
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Non-equilibrium cobalt(III) “click” capsules†
P. R.
Symmers
a,
M. J.
Burke
a,
D. P.
August
a,
P. I. T.
Thomson
a,
G. S.
Nichol
a,
M. R.
Warren
b,
C. J.
Campbell
a and
P. J.
Lusby
*a
aEaStCHEM School of Chemistry, University of Edinburgh, The King's Buildings, David Brewster Road, Edinburgh EH9 3FJ, UK. E-mail: Paul.Lusby@ed.ac.uk
bDiamond Light Source Ltd, Diamond House, Harwell Science and Innovation Campus, Didcot, Oxfordshire OX11 0DE, UK
First published on 7th October 2014
Abstract
Cobalt(III) tetrahedral capsules have been prepared using an assembly-followed-by-oxidation protocol from a cobalt(II) precursor and a readily derivatizable pyridyl-triazole ligand system. Experiments designed to probe the constitutional dynamics show that these architectures are in a non-equilibrium state. A preliminary investigation into the host–guest chemistry of a water-soluble derivative shows it can bind and differentiate a range of different neutral organic molecules. The stability of this ensemble also permits the study of guest-binding at high salt concentrations.
Introduction
The chemistry of molecular container species continues to thrive, not least because of applications from storage, sensing and separation, through drug delivery to catalysis.1 In the last twenty years, self-assembled systems have appeared, a few that rely on exclusively weak non-covalent interactions such as hydrogen bonding2 and many which use metal–ligand interactions.3 Whilst using coordination complexes as structural elements greatly increases the palate of molecular building blocks, the real advantage of these systems is that the reversibility of these interactions facilitates thermodynamic self-assembly, often producing discrete architectures in quantitative yield. However, this same facet can be viewed as a double-edged sword, with the dynamics of these systems providing a hurdle to many potential applications.4 A strategy that has been used to generate inert coordination based systems is to use metal–ligand interactions that are substitutionally non-labile at room temperature and only become dynamic when heated.5 The problem with this method is that (a) longer reaction times and templates are often required,5b leading to lower yields and/or kinetically trapped intermediates6 and (b) it invariably requires the use of more expensive/more toxic third-row transition metals. An alternative way to circumvent these problems is to alter the characteristics of the transition metal center following self-assembly, most obviously through a change in the oxidation state. In this regard, cobalt would appear an ideal choice, because although Co(II) is labile, it can be readily oxidized without a change in the coordination geometry preference to give inert Co(III).7 Herein we report the synthesis of highly cationic Co(III)4L612+ tetrahedral capsules8 using an assembly-followed-by-oxidation protocol. These systems have the characteristics of fully covalent capsules9 in that they appear constitutionally non-dynamic, as evidenced by scrambling experiments. Host–guest studies with a water soluble derivative have revealed that the capsule can bind a range of neutral organic guests, and is further able to differentiate structurally similar molecules. The kinetic inertness of this system has also allowed the study of guest binding at high salt concentrations.Results and discussion
