their lability in the presence of intracellular esterases.1 This reactivity is the basis for the clinical utility of the many prodrugs that are unmasked by enzyme-catalyzed ester hydrolysis.2–5Inspired by prodrugs, we envisioned esterification as a means to modify proteins covalently but reversibly with desirable pendants, such as pharmacokinetic enhancing, cell-targeting, or cell-penetration moieties. The acetylation of serine and threonine residues is a natural post-translational modification.6,7 There is not, however, an efficient means to generate esters from protein carboxyl or hydroxyl groups by chemical synthesis. Carbodimides and other reagents can be used to activate protein carboxyl groups, but solvent water and protein amino, hydroxyl, and sulfhydryl groups compete effectively with exogenous alcohols for the ensuing activated acyl groups.8–10 Accordingly, we sought a different strategy—one that enlists O-alkylation rather than acyl transfer.
Diazo compounds are in widespread use in synthetic organic chemistry.11–13 The simplest—diazomethane—readily converts carboxylic acids into methyl esters with molecular nitrogen as the only byproduct. Diazomethane suffers, however, from non-specific reactivity. For example, diazomethane is known to react with water and with the side chains of lysine and tyrosine residues.14 Other non-stabilized diazo compounds or stabilized metal carbenoids are capable of esterifying carboxylic acids,15–20 but their high reactivity likewise limits utility with biomolecules.
We were aware of precedent for the use of stabilized diazo compounds in a biochemical context. In the 1960s, 2-diazoacetamide,21,22N-(diazoacetyl)glycinamide,23 and diphenyldiazomethane24 were used to identify the most reactive carboxylic acid groups in proteins. These efforts required adding a vast molar excess (>103-fold) of the diazo compound and tedious monitoring of reaction pH, all to achieve modest labelling. We suspected that the problem was non-specific reactivity, as with diazomethane.14 Here, we report on the reactivity of still more stabilized diazo compounds with relevant carboxylic acids.
Results and discussion
We began by screening the reactivity of diazo compounds 1 and 2 with small-molecule carboxylic acids of varying acidity and bearing a variety of reactive functional groups. These diazo compounds were accessed from their corresponding azide by a deimidogenation reaction that uses a phosphinoester to convert an azide into a diazo-compound.25,26 In acetonitrile, both diazo compounds were unreactive toward some nucleophiles common in biomolecules: