
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery of potent inhibitors of human β-tryptase from pre-equilibrated dynamic combinatorial libraries†
Qian-Qian
Jiang
,
Wilhelm
Sicking
,
Martin
Ehlers
and
Carsten
Schmuck
*
Institut für Organische Chemie, Universität Duisburg-Essen, Universitätsstraße 7, 45141 Essen, Germany. E-mail: carsten.schmuck@uni-due.de
First published on 8th December 2014
Abstract
Pre-equilibrated dynamic combinatorial libraries based on acyl hydrazone interchange of peptide-derived hydrazides and di- and tri-aldehydes have been used to discover potent inhibitors with nanomolar affinities for β-tryptase. To identify potent inhibitors the activity of the full library containing 95 members was compared with those of sub-libraries in which individual building blocks were missing. The most active library members contain a rigid central aromatic scaffold with three cationic peptide arms. The arms of the best inhibitors also contained a tailor-made GCP oxoanion binding motif attached to a lysine side chain. The most potent tri-armed hydrazones with peptide arms GKWR or GKWK(GCP) were shown to inhibit β-tryptase (Kica. 10–20 nM) reversibly, non-competitively and selectively (compared to related serine proteases, e.g. trypsin and chymotrypsin), most likely by binding to the protein surface, also in agreement with molecular modelling calculations. These new inhibitors are one order of magnitude more efficient than related tetravalent inhibitors obtained from previous work on a split-mix-combinatorial library and were identified with significantly less effort, demonstrating the usefulness of this approach for the identification of enzyme inhibitors in general.
Introduction
Dynamic combinatorial chemistry (DCC) uses reversible covalent or non-covalent interactions between a set of molecular building blocks to generate a mixture of all possible combinations of the available components under thermodynamic control.1 By adding a target to such a dynamic combinatorial library (DCL), the composition of the DCL will adjust to minimize the free energy of the overall system, resulting in the amplification of efficient target binders. Since the introduction of this concept in the mid-1990s,2 a number of reversible covalent reactions have been developed for the preparation of DCLs. These include the reversible formation of disulfides, imines and hydrazones.3 Besides its application in supramolecular chemistry and metal complex formation,4 DCC has also been applied successfully for the identification of ligands and inhibitors for enzymes and other biomolecules.5,6 For example, Otto and Sanders have employed disulfide exchange for a series of structurally diverse dithiol building blocks to generate diverse DCLs of the corresponding macrocycles for the identification of receptors for different supramolecular guests.4a,b Using hydrazone-based DCLs, Sanders et al. have identified linear receptors for dihydrogen phosphate ions4c and macrocyclic receptors for heavy metal ions.4d Although a powerful and promising concept, DCC nevertheless still has its limitations, as not every reaction is compatible with the physiological conditions dictated by a bio-molecular target. So far, disulfide5 and imine exchanges6 have been used successfully for DCC in biological systems. As imines are inherently unstable in aqueous media, they are often reduced to amines after the amplification process prior to analysis in order to prevent screening and isolation problems. In contrast to imines, hydrazones are stable, allowing for their isolation and analysis in aqueous solutions of around neutral pH, however, their formation requires acidic conditions (optimal rate at approximately pH 4.5 (ref. 8a) or below), which are not compatible with most biological targets. Therefore, Lehn et al.7 introduced pre-equilibrated DCLs based on reversible hydrazone formation in order to identify efficient binders for a given biomolecule using a deconvolution strategy. Firstly, equilibration of the library is carried out in the absence of the biological target. Secondly, the conditions are adjusted to the correct physiological conditions required by the biological assay, thus freezing-out the library composition. Thirdly, the best binder can then be identified by comparing the screening results for the full library with those of different sub-libraries, in which a different building block is missing in each one.8 When a building block, which is part of an active library member, is missing, the corresponding sub-library will be less active than the full library. By using this deconvolution strategy, Lehn et al. have successfully established DCLs for targeting proteins such as acetylcholinesterase,7a HPr kinase/phosphatase7b and the plant lectin concanavalin A (Con A).7cIn this article we describe the application of pre-equilibrated DCLs for the identification of inhibitors that bind with low nanomolar affinities to β-tryptase. As our data show, these ligands bind reversibly and in a non-competitive manner to the protein surface. β-Tryptase, which is the predominant serine protease in human mast cells, is responsible for several allergic and inflammatory disorders.9 It consists of four identical monomers stabilized by heparin.10 A large variety of β-tryptase inhibitors have been reported in the literature. These range from biomacromolecules such as LDTI (leech-derived tryptase inhibitor),11 lactoferrin,12 myeloperoxidase (MPO),13 TDPI (tick-derived protease inhibitor)14 and MCoTI-II (cyclic cysteine knotted miniprotein) derived inhibitors15 to small synthetic inhibitors.16,17 The synthetic inhibitors reported so far, however, are classical competitive inhibitors which bind to one or two of the four active sites of the enzyme at the same time. We have recently reported18a a new strategy to inhibit β-tryptase by protein surface binders identified from the screening of a focused combinatorial split-mix library of 216 tetravalent peptide ligands derived from a dendritic lysine trimer with four tetrapeptide arms attached. The most efficient ligands were found to bind with a low micromolar to high nanomolar affinity to the central pore around the entrance to the active sites of the enzyme, thus preventing access of the substrate to the active sites. The enzyme was thus reversibly and non-competitively inhibited. However, the synthesis and screening of such a focused, solid phase bound combinatorial library still requires significant effort. We now demonstrate that significantly more potent inhibitors can be found (with even less effort) through the screening and deconvolution of pre-equilibrated DCLs based on reversible hydrazone formation between tetrapeptide hydrazides and di-/tri-aldehydes.