Design strategy and synthesis
The ligand system, L, that we targeted to explore the assembly-followed-by-oxidation protocol is constructed in a modular fashion (see the ESI†), using the popular copper catalyzed azide-alkyne cycloaddition (Cu-AAC) reaction (Scheme 1).10 Our motives for targeting this system were multiple. Firstly, the resultant N,N-donor pyridyl-triazole units are more synthetically accessible than, for example, a classic 2,2′-bipy motif.11 Secondly, this motif facilitates exo-functionalization of the capsule with different chemical groups thus facilitating various applications.12 Thirdly, the ligand itself is constitutionally robust, which is essential for creating non-equilibrium capsules based on substitutionally inert transition metal ions. In this regard, it can be viewed as an alternative approach to the very elegant work to recently come out of Jonathan Nitschke's laboratory.1c,8e,g,h,14b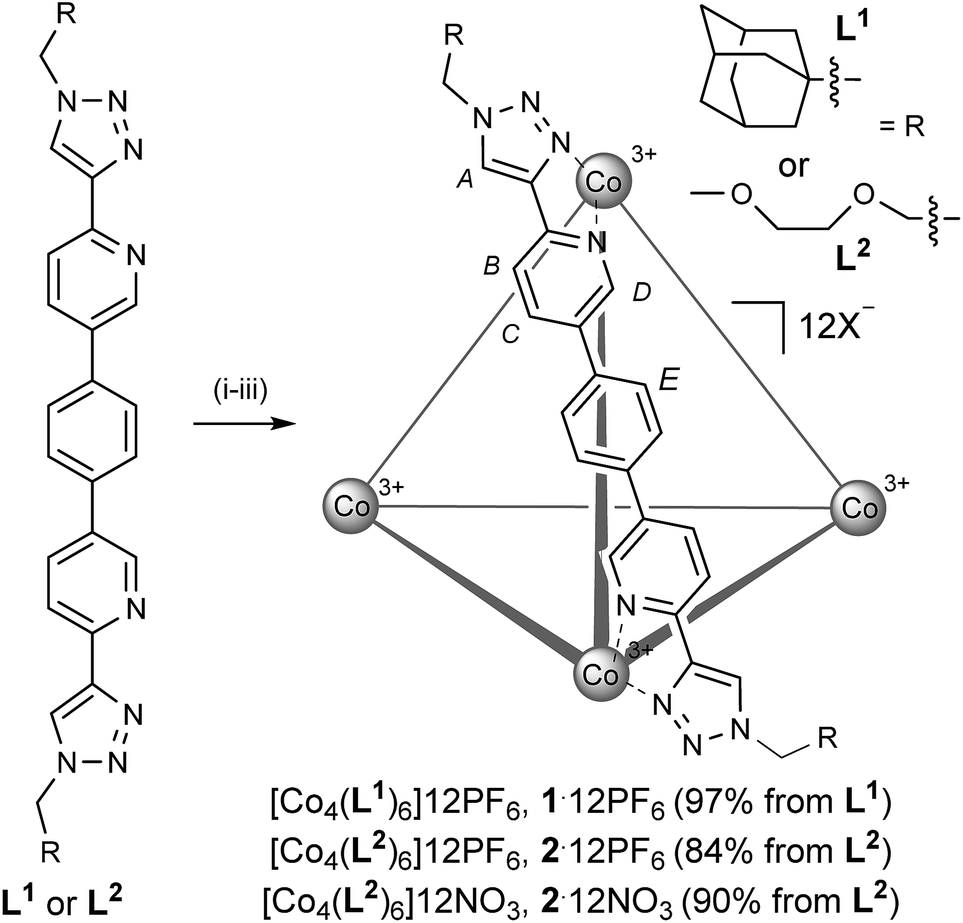 | ||
| Scheme 1 Synthesis of non-equilibrium Co(III) capsules: (i) Co(ClO4)2·6H2O, CH3CN, 50 °C, 17 h; (ii) cerium ammonium nitrate, CH3CN, RT, 2.5 h; (iii) (a) NH4PF6, H2O, CH3CN, RT, 0.5 h or (b) CG-400 resin, H2O, CH3CN, RT, 2.5 h and then AgNO3, H2O, RT, 16 h. | ||
Although L1 showed poor solubility in all solvents, when it was reacted with Co(ClO4)2·6H2O in CH3CN, dissolution occurred over several hours at 323 K (Scheme 1, step (i)). When a small portion of this reaction was analyzed, the broadness and the position of the chemical shifts in the 1H NMR spectrum were strongly indicative of a Co(II) species, while n-ESI-MS (nanoelectrospray mass spectrometry) showed predominant peaks that could be ascribed to [[Co2II(L1)3]nClO4](4−n)+ but, interestingly, no obvious indication of a Co(II)4(L1)6 species. Subsequent slow addition of cerium ammonium nitrate (Scheme 1, step (ii)) resulted in an orange precipitate that was isolated by filtration. This intermediate mixed counteranion species was then treated with NH4PF6 (Scheme 1, step (iii) (a)) to give an orange product in 97% from L1. The 1H NMR spectrum of this revealed the formation of a single, highly symmetric, diamagnetic species, while analysis by n-ESI-MS showed a series of highly charged species that matched the predicted isotopic distribution for [[Co4(L1)6]nPF6](12−n)+ (see the ESI†).
Single crystals of [Co4(L1)6]12PF6, 1·12PF6, suitable for XRD were grown from diisopropyl ether diffusion into saturated acetonitrile solutions. However, these crystals suffered severely from immediate and rapid solvent loss when removed from the mother liquor, such that early attempts to collect data resulted in only poorly resolved structures. Using the combination of capillary mounting in the mother liquor and a synchrotron radiation source (see the ESI†), a fully refined structure was finally obtained, which confirms a homochiral, M4L6 tetrahedral species (Fig. 1).13 Notably, only two PF6− counteranions per asymmetric unit (i.e. per metal ion) could be identified, however, the Co–N bond lengths for the two crystallographically distinct Co environments range from 1.881(8)–2.037(8) Å (see the ESI†), completely consistent with a Co(III) structure (as is all the other characterization data).