amino, hydroxyl, and sulfhydryl groups. Both, however, did react with carboxylic acids to give comparable yields of ester product (Scheme 1). In contrast to diazo compounds 1 and 2, diethyl 2-diazomalonate was unreactive with a–h. Tellingly, β-alanine (h) proved unreactive with all three diazo compounds under any tested condition. This acid alone exists as a zwitterion, and its lack of reactivity is consistent with esterification occurring via a diazonium-carboxylate salt (Fig. 1).27,28 Moreover, we noted that a carboxylic acid (a–g) is acidic enough to promote the reaction, but a phenolic hydroxyl (d), sulfhydryl (f), or ammonium group (h) is not.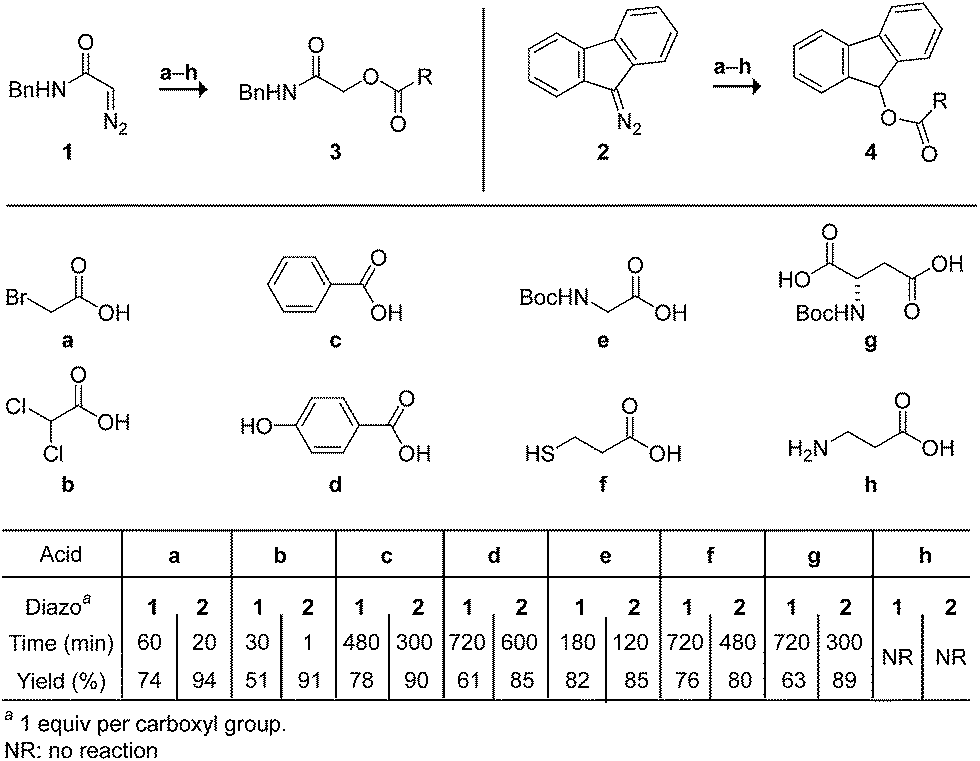 | ||
| Scheme 1 Chemoselective esterification in acetonitrile. | ||
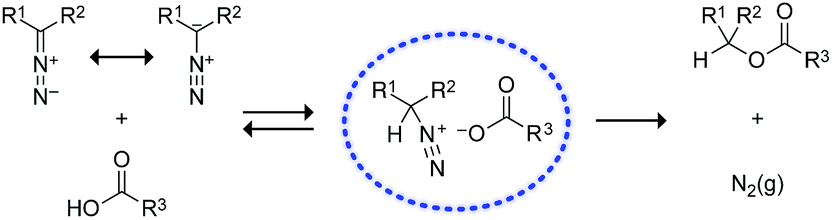 | ||
| Fig. 1 Putative mechanism for the O-alkylation of carboxylic acids by diazo compounds.27,28 | ||
Next, we investigated the analogous reactions in an aqueous environment (Scheme 2). Whereas diazo compound 1 was competent for esterification in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of acetonitrile and 10 mM MES–NaOH buffer, pH 5.5, the major product was alcohol 5 formed when water attacks the diazonium ion. On the other hand, diazo compound 2 gave primarily the desired ester 4 in all cases. Interestingly, the ester:alcohol product ratios with 1 were variable, whereas those with 2 were approximately two.29 These data are consistent with the nascent diazonium-carboxylate salt being maintained in a solvent cage by Coulombic forces (Fig. 1), as described by others.28,30 Carrying out the reaction in a 1:1 mixture of acetonitrile and 10 mM MES–NaOH buffer resulted in greatly deleterious partitioning with 1 but not 2. Diethyl 2-diazomalonate was found to be unreactive in these aqueous conditions, as in neat acetonitrile.
:1 mixture of acetonitrile and 10 mM MES–NaOH buffer, pH 5.5, the major product was alcohol 5 formed when water attacks the diazonium ion. On the other hand, diazo compound 2 gave primarily the desired ester 4 in all cases. Interestingly, the ester:alcohol product ratios with 1 were variable, whereas those with 2 were approximately two.29 These data are consistent with the nascent diazonium-carboxylate salt being maintained in a solvent cage by Coulombic forces (Fig. 1), as described by others.28,30 Carrying out the reaction in a 1:1 mixture of acetonitrile and 10 mM MES–NaOH buffer resulted in greatly deleterious partitioning with 1 but not 2. Diethyl 2-diazomalonate was found to be unreactive in these aqueous conditions, as in neat acetonitrile.