Results and discussion
Design and synthesis of the libraries
Five peptide-derived hydrazides (A–E) and five different commercially available di- and tri-aldehydes (1–5) were chosen as building blocks to generate the library (Scheme 1). The peptide sequences of hydrazides A (GKWR) and B (GKWK) were based on the results of our earlier work,18 which showed that a combination of cationic and aromatic amino acids in the arms provided the most efficient inhibitors of β-tryptase. Hydrazides C and D were derived from B by attaching an artificial arginine analogue to one of the two lysine side chains. This guanidiniocarbonyl pyrrole moiety (GCP) was developed in our group as an efficient binding site for oxoanions19 and has already been successfully used as a building block for artificial receptors for oligopeptides,20 oligonucleotides and nucleic acids.21 Just recently we also found that GCP-modified peptides can be highly efficient gene carriers for artificial cell transfection.22 Hydrazide E (EFAG) was chosen as a negative control, as the negatively charged peptide should be unfavorable for the inhibition of β-tryptase.18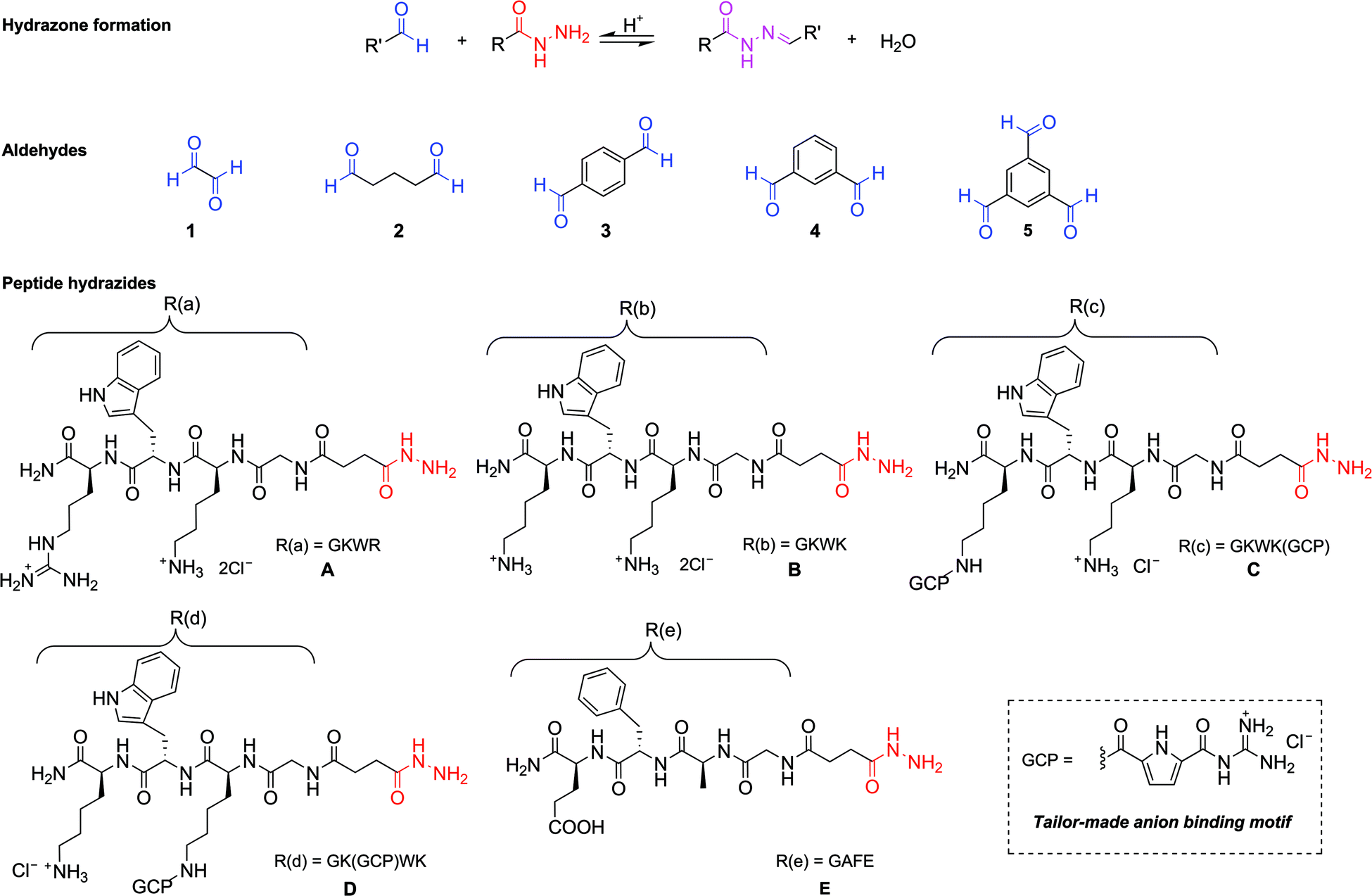 | ||
| Scheme 1 Molecular structures of the aldehydes (1–5) and peptide-derived hydrazides (A–E, including the short names of the peptide arms) selected as the building blocks for the hydrazone-based dynamic combinatorial libraries. The five quasi-peptide arms are abbreviated as R with related compound labels R(a)–R(e). | ||
All hydrazides were synthesized by microwave assisted solid phase peptide synthesis (SPPS) on a Fmoc Rink amide resin (Scheme 2, using hydrazide C as an example). Fmoc deprotection was performed by treatment with 20% piperidine/DMF for 1.5 min at rt and 5 min under microwave irradiation (20 W, Tmax = 60 °C). Coupling steps were carried out under an argon atmosphere with PyBOP and DIPEA in DMF under microwave irradiation (20 W, Tmax = 60 °C) for 20 min.