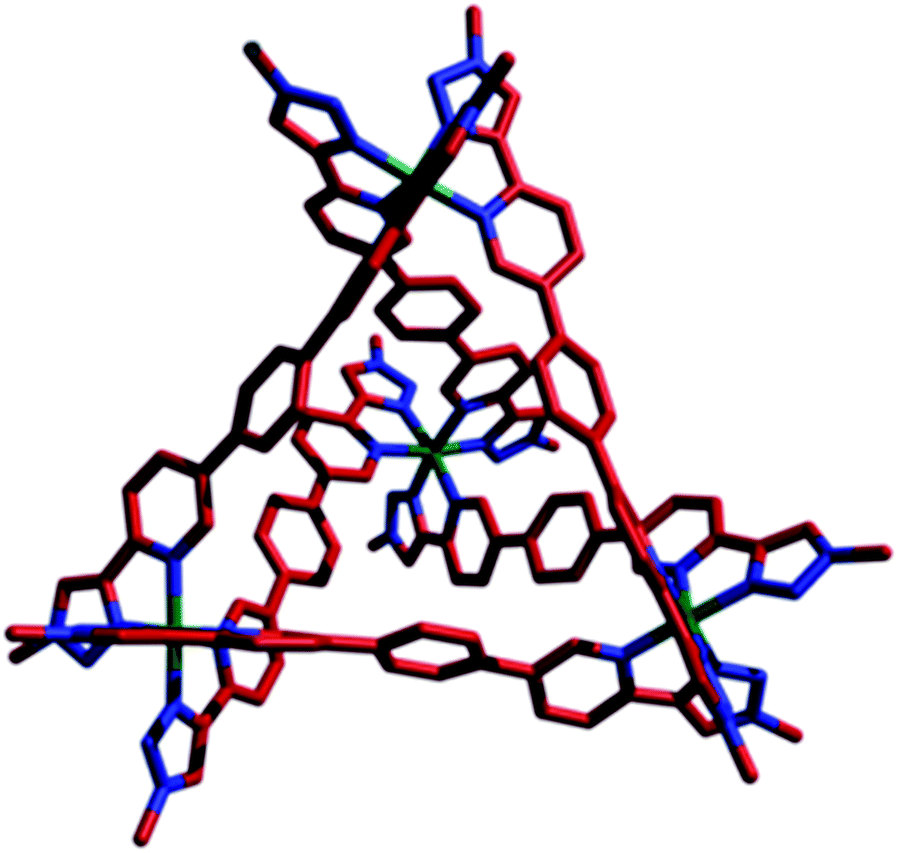 | ||
| Fig. 1 X-ray crystal structure of 1·12PF6. For clarity, PF6 counteranions and the peripheral adamantyl groups have been removed. Color code: Co, green; C, red; N, blue. | ||
2·12PF6 has also been accessed using the same assembly-followed-by-oxidation and anion metathesis conditions (see the ESI†). Most probably because of the conformational flexibility of the peripheral PEG groups, we have been unable to obtain XRD-quality crystals for this compound. However, a comparison of the 1H NMR DOSY spectra of 1·12PF6 and 2·12PF6 showed very similar diffusion coefficients under the same conditions (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D = −9.33 and −9.34 m2 s−1, respectively), thus indicating that the assembly-followed-by-oxidation protocol with L2 also gives an M4L6 species. A preliminary electrochemical investigation has also be carried out using 2·12PF6 in CH3CN (see the ESI†), which shows a reversible reduction at −791 mV (vs. SCE). This single chemically-reversible reduction, which we attribute to the Co(III)/Co(II) couple, shows that the metal centers behave independently, and is fully chemically-reversible irrespective of scan rate, down to 50 mV s−1. This would indicate that the tetrahedral capsule 28+ appears stable and does not undergo rearrangement (as perhaps could be expected with coordinatively flexile, high spin d7 metal vertices14). Electrochemical experiments also show an irreversible reduction in the region of −1300–1500 mV. These have previously been observed for pyridyl-triazole complexes, and are a result of ligand-based reduction.15
D = −9.33 and −9.34 m2 s−1, respectively), thus indicating that the assembly-followed-by-oxidation protocol with L2 also gives an M4L6 species. A preliminary electrochemical investigation has also be carried out using 2·12PF6 in CH3CN (see the ESI†), which shows a reversible reduction at −791 mV (vs. SCE). This single chemically-reversible reduction, which we attribute to the Co(III)/Co(II) couple, shows that the metal centers behave independently, and is fully chemically-reversible irrespective of scan rate, down to 50 mV s−1. This would indicate that the tetrahedral capsule 28+ appears stable and does not undergo rearrangement (as perhaps could be expected with coordinatively flexile, high spin d7 metal vertices14). Electrochemical experiments also show an irreversible reduction in the region of −1300–1500 mV. These have previously been observed for pyridyl-triazole complexes, and are a result of ligand-based reduction.15
In order to assess whether these assemblies are in a non-equilibrium state, we have combined equimolar quantities of 1·12PF6 and 2·12PF6 in CD3CN and monitored this mixed solution as a function of time using both 1H NMR spectroscopy and n-ESI-MS. Similar experiments have previously been used to demonstrate that metallosupramolecular species are constitutionally dynamic; even for systems which exhibit pronounced kinetic stability, brought about by the cooperative effects of multiple metal–ligand interactions, entropy-driven scrambling of components still happens at room temperature over a few days.16 In contrast, we observe no ligand exchange after a week at room temperature. Only through prolonged heating of the same sample, first at 50 °C (1 week), then at 60 °C (1 week) and then finally at 70 °C, could any mixed component species be identified, but even then the 1H NMR spectrum remained largely unchanged and only minor peaks were observed by MS (see the ESI†). This indicates that these Co(III) tetrahedra are constitutionally non-dynamic.