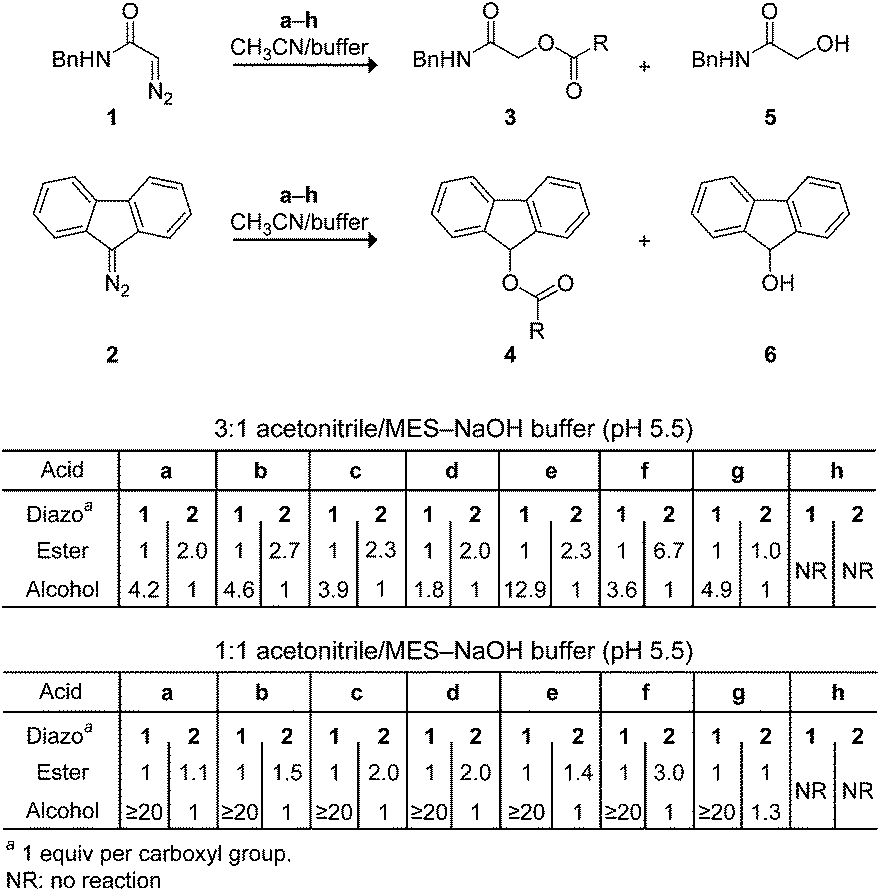 | ||
| Scheme 2 Chemoselective esterification in an aqueous environment. | ||
What is the basis for this differential reactivity? The pKa values of relevant acids have been measured in dimethyl sulfoxide.31 These pKa values correlate with the observed reactivity via the mechanism of Fig. 1. Specifically, the conjugate base of diethylmalonate (pKa 16.4)32 is weak, and diethyl 2-diazomalonate cannot abstract a proton from a carboxylic acid. On the other hand, the conjugate base of diethylacetamide (pKa 35)33 is strong, and diazo compound 1 lacks chemoselectivity in an aqueous environment (Scheme 2). Only the conjugate base of fluorene (pKa 22.6)34 is matched to the task, enabling diazo compound 2 access to the mechanism of Fig. 1 with high chemoselectivity.
We used thioacetic acid to probe the mechanism of esterification. Its acidic proton resides primarily on sulfur,35 but an intermediate diazonium-thiocarboxylate salt could lead to either a thioester or thionoester product. In anhydrous acetonitrile, complete selectivity for thioester formation was obtained with diazo compound 2 (Scheme 3). This selectivity rules out a cyclic transition state reminiscent of an ene reaction,36,37 which would provide the thionoester as the product. On the other hand, diazo compound 1 produced a mixture of thio- and thionoester products. In an aqueous environment, these same reactants led exclusively to a thio- rather than a thionoester, along with the hydrolysis product from diazo compound 1.38 The lack of thionoester formation can be attributed to the greater nucleophilicity of sulfur on the diazonium salt and the greater ability of oxygen to form a hydrogen bond with solvent water,39 which would decrease its nucleophilicity still further.