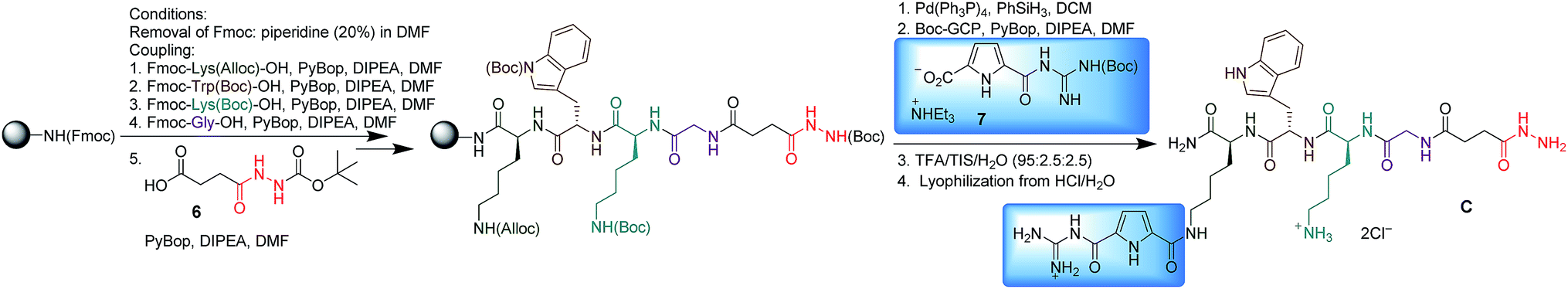 | ||
| Scheme 2 Microwave assisted solid phase peptide synthesis of hydrazide C. | ||
Completion of each coupling and Fmoc deprotection was monitored by a Kaiser test. Following this procedure, after Fmoc deprotection, the first amino acid, Fmoc-Lys(Alloc)-OH, was attached to the resin, followed by Fmoc-Trp(Boc)-OH, Fmoc-Lys(Boc)-OH, Fmoc-Gly-OH and Boc-butanedionic monohydrazide 6. Then the Alloc group was removed with Pd(PPh3)4 in the presence of PhSiH3 in DCM for 10 min at 20 W and a maximum temperature of 30 °C. Finally, coupling of the Boc-protected GCP moiety (7) was performed using the same protocol as for the previous peptide coupling steps. The product was cleaved from the resin using a mixture of TFA/H2O/triisopropylsilane (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5:2.5) and purified by RP18-MPLC. The trifluoroacetate counterions were replaced with chlorides by lyophilization to give C as the hydrochloride salt. Hydrazides A, B, D and E were synthesized accordingly (for details, see ESI†).
:2.5:2.5) and purified by RP18-MPLC. The trifluoroacetate counterions were replaced with chlorides by lyophilization to give C as the hydrochloride salt. Hydrazides A, B, D and E were synthesized accordingly (for details, see ESI†).
With the individual building blocks in hand, the dynamic combinatorial library was generated by simply mixing the hydrazides with the aldehydes in an acetate buffer at pH 4.0 containing 10% DMSO (to dissolve the aromatic aldehydes). Besides the full-library (containing all ten building blocks), ten sub-libraries, in which a different building block was missing in each, were also generated that way. The final concentration of the libraries amounted to 0.5 mM in total for both the hydrazide and aldehyde groups. Together with a reference sample, consisting of an acetate buffer containing 10% DMSO and none of the building blocks, this series of 12 samples was sufficient to screen all of the library members. The different libraries were equilibrated at pH = 4 at ambient temperature for three days.7 The formation of the corresponding hydrazones was confirmed by analytical HPLC and mass spectrometry (for details, see ESI†).