Host–guest chemistry
Water-soluble systems have featured prominently as solution container compounds,1b,c,d,f,g,5b,17 principally because the hydrophobic effect is a powerful driving force for the encapsulation of a wide range of molecules. For charged metallosupramolecular capsules, dissolution in water or other polar media also results in solvation of the associated counteranions (or countercations in the case of Raymond's anionic Ga(III) tetrahedra1c,8b,c), which can occupy the cavity8d,f,g,14b,18 and block different guests from binding. While 2·12PF6 is insoluble in water, we were encouraged that the intermediate 212+ with mixed ClO4–NO3 counteranions (i.e. the species obtained directly from step (ii)) is soluble in 1:1 CD3CN:D2O. To further increase aqueous solubility, this species was first treated with CG-400 resin and then with AgNO3 to give 2·12NO3 (Scheme 1, step (iii) (b)). All the spectroscopic evidence (MS, 1H NMR, DOSY, see the ESI†) indicates that anion exchange takes place without perturbation to the tetrahedral framework, and furthermore, the resulting compound is soluble in water at 2.5 mM. It is interesting to note that the use of nitrate counteranions to water-solubilize coordination capsules has largely been limited to those systems which possess 2nd and 3rd row transition metals (most commonly Pd and Pt), probably a reflection of the softer bonding characteristics in comparison to the 1st row elements, which (in addition to nitrate-hydration) ensures outer-sphere coordination is thermodynamically preferred. Despite the oxophilic nature of Co(III), 2·12NO3 appears indefinitely stable as a 2.5 mM solution in D2O, further highlighting that these species exist in an out-of-equilibrium state.19
To predict the size of guest molecules that 212+could bind, calculations were carried out using the atomic coordinates from the X-ray structure of 112+,20 which revealed the volume of the empty cavity is 358 Å3 (see the ESI†). Application of the guidelines for suitable guests laid down by Rebek21 would indicate that molecules with volumes of 164–229 Å3 should likely be ideal. However, an initial exploration of hydrocarbons close to this size range (2-methylnaphthalene, 168 Å3; biphenyl, 183 Å3; fluorene, 189 Å3; phenanthrene, 201 Å3; anthracene, 201 Å3; pyrene, 220 Å3, n-dodecane, 235 Å3) showed no evidence for encapsulation. Instead, when excess triisopropylsilyl alcohol (TIPSOH) was added to a sample of 2·12NO3, 1H NMR spectroscopy revealed the appearance of a new set of capsule signals (Fig. 2b) alongside those of free 212+ (Fig. 2a). In addition, a set of upfield-shifted signals relative to free TIPSOH with equimolar intensity relative to the new capsule resonances, strongly suggest that one silyl guest is encapsulated within 212+, and that exchange in and out of the cavity is slow on the NMR timescale. Further evidence for this encapsulation is provided by 1H NMR DOSY, which shows that the encapsulated TIPSOH species diffuses at the same rate as both the free and bound cage. Based on the molar ratios at equilibrium, the Ka of TIPSOH for 212+ has been calculated to be ca. 1400 M−1, while EXSY gives the activation barrier for exchange of this guest as 17.3 kcal mol−1 (see the ESI†). Interestingly, the volume of TIPSOH (220 Å3) is quite a lot larger than 55% of the empty cavity, however, this could quite easily be a result of the relatively large portals into which the guest can protrude.
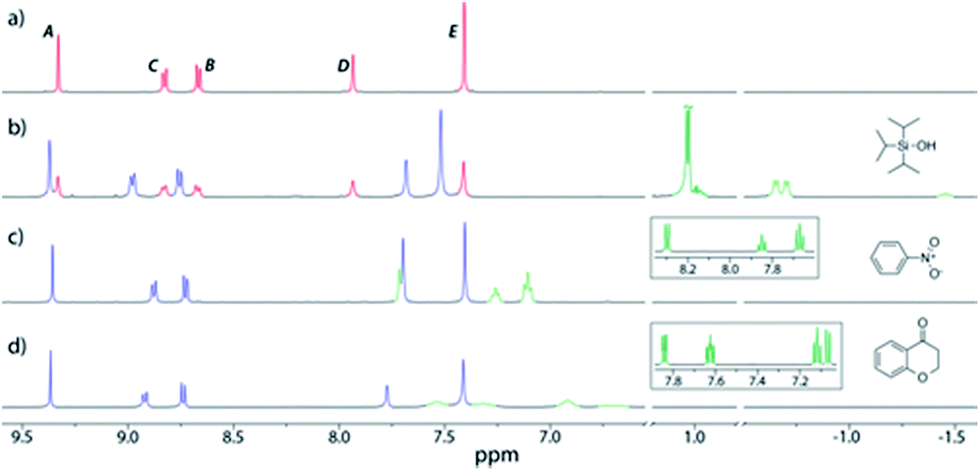 | ||
| Fig. 2 Partial 1H NMR spectra (500 MHz, D2O, 298 K) of (a) 2·12NO3; (b) 2·12NO3 with excess TIPSOH; (c) 2·12NO3 with excess nitrobenzene; (d) 2·12NO3 with excess chromanone. Color code: free capsule signals, red; bound capsule, blue; bound guest, pale green; free guest, dark green. The insets in spectra (c) and (d) show the partial 1H NMR spectra of free guests in D2O. The assignments correspond to the lettering in Scheme 1. | ||
The effect of ionic strength on hydrophobically-driven guest encapsulation within synthetic host systems has been rarely studied,22 with respect to metallosupramolecular systems, this may be a result of instability towards high concentrations of salt. However, 2·12NO3 is stable even in solutions of 5 M NaNO3 in D2O. Furthermore, an increasing affinity of the guest in 0.1 M, 1 M and 5 M NaNO3 D2O solutions is clearly observed through the decrease in % unbound species, such that at the highest salt concentration, free 212+ is beyond the spectroscopic detection limit (see the ESI†). Through dilution experiments, it has been calculated that the affinity of the TIPSOH guest increases nearly four-fold in 5 M NaNO3 solution to 4700 M−1.