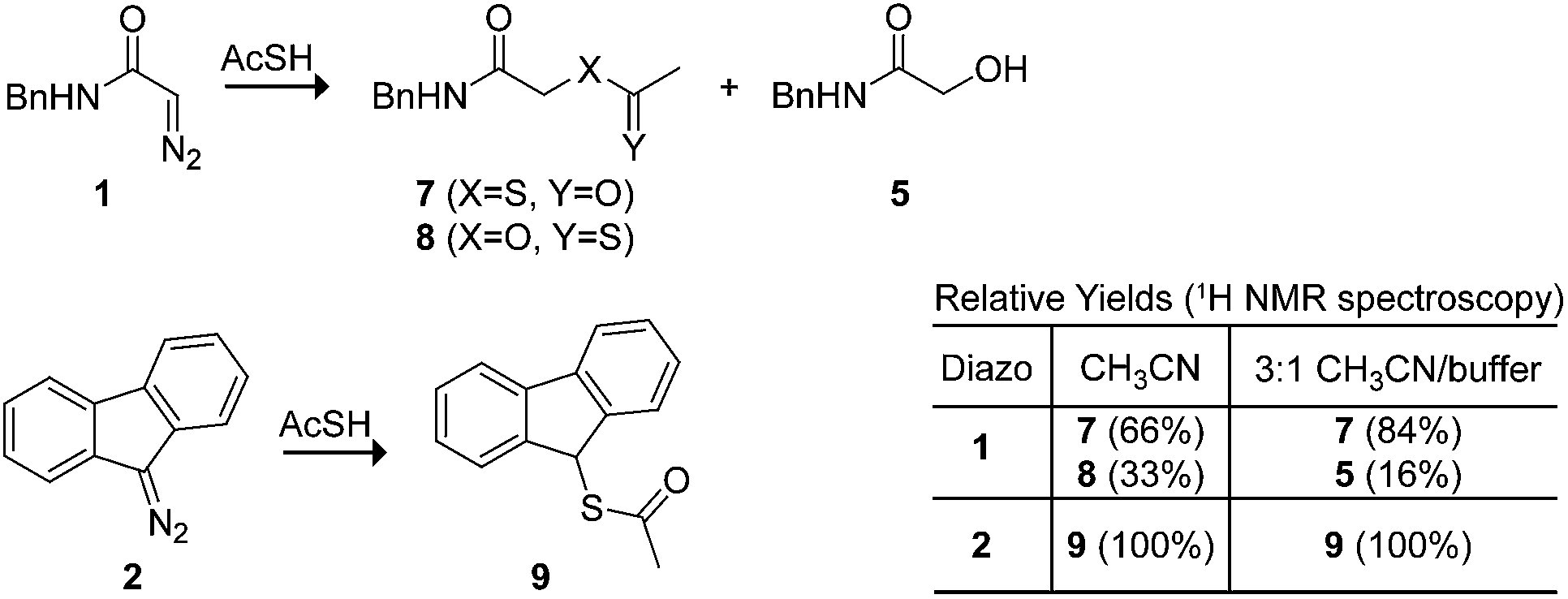 | ||
| Scheme 3 Thio- versus thionoester formation. | ||
Finally, we assessed the ability of diazo compounds 1 and 2 to esterify the carboxyl groups present in a model protein, bovine pancreatic ribonuclease (RNase A; Fig. 2A). The catalytic activity of this well-characterized enzyme provides an extremely sensitive measure of native structure and function.40 RNase A was incubated for 4 h at 37 °C in a 1:1 mixture of 10 mM MES–NaOH buffer, pH 5.5, and acetonitrile containing a diazo compound (10 equiv.). Under these conditions, diazo compound 1 proved incapable of labelling the protein. Only in the presence of 200 equiv. was any esterification observed with this reagent, consistent with its tendency towards hydrolysis in an aqueous environment (Scheme 2). In contrast, diazo compound 2 esterified, on average, three of the eleven carboxylates (Fig. S1†). Using trypsin digestion coupled with mass spectrometry, we were able to identify the esterified residues. Diazo compound 2 (10 equiv.) labelled Asp14, Glu49, Glu111, and Asp121, almost exclusively. Diazo compound 1 (200 equiv.) labelled a similar subset of residues (Glu9, Asp14, Glu49, Glu111, and the C terminus). The basis for reaction with these particular residues is not clear, but likely integrates the reactivity (i.e., high pKa) and accessibility of their carboxyl groups.