The theoretical maximum number of hydrazones within the library, N, can be calculated using the following equation:7
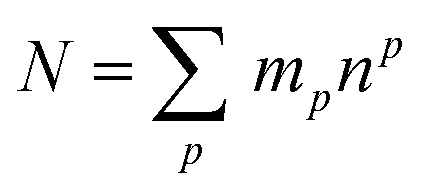
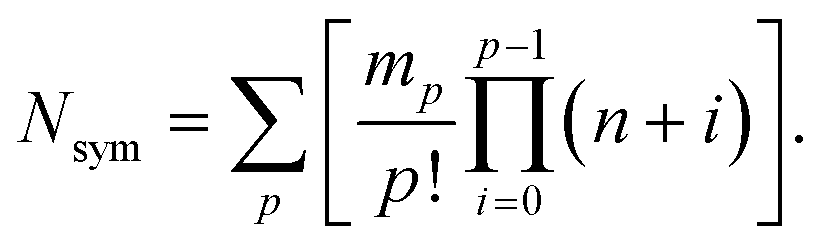
Thus, the full library contains a maximum of 95 different members. As for the sub-libraries, the library size is reduced accordingly depending on the functionality degree of the building block which is removed. For example, the number of compounds is reduced to 60 upon removal of a hydrazide, whereas it is reduced to 80 or 60 upon removal of a di-aldehyde or tri-aldehyde, respectively. However, we cannot conclude that the library used here does indeed contain all of these compounds and even if it does, we do not know the relative amounts in which they are present within the mixture. If the actual composition of the library deviates from the theoretical one, it is of course possible that the inhibitors identified from the analysis of the pre-equilibrated library do not include the best possible inhibitor that could be formed from the building blocks used. This limitation always applies to the use of pre-equilibrated libraries and should accordingly be kept in mind.
Enzyme screening of the DCLs
After equilibration, the resulting mixtures of acyl hydrazones were tested for their inhibition of β-tryptase. By adjusting the pH to 7.4, any further changes in the composition of the library were prevented. The enzyme inhibition experiments were conducted using heparin-stabilized human rhSkin β-tryptase in a high throughput assay performed in Tris-HCl buffer (50 mM, pH 7.4) according to established procedures.18 The enzyme activity was screened in the presence of the libraries by using chromogenic substrate Toc-Gly-Pro-Arg-AMC, which is cleaved by the enzyme and thus leads to the release of free AMC. This results in a strong increase in fluorescence emission at 460 nm (380 nm excitation) and allows for monitoring of the rate of the enzymatic hydrolysis.Firstly, the IC50 values of the ten individual building blocks (1–5 and A–E) were measured to see whether they also inhibit β-tryptase. As shown in Table 1, the four cationic peptide-derived hydrazides A–D inhibit β-tryptase, but only in a micromolar range. Hydrazide A containing the peptide sequence GKWR showed the most efficient inhibition of the enzyme with an IC50-value of 13 μM. The two peptide-derived hydrazides C and D containing the artificial GCP group have IC50 values in the same range (ca. 24 μM), whereas hydrazide B, which consists of the same peptide sequence as C or D but with no GCP group attached to the lysine side chain, is significantly less efficient (IC50: 63 μM). As expected, negatively charged hydrazide E did not inhibit the enzyme at concentrations as high as 1 mM. The five aldehydes also showed no inhibition even at concentrations of 10 mM.
| Hydrazide | IC50 | Aldehyde | IC50 |
|---|---|---|---|
| A | 12.65 ± 0.61 | 1 | |
| B | 62.63 ± 6.86 | 2 | |
| C | 23.58 ± 1.17 | 3 | >10000 |
| D | 24.50 ± 2.87 | 4 | |
| E | >1000 | 5 |
The screening of the pre-equilibrated libraries was conducted using the same assay and a concentration of 1.25 μM for the individual building blocks. Preliminary tests showed that this concentration is sufficiently low to allow for significant discrimination between the full library and the sub-libraries. The results obtained from the enzyme screening are shown in Fig. 1A. As only the relative changes in activity in going from the full library to the sub-libraries are relevant, we normalized from no inhibition of the enzyme (100% normalized activity) to the inhibition value of the full library (0% normalized activity). These data show that hydrazides A–D, featuring peptide sequences with a combination of cationic and aromatic amino acids, are required for efficient enzyme inhibition. Hydrazide C (GKWK(GCP)) is the most active building block; its removal restores the enzyme activity by 47% relative to the inhibition by the full library. Hydrazides A (GKWR) and B (GKWK) are less, but similarly active (ca. 20–25% recovery of enzyme activity), while D (GK(GCP)WK) is the least active in this series of positively charged peptide hydrazides (13% recovery). These results also show that the artificial GCP group can significantly increase the potency of the inhibitors relative to both lysine and arginine (compare C to either A or B), however, its effect depends on its position within the hydrazide (compare C and D). Hydrazones containing negatively charged hydrazide E (GAFE) are completely inefficient as inhibitors. As one would expect, the removal of the completely inactive constituent E from the full library actually increases the inhibitory activity of the sub-library, as the concentration of active inhibitors increases.