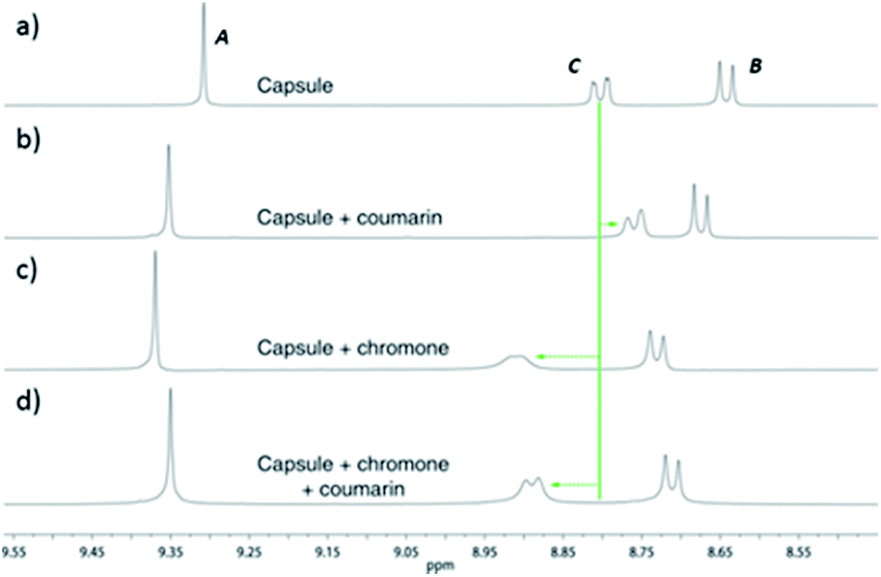
In addition to TIPSOH, we have also found that a range of other organic molecules act as guests for 212+ (Fig. 3). In contrast, these exhibit exchange fast on the NMR timescale, such that a single set of resonances are observed for both guest and host, for example, Fig. 2c and d, shows the 1H NMR spectra of 2·12NO3 in the presence of excess nitrobenzene and chromanone. In these examples, the guest's signals are significantly upfield shifted with respect to the free species in the same solvent, consistent with being encapsulated and experiencing (time-averaged) shielding effects from the capsules' aromatic struts. Furthermore, for the majority of these guests, the direction in which the capsules' signals HA–E shift are consistent, also similar to what is observed for TIPSOH encapsulation, thus indicating that guests bind in a conserved fashion within 212+ (or otherwise cause a similar binding-induced re-organization). Interestingly, the molecules that act as guests could collectively be described as weakly amphiphilic. These general observations points to a mode of binding in which a specific guest functional group–cage interaction(s) is(are) supplemented by the hydrophobic effect.17d A preliminary investigation into the relative affinities of some of the guests shown in Fig. 3 reveal that the regioisomeric compounds coumarin and chromone possess binding constants with a ten-fold difference, 120 M−1 and 1200 M−1, respectively (see the ESI†). This data is also supported by a competition binding experiment involving these two guests. Whereas coumarin-only binding causes an upfield shift in the HC environment (Fig. 4b) with respect to free 212+ (Fig. 4a), chromone encapsulation causes the same signal to become deshielded (Fig. 4c). In the presence of a 1:1 mixture of both analytes (Fig. 4d), this same signal is similarly deshielded, indicating the capsule is able to preferentially bind chromone in the presence of coumarin, showing that the capsule can differentiate molecules based on shape or the relative positioning of functional groups and not solely on the basis of more bulk descriptors.