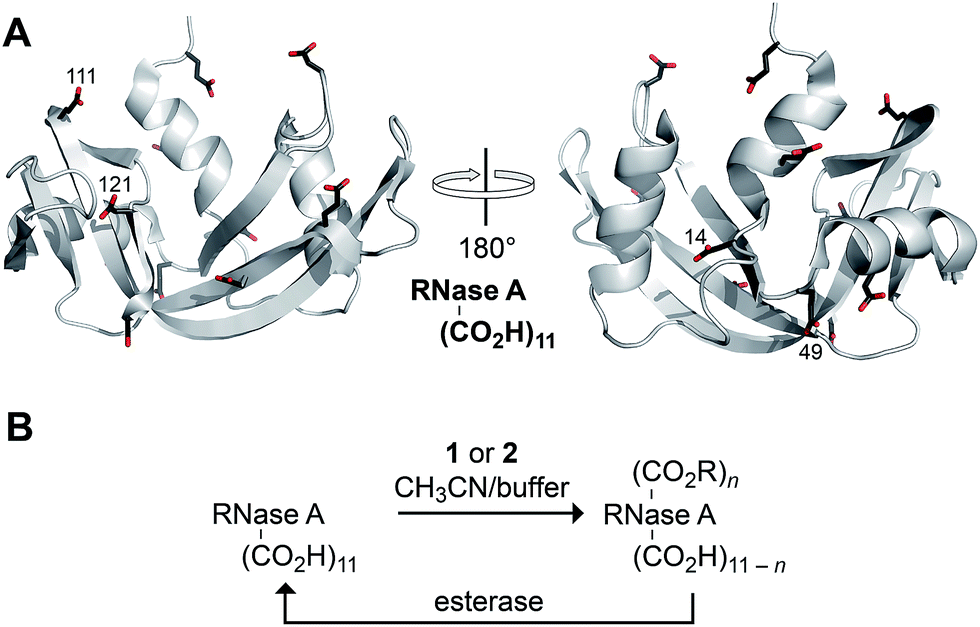 | ||
| Fig. 2 (A) Three-dimensional structure of RNase A (PDB entry 6rsa) showing its eleven carboxyl groups. The four residues labeled primarily by diazo compound 2 are indicated explicitly. (B) Scheme for the bioreversible esterification of RNase A with diazo compound 2. | ||
Most importantly, we probed the bioreversibility of our protein esterification reaction (Fig. 2B). To enhance the relevance of this experiment, we sought to employ esterases endogenous to human cells rather than commercial enzymes. Accordingly, we used recombinant DNA technology to add an 8-residue FLAG tag to the N-terminus of RNase A, and we esterified the ensuing FLAG–RNase A with diazo compound 2 as described above. We found labelling to be comparable to that of wild-type RNase A (Fig. S12†), and we observed that the enzyme lost half of its catalytic activity upon esterification (Fig. S13†).41 Then, we incubated the esterified protein with a HeLa cell extract. We recovered the enzyme from this complex mixture with an anti-FLAG antibody immobilized on magnetic beads. We could not detect any esters in the extract-treated enzyme with mass spectrometry (Fig. S12†),42 and we found that the enzyme had recovered all of its catalytic activity (Fig. S13†). These data indicate that the protein esterification reaction was fully bioreversible.
Finally, we sought to demonstrate the generality of our esterification method by employing a second protein substrate with a different tag for its purification. Specifically, we applied diazo compound 2 (10 equiv.) and the conditions described above to the mCherry variant of red fluorescent protein (RFP) from Discosoma sp. We found that His6–RFP became esterified with 1–3 fluorenyl groups. The protein did not lose its fluorescence during the esterification reaction. As with RNase A, the esterification of RFP was bioreversible. Incubation with a HeLa cell extract followed by recovery with a Ni-affinity resin, yielded unmodified His6–RFP (Fig. S13†). Success with both FLAG-labeled RNase A and His6-labeled RFP indicates that our esterification method has a broad scope.