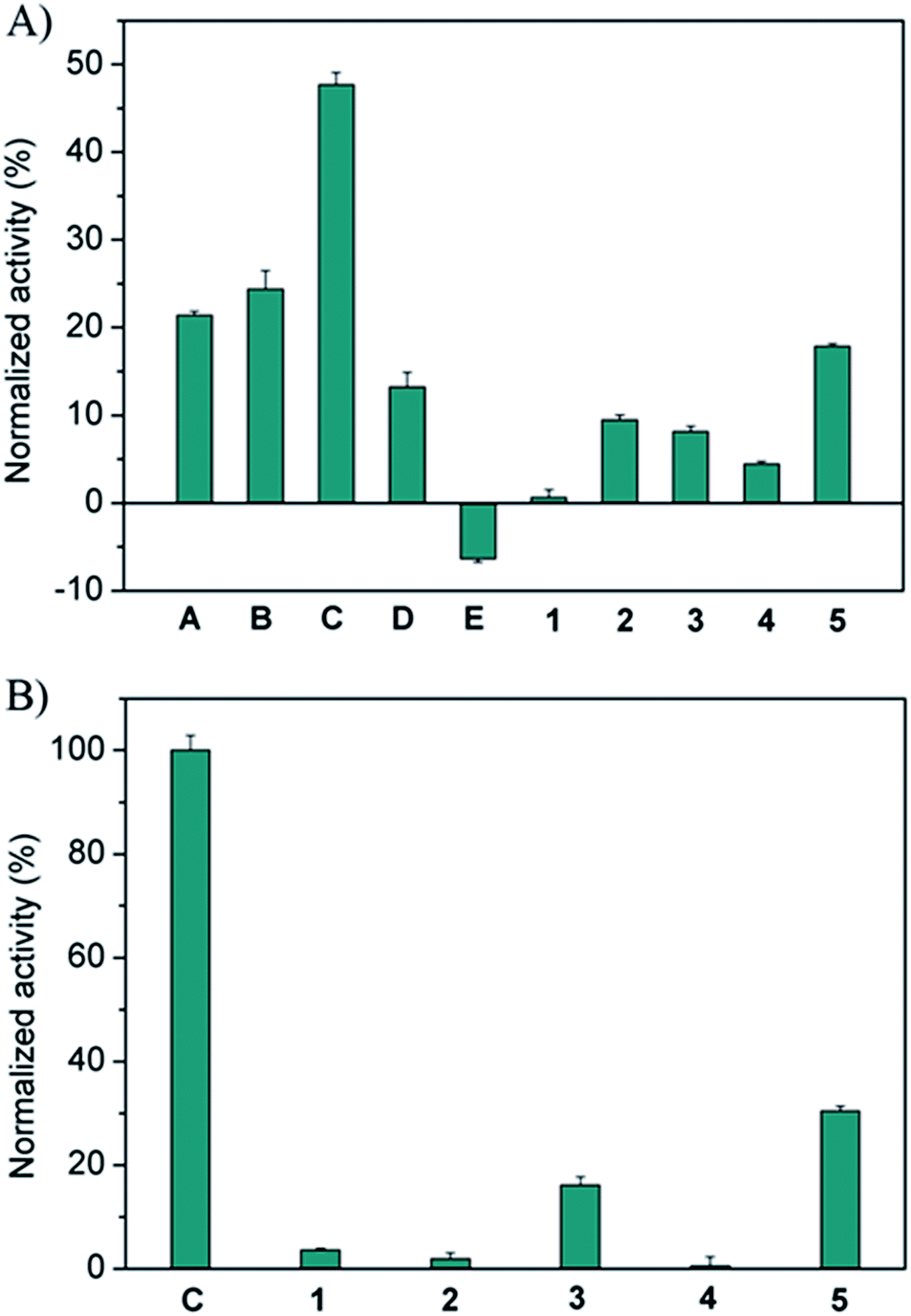 | ||
| Fig. 1 Normalized relative enzyme activity of sub-libraries, compared to the full library. Columns represent the sub-libraries lacking the building block stated below. (A) DCL containing five hydrazides and five aldehydes. (B) DCL containing only one hydrazide C and five aldehyde building blocks. Error bars represent the standard deviation of triplicate measurements. | ||
For the aldehyde scaffolds it can be seen that tri-aldehyde 5 is the most important one (18% recovery), while di-aldehydes 1–4 showed smaller effects (ca. 5–10% recovery), with 1 being the least efficient building block for inhibition. To further study the role of the scaffolds, a second but size-reduced library containing all five aldehydes and only the most active hydrazide C, and the corresponding sub-libraries (in which a different aldehyde is missing in each), were generated and analyzed as described above. The results shown in Fig. 1B confirm that the aromatic and rigid scaffolds 5 and 3 are the most active building blocks (ca. 15–30% recovery), at least for hydrazide C. Moreover, the removal of hydrazide C leads to full recovery of enzyme activity, once again confirming that the aldehydes themselves have no inhibitory activity.