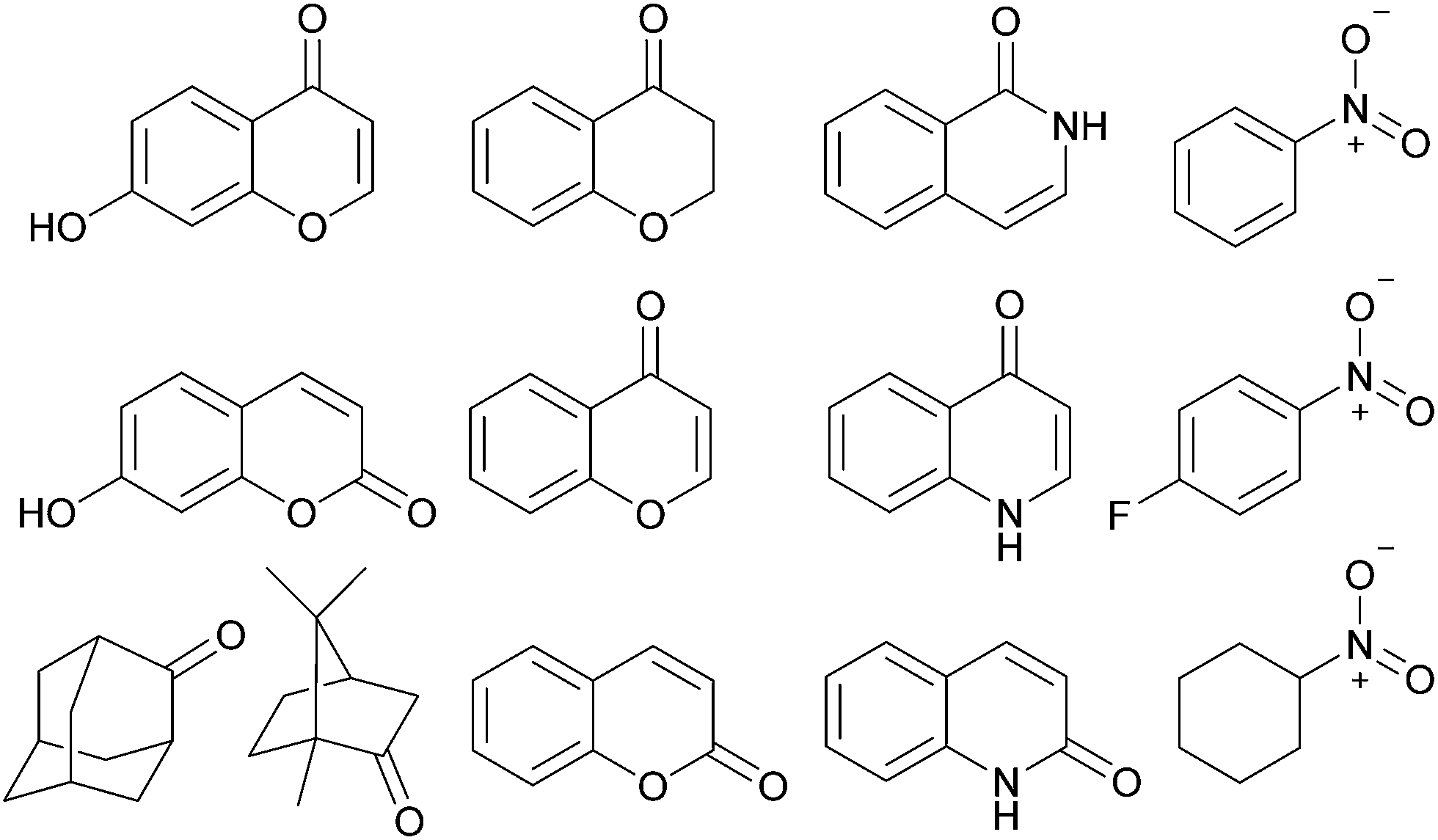 | ||
| Fig. 3 Guest molecules for 212+ that exhibit fast exchange on the NMR timescale. | ||
 | ||
| Fig. 4
1H NMR spectra (500 MHz, D2O, 298 K) showing the preferential binding of chromone in the presence of coumarin. (a) 2·12NO3 only; (b) 2·12NO3 in the presence of coumarin; (c) 2·12NO3 in the presence of chromone; (d) 2·12NO3 in the presence of 1:1 coumarin and chromone. | ||
Conclusions
Coordination capsules almost always provide an opportunity to explore chemical equilibria, both at the level of the architecture self-assembly process and also due to their reversible interactions with guest molecules. Here we have reported a rare example of a coordination capsule which is not in equilibrium with its disassembled state. Similarly rare are coordination capsules which exhibit non-equilibrium guest binding properties.23 The development of metal-based (and fully organic) assemblies that are both constitutionally non-dynamic and also possess non-reversible guest binding properties,24 coupled with stimuli-responsive release mechanisms, could lead to improved function for a range of applications. As is the case in the field of synthetic molecular machines,25 we envisage that systems able to operate far away from equilibrium will be able to perform tasks not currently possible for their thermodynamic equivalents.Acknowledgements
This work was supported by the EPSRC and the Royal Society. P.J.L. is a Royal Society University Research Fellow.Notes and references
- For recent examples, see: (a) T. Mitra, K. E. Jelfs, M. Schmidtmann, A. Ahmed, S. Y. Chong, D. J. Adams and A. I. Cooper, Nat. Chem., 2013, 5, 276 CrossRef CAS PubMed; (b) F. Schmitt, J. Freudenreich, N. P. E. Barry, L. Juillerat-Jeanneret, G. Süss-Fink and B. Therrien, J. Am. Chem. Soc., 2012, 134, 754 CrossRef CAS PubMed; (c) P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697 CrossRef CAS PubMed; (d) N. Saleh, A. L. Koner and W. M. Nau, Angew. Chem., Int. Ed., 2008, 47, 5398 CrossRef CAS PubMed; (e) M. Dionisio, J. M. Schnorr, V. K. Michaelis, R. G. Griffin, T. M. Swager and E. Dalcanale, J. Am. Chem. Soc., 2012, 134, 6540 CrossRef CAS PubMed; (f) C. Zhao, Q.-F. Sun, W. M. Hart-Cooper, A. G. DiPasquale, F. D. Toste, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2013, 135, 18802 CrossRef CAS PubMed; (g) T. Murase, Y. Nishijima and M. Fujita, J. Am. Chem. Soc., 2012, 134, 162 CrossRef CAS PubMed; (h) T. V. Nguyen, H. Yoshida and M. S. Sherburn, Chem. Commun., 2010, 46, 5921 RSC.