Concluding remarks
We find that a diazo compound can effect the efficient O-alkylation of carboxylic acids in an aqueous environment near neutral pH, and that this reactivity extends to proteins. The esters in the ensuing protein can be hydrolysed by esterases that are endogenous to human cells, thereby recreating the wild-type protein and avoiding a compromise to function or the display of an epitope. By enabling the facile semisynthesis of “proproteins” containing transitory pharmacokinetic enhancing, cell-targeting, or cell-penetration moieties, we envision that O-alkylation of carboxylic acids by diazo compounds could broaden the utility of antibodies, enzymes, hormones, and other proteins in chemical biology and pharmacology.Acknowledgements
This work was supported by grant R01 GM044783 (NIH). N.A.M. was supported by postdoctoral fellowship F32 GM096712 (NIH). K.A.A. was supported by a PhRMA predoctoral fellowship and by Molecular and Cellular Pharmacology Training Grant T32 GM008688 (NIH). A.K.F.D. was supported by a Hilldale Undergraduate/Faculty Research Fellowship. J.E.L. was supported by an NSF Graduate Research Fellowship. This work made use of the National Magnetic Resonance Facility at Madison, which is supported by grant P41 GM103399 (NIH), and the Biophysics Instrumentation Facility, which was established with grants BIR-9512577 (NSF) and S10 RR013790 (NIH).Notes and references
- L. Tian, Y. Yang, L. M. Wysocki, A. C. Arnold, A. Hu, B. Ravichandran, S. M. Sternson, L. L. Looger and L. D. Lavis, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4756–4761 CrossRef CAS PubMed.
- B. Testa and J. M. Mayer, Hydrolysis in Drug and Prodrug Metabolism: Chemistry, Biochemistry, and Enzymology, Academic Press, New York, NY, 2003 Search PubMed.
- Prodrugs: Challenges and Rewards, ed. V. J. Stella, R. T. Borchardt, M. J. Hageman, R. Oliyai, M. Maag and J. W. Tilley, Springer, New York, NY, 2007 Search PubMed.
- L. D. Lavis, ACS Chem. Biol., 2008, 3, 203–206 CrossRef CAS PubMed.
- K. M. Huttunen, H. Raunio and J. Rautio, Pharmacol. Rev., 2011, 63, 750–771 CrossRef CAS PubMed.
- S. Mukherjee, G. Keitany, Y. Li, Y. Wang, H. L. Ball, E. J. Goldsmith and K. Orth, Science, 2006, 312, 1211–1214 CrossRef CAS PubMed.
- S. Mukherjee, Y.-H. Hao and K. Orth, Trends Biochem. Sci., 2007, 32, 210–216 CrossRef CAS PubMed.
- Bioconjugation, ed. M. Aslam and A. Dent, Macmillan Reference, London, UK, 1998 Search PubMed.
- R. L. Lundblad, Chemical Reagents for Protein Modification, CRC Press, Boca Raton, FL, 3rd edn, 2005 Search PubMed.
- G. T. Hermanson, Bioconjugate Techniques, Academic Press, New York, NY, 2nd edn, 2008 Search PubMed.
- M. Regitz and G. Maas, Diazo Compounds: Properties and Synthesis, Academic Press, New York, NY, 1986 Search PubMed.
- A. Padwa and D. J. Austin, Angew. Chem., Int. Ed., 1994, 33, 1797–1815 CrossRef.
- Y. Zhang and J. B. Wang, Eur. J. Org. Chem., 2011, 1015–1026 CrossRef.
- A. C. Chibnall, J. L. Mangan and M. W. Rees, Biochem. J., 1958, 68, 114–118 CrossRef CAS PubMed.
- M. P. Doyle, Chem. Rev., 1986, 86, 919–939 CrossRef CAS.
- T. Shinada, T. Kawakami, H. Sakai, I. Takada and Y. Ohfune, Tetrahedron Lett., 1998, 39, 3757–3760 CrossRef CAS.
- M. E. Furrow and A. G. Myers, J. Am. Chem. Soc., 2004, 126, 12222–12223 CrossRef CAS PubMed.
- S. Bertelsen, M. Nielsen, S. Bachmann and K. A. Jorgensen, Synthesis-Stuttgart, 2005, 2234–2238 CAS.
- K. De, J. Legros, B. Crousse and D. Bonnet-Delpon, J. Org. Chem., 2009, 74, 6260–6265 CrossRef CAS PubMed.
- L. Dumitrescu, K. Azzouzi-Zriba, D. Bonnet-Delpon and B. Crousse, Org. Lett., 2011, 13, 692–695 CrossRef CAS PubMed.