Evaluation of the active compounds from the DCL
In order to validate the results of the initial DCL screening, a set of representative library members, shown in Scheme 3, based on scaffolds 5 and 3 and the five hydrazides were individually synthesized and their inhibitory activities were determined using standard methods (for details, see ESI†). Acyl hydrazones of type 5(A–E)3 and 3(A–C)2 have three or two identical arms, respectively, whereas 3(AC) features two different arms.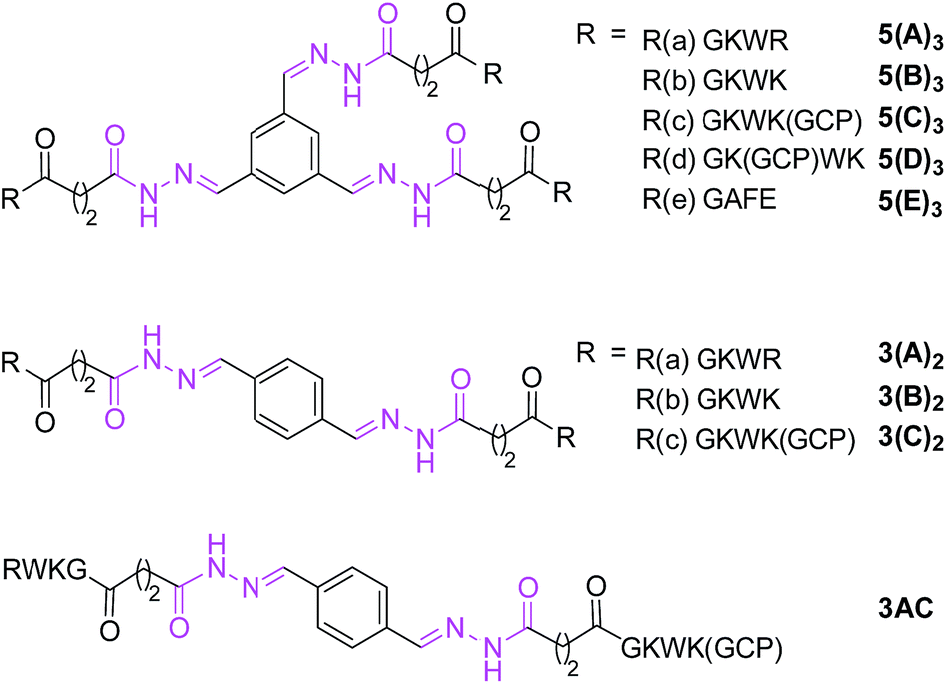 | ||
| Scheme 3 Molecular structures of individually synthesized acyl hydrazones (also see Scheme 1). | ||
The inhibition data for the interactions of the acyl hydrazones with β-tryptase are summarized in Table 2. As these compounds act as non-competitive inhibitors (v. i.), IC50 and Ki values are identical. A representative inhibition profile for 5(C)3 is shown in Fig. 2. Inhibitors 5(A–E)3, 3(A–C)2 and 3(AC) derived from the combination of the most active hydrazides A–C and aldehydes 5 or 3 have Ki values in the low nanomolar range. Hence, they are three orders of magnitude more potent as inhibitors than the individual hydrazides themselves, which have Ki-values of between 13 and 63 μM (Table 1). The two most active inhibitors 5(A)3 and 5(C)3, containing hydrazides with GKWR and GKWK(GCP) motifs, have Ki-values of 12 and 22 nM, respectively. As expected, hydrazone 5(E)3 containing the negatively charged hydrazide E showed no inhibition of β-tryptase at all. Interestingly, compound 5(D)3 which contains a GK(GCP)WK motif showed a biphasic inhibition profile (Fig. 3), suggesting high and low affinity binding modes with Ki values of 114 nM and 30 μM, respectively.23 For the high affinity binding mode, which accounts for only ca. 25–30% inhibition of the enzyme, 5(D)3 is similarly active (Ki = 114 nM) to 5(B)3 (Ki = 105 nM), but much less active than 5(C)3 (Ki = 22.5 nM) which contains the isomeric hydrazide C. The low affinity binding mode shows the same activity as the individual hydrazide D (cf.Table 1, IC50ca. 25 μM).
| Compound | β-Tryptase | Trypsin | α-ChT |
|---|---|---|---|