- (a) R. Wyler, J. de Mendoza and J. Rebek, Angew. Chem., Int. Ed. Engl., 1993, 32, 1699 CrossRef; (b) L. R. MacGillivray and J. L. Atwood, Nature, 1997, 389, 469 CrossRef CAS.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810 CrossRef CAS PubMed.
- B. Kang, J. W. Kurutz, K.-T. Youm, R. K. Totten, J. T. Hupp and S. T. Nguyen, Chem. Sci., 2012, 3, 1938 RSC.
- (a) M. Fujita, F. Ibukuro, K. Yamaguchi and K. Ogura, J. Am. Chem. Soc., 1995, 117, 4175 CrossRef CAS; (b) F. Ibukuro, T. Kusukawa and M. Fujita, J. Am. Chem. Soc., 1998, 120, 8561 CrossRef CAS . Light and solvent effects have also been used to labilize normally inert Pt–N interactions, see:; (c) K. Yamashita, M. Kawano and M. Fujita, J. Am. Chem. Soc., 2007, 129, 1850 CrossRef CAS PubMed; (d) D. Fujita, A. Takahashi, S. Sato and M. Fujita, J. Am. Chem. Soc., 2011, 133, 13317 CrossRef CAS PubMed . For a review on chemical locking in self-assembly, see:; (e) J. A. Thomas, Chem. Soc. Rev., 2007, 36, 856 RSC.
- (a) S. Tashiro, M. Tominaga, T. Kusukawa, M. Kawano, S. Sakamoto, K. Yamaguchi and M. Fujita, Angew. Chem., Int. Ed., 2003, 42, 3267 CrossRef CAS PubMed; (b) C. R. K. Glasson, G. V. Meehan, J. K. Clegg, L. F. Lindoy, J. A. Smith, F. R. Keene and C. Motti, Chem. Eur. J., 2008, 14, 10535 CrossRef CAS PubMed.
- (a) L. J. Charbonniere, G. Bernardinelli, C. Piguet, A. M. Sargeson and A. F. Williams, J. Chem. Soc., Chem. Commun., 1994, 1419 RSC; (b) E. C. Constable, K. Harris, C. E. Housecroft and M. Neuburger, Dalton Trans., 2011, 40, 1524 RSC; (c) J.-F. Ayme, J. Lux, J.-P. Sauvage and A. Sour, Chem. Eur. J., 2012, 18, 5565 CrossRef CAS PubMed; (d) D. A. Leigh, P. J. Lusby, A. M. Z. Slawin and D. B. Walker, Chem. Commun., 2012, 48, 5826 RSC.
- For examples of M4L6 tetrahedral capsules, see: (a) R. W. Saalfrank, A. Stark, K. Peters and H. G. von Schnering, Angew. Chem., Int. Ed. Engl., 1988, 27, 851 CrossRef; (b) T. Beissel, R. E. Powers and K. N. Raymond, Angew. Chem., Int. Ed. Engl., 1996, 35, 1084 CrossRef CAS; (c) D. L. Caulder, R. E. Powers, T. N. Parac and K. N. Raymond, Angew. Chem., Int. Ed., 1998, 37, 1840 CrossRef CAS; (d) J. S. Fleming, K. L. V. Mann, C.-A. Carraz, E. Psillakis, J. C. Jeffery, J. A. McCleverty and M. D. Ward, Angew. Chem., Int. Ed., 1998, 37, 1279 CrossRef CAS; (e) P. Mal, D. Schultz, K. Beyeh, K. Rissanen and J. R. Nitschke, Angew. Chem., Int. Ed., 2008, 47, 8297 CrossRef CAS PubMed; (f) C. R. K. Glasson, J. K. Clegg, J. C. McMurtrie, G. V. Meehan, L. F. Lindoy, C. A. Motti, B. Moubaraki, K. S. Murray and J. D. Cashion, Chem. Sci., 2011, 2, 540 RSC; (g) Y. R. Hristova, M. M. J. Smulders, J. K. Clegg, B. Breiner and J. R. Nitschke, Chem. Sci., 2011, 2, 638 RSC; (h) J. L. Bolliger, A. M. Belenguer and J. R. Nitschke, Angew. Chem., Int. Ed., 2013, 52, 7958 CrossRef CAS PubMed.