- A. L. Grossberg and D. Pressman, J. Am. Chem. Soc., 1960, 82, 5478–5482 CrossRef CAS.
- M. S. Doscher and P. E. Wilcox, J. Biol. Chem., 1961, 236, 1328–1337 CAS.
- J. P. Riehm and H. A. Scheraga, Biochemistry, 1965, 4, 772–782 CrossRef CAS PubMed.
- G. R. Delpierre and J. S. Fruton, Proc. Natl. Acad. Sci. U. S. A., 1965, 54, 1161–1167 CrossRef CAS.
- E. L. Myers and R. T. Raines, Angew. Chem., Int. Ed., 2009, 48, 2359–2363 CrossRef CAS PubMed.
- H.-H. Chou and R. T. Raines, J. Am. Chem. Soc., 2013, 135, 14936–14939 CrossRef CAS PubMed.
- J. F. McGarrity and T. Smyth, J. Am. Chem. Soc., 1980, 102, 7303–7308 CrossRef CAS.
- I. Szele, M. Tencer and H. Zollinger, Helv. Chim. Acta, 1983, 66, 1691–1703 CrossRef CAS.
- The 4 to 6 ratio for BocAspOH (g) was 1:1 due in part to the need for two O-alkylation reactions to produce the ester product (4).
- The additional enhancement in selectivity achieved with 3-mercaptopropanoic acid was likely due to coordination between the pendant thiol functionality and the intermediate diazonium ion, hindering external attack from water (J. R. Larson and N. D. Epiotis, J. Am. Chem. Soc., 1981, 103, 410–416).
- R. W. Taft and F. G. Bordwell, Acc. Chem. Res., 1988, 21, 463–469 CrossRef CAS.
- W. N. Olmstead and F. G. Bordwell, J. Am. Chem. Soc., 1980, 45, 3299–3305 CAS.
- M. L. Hlavinka, J. F. Greco and J. R. Hagadorn, Chem. Commun., 2005, 5304–5306 RSC.
- G. de Almeida, E. M. Sletten, H. Nakamura, K. K. Palaniappan and C. R. Bertozzi, Angew. Chem., Int. Ed., 2012, 51, 2443–2447 CrossRef CAS PubMed.
- C. Öġreir and M. Özkütük, Guyana J. Sci., 2012, 25, 343–354 Search PubMed.
- K. Alder, F. Pascher and A. Schmitz, Ber. Dtsch. Chem. Ges., 1943, 76, 27–53 CrossRef.
- S. M. Bachrach and S. L. Jiang, J. Org. Chem., 1997, 62, 8319–8324 CrossRef CAS PubMed.
- O- to S-alkyl transfer as in the high-temperature Newman–Kwart rearrangement of a thionocarbamate or Schönberg rearrangement of a thionocarbonate is unlikely for a thionoester at room temperature.
- P. Zhou, F. F. Tian, F. L. Lv and Z. C. Shang, Proteins, 2009, 76, 151–163 CrossRef CAS PubMed.
- R. T. Raines, Chem. Rev., 1998, 98, 1045–1066 CrossRef CAS PubMed.
- Notably, Glu111 (A. Tarragona-Fiol, H. J. Eggelte, S. Harbron, E. Sanchez, C. J. Taylorson, J. M. Ward and B. R. Rabin, Protein Eng., 1993, 6, 901–906) and Asp121 (L. W. Schultz, D. J. Quirk and R. T. Raines, Biochemistry, 1998, 37, 8886–8898) have been implicated in the catalytic activity of RNase A.
- We detected no evidence (e.g., Δm/z = −18) for the intramolecular attack of a lysine Nε on a nascent ester, consistent with carboxyl groups near lysine and other cationic residues having pKa values that are too low for participation in the mechanism in Fig. 1.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical data, and spectral data for novel compounds. See DOI: 10.1039/c4sc01768d |
| ‡ These authors contributed equally to this work. |
| § Present address for Prof. N. A. McGrath: Department of Chemistry, University of Wisconsin–La Crosse, 1725 State Street, La Crosse, WI 54601 USA. |
| ¶ Present address for Dr Jo E. Lomax: Science Exchange, 555 Bryant Street, #939, Palo Alto, CA 94301 USA. |
| This journal is © The Royal Society of Chemistry 2015 |