| a A biphasic behaviour was observed with two binding modes, one with higher affinity (Ki = 114.0 ± 19.3 nM) and one with lower affinity (Ki = 29.93 ± 1.44 μM). | |||
| 5(A)3 | 12.5 ± 0.8 | ||
| 5(B)3 | 104.9 ± 31.0 | ||
| 5(C)3 | 22.5 ± 2.1 | ||
| 5(D)3 | 114/29.900a | ||
| 5(E)3 | >100.000 | >100.000 | >100.000 |
| 3(A)2 | 65.6 ± 2.3 | ||
| 3(B)2 | 391.8 ± 24.4 | ||
| 3(C)2 | 78.0 ± 3.1 | ||
| 3(AC) | 120.8 ± 3.9 | ||
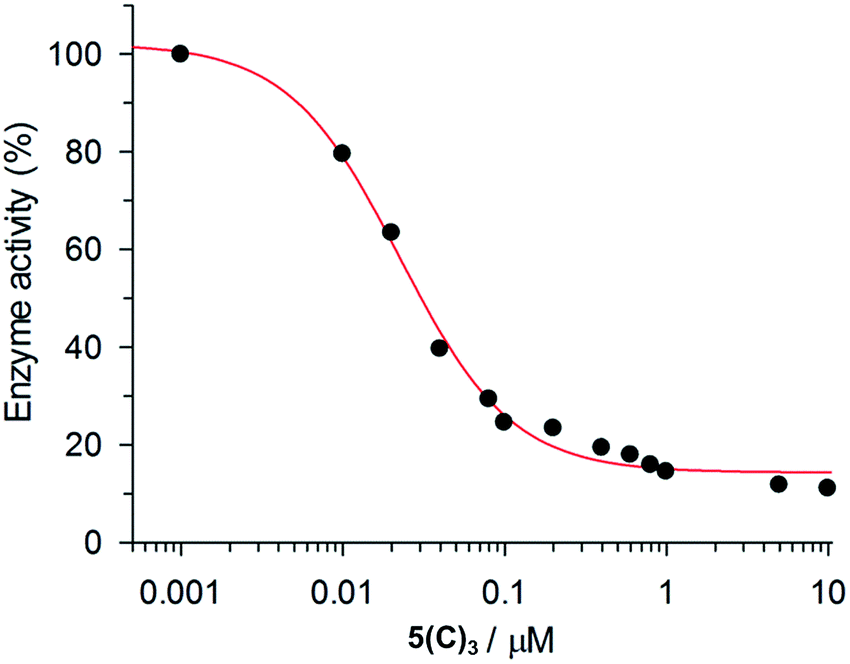 | ||
| Fig. 2 Monophasic inhibition of β-tryptase by 5(C)3. | ||
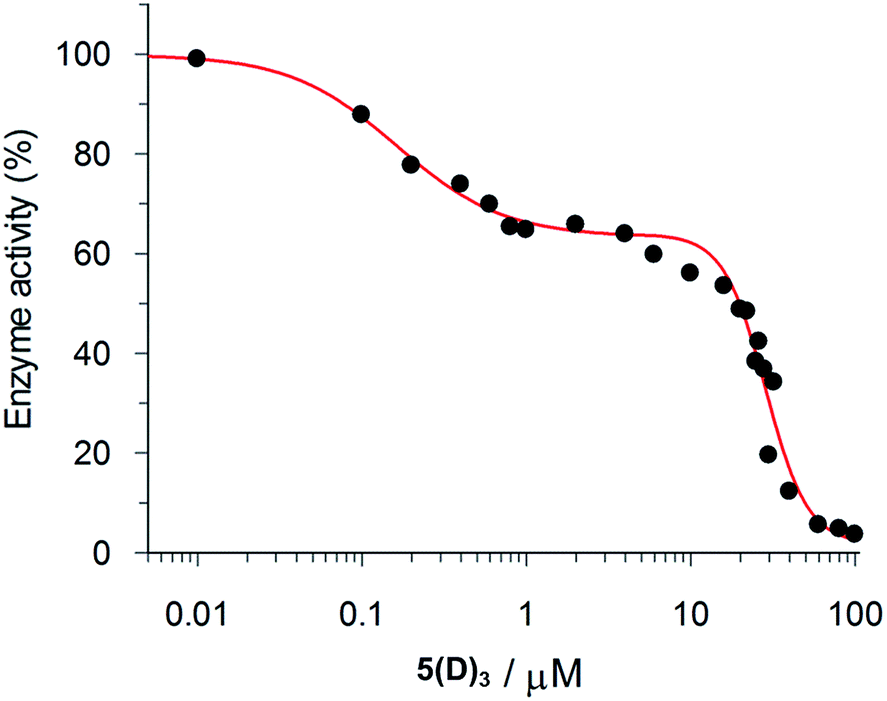 | ||
| Fig. 3 Biphasic inhibition of β-tryptase by 5(D)3. The IC50 values for the high affinity (114.0 ± 19.3 nM) and low affinity (29.93 ± 1.44 μM) binding processes were determined by the GraFit program. | ||
This result is in agreement with the screening of the pre-equilibrated libraries (Fig. 1a). It is, however, somehow surprising: while individual hydrazides C and D, which only differ in their peptide sequences, show similar inhibitory activities (Table 1), hydrazones 5(C)3 and 5(D)3 are vastly different. This simple change in the position of the GCP group significantly affects (i) the affinity of the ligand (with 5(C)3 being five times more potent than 5(D)3) and (ii) its inhibition profile (single mode for 5(C)3vs. biphasic behaviour for 5(D)3). These differences most likely originate from the different binding modes observed for 5(C)3 and 5(D)3.
Of course the pre-equilibrated library also contained mixed hydrazones with two or three different hydrazides attached to one scaffold. Therefore, heterodimeric compound 3(AC) was also synthesized and its activity was determined. The Ki value of 121 nM showed that 3(AC) is also quite an efficient inhibitor, but that it is less active than the two corresponding homodimeric species 3(A)2 and 3(C)2 (Ki values of 65.6 and 78 nM, respectively). Therefore, we did not synthesize any further heterotrimeric compounds based on the tri-aldehyde. However, it is still possible that one of the mixed species could be an even more potent inhibitor than any of the homomeric compounds discussed here.