- Excluding those that possess dynamic covalent bonds.
- (a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS; (b) C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef.
- (a) Y. Li, J. C. Huffman and A. H. Flood, Chem. Commun., 2007, 2692 RSC; (b) J. D. Crowley and P. H. Bandeen, Dalton Trans., 2010, 39, 612 RSC; (c) S. K. Vellas, J. E. M. Lewis, M. Shankar, A. Sagatova, J. D. A. Tyndall, B. C. Monk, C. M. Fitchett, L. R. Hanton and J. D. Crowley, Molecules, 2013, 18, 6383 CrossRef CAS PubMed.
- J. E. M. Lewis, A. B. S. Elliott, C. J. McAdam, K. C. Gordon and J. D. Crowley, Chem. Sci., 2014, 5, 1833 RSC.
- ESI.†.
- (a) I. S. Tidmarsh, T. B. Faust, H. Adams, L. P. Harding, L. Russo, W. Clegg and M. D. Ward, J. Am. Chem. Soc., 2008, 130, 15167 CrossRef CAS PubMed; (b) I. A. Riddell, M. M. J. Smulders, J. K. Clegg, Y. R. Hristova, B. Breiner, J. D. Thoburn and J. R. Nitschke, Nat. Chem., 2012, 4, 751 CrossRef CAS PubMed.
- T. Y. Kim, A. B. S. Elliott, K. J. Shaffer, C. J. McAdam, K. C. Gordon and J. D. Crowley, Polyhedron, 2013, 52, 1391 CrossRef CAS.
- S. Sato, Y. Ishido and M. Fujita, J. Am. Chem. Soc., 2009, 131, 6064 CrossRef CAS PubMed.
- (a) F. Cramer, W. Saenger and H.-C. Spatz, J. Am. Chem. Soc., 1967, 89, 14 CrossRef CAS; (b) P. V. Demarco and A. L. Thakkar, J. Chem. Soc. D, Chem. Commun., 1970, 2 CAS; (c) F. Diederich and K. Dick, J. Am. Chem. Soc., 1984, 106, 8024 CrossRef CAS; (d) M. Whitehead, S. Turega, A. Stephenson, C. A. Hunter and M. D. Ward, Chem. Sci., 2013, 4, 2744 RSC; (e) K.-D. Zhang, D. Ajami and J. Rebek, J. Am. Chem. Soc., 2013, 135, 18064 CrossRef CAS PubMed.
- (a) G. H. Clever, S. Tashiro and M. Shionoya, Angew. Chem., Int. Ed., 2009, 48, 7010 CrossRef CAS PubMed; (b) O. Chepelin, J. Ujma, X. Wu, A. M. Z. Slawin, M. B. Pitak, S. J. Coles, J. Michel, A. C. Jones, P. E. Barran and P. J. Lusby, J. Am. Chem. Soc., 2012, 134, 19334 CrossRef CAS PubMed; (c) S. Freye, R. Michel, D. Stalke, M. Pawliczek, H. Frauendorf and G. H. Clever, J. Am. Chem. Soc., 2013, 135, 8476 CrossRef CAS PubMed.
- Were 2·12NO3 constitutionally dynamic, the affinity of nitrate anions for Co(III) and the low-aqueous solubility of L2 would drive the equilibrium towards the disassembled state.
- Making the assumption that the peripheral groups do not substantially alter the size of the cavity.
- S. Mecozzi and J. Rebek, Chem. Eur. J., 1998, 4, 1016 CrossRef CAS.
- C. L. D. Gibb and B. C. Gibb, J. Am. Chem. Soc., 2011, 133, 7344 CrossRef CAS PubMed.
- N. Takeda, K. Umemoto, K. Yamaguchi and M. Fujita, Nature, 1999, 398, 794 CrossRef CAS.
- For a (non-self-assembled) constitutionally fixed organic capsule that exhibits non-equilibrium guest binding, see: D. J. Cram, M. E. Tanner and R. Thomas, Angew. Chem., Int. Ed. Engl., 1991, 30, 1024 CrossRef.
- (a) B. L. Feringa, Acc. Chem. Res., 2001, 34, 504 CrossRef CAS PubMed; (b) V. Serreli, C.-F. Lee, E. R. Kay and D. A. Leigh, Nature, 2007, 445, 523 CrossRef CAS PubMed; (c) H. Li, C. Cheng, P. R. McGonigal, A. C. Fahrenbach, M. Frasconi, W.-G. Liu, Z. Zhu, Y. Zhao, C. Ke, J. Lei, R. M. Young, S. M. Dyar, D. T. Co, Y.-W. Yang, Y. Y. Botros, W. A. Goddard, M. R. Wasielewski, R. D. Astumian and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135, 18609 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, X-ray crystallography results, CV, volume calculations, scrambling and guest-binding experiments. CCDC 1014311. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc03036b |
| This journal is © The Royal Society of Chemistry 2015 |