Molecular modelling studies
To obtain further insight into the origins of these unexpected differences between 5(C)3 and 5(D)3, molecular mechanics calculations were performed. The calculations first of all confirmed that both molecules can indeed bind to the surface of β-tryptase, blocking access to its central pore where the active sites are situated (Fig. 4). The binding interactions, as shown in Fig. 4C and D, indicate that the inhibitors bind to two different types of binding sites on the protein surface: the monomer–monomer interfaces and binding sites close to the active sites. This behaviour is comparable to those of our less potent four-armed inhibitors, reported previously.18b The calculations indicate that both inhibitors interact with a significant number of anionic residues inside and around the entrance to the central pore, mainly forming H-bond assisted ion pairs to glutamate and aspartate. However, a closer look at the calculated structures indicates that inhibitor 5(C)3 blocks the pore completely, whereas 5(D)3 does so only partially, thus leaving more open space for substrates to reach the active sites, which in turn would explain the lower inhibition of β-tryptase by this ligand.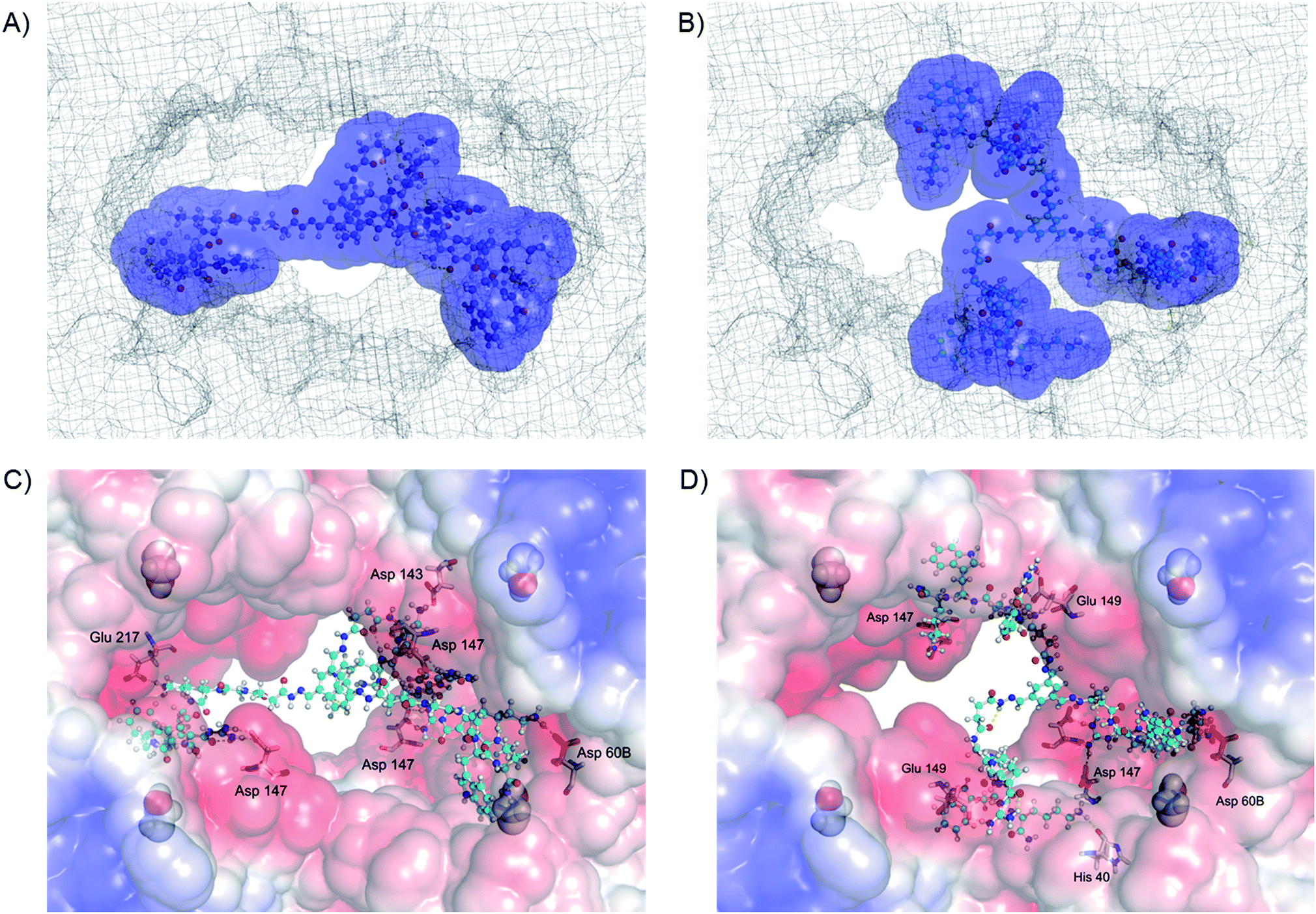 | ||
| Fig. 4 Calculated binding modes of (A) inhibitor 5(C)3 (total energy of the depicted complex: −53717.2 kJ mol−1) and (B) inhibitor 5(D)3 (total energy of the depicted complex: −53683.9 kJ mol−1) to the surface (shown as mesh) of β-tryptase (PDB code: 1 A0L). The inhibitors are shown with their accessible surface area (asa; used solvent radius 1.4 Å for water). Close-ups of the interactions of (C) 5(C)3 (shown as balls and sticks) and (D) 5(D)3 (shown as balls and sticks) with the enzyme surface. Key protein residues (Glu, Asp and His) that interact with the inhibitors are displayed as sticks. The red, blue and grey colors on the enzyme surface indicate the negative, positive and neutral electrostatic potentials, respectively. A number of anionic residues are located around the entrance to the central pore in which the active sites are situated. Catalytic serine residues are shown using the space-filling model. | ||
As 5(D)3 and 5(C)3 are constitutional isomers, we also calculated their relative interaction energies with the protein.24 The complex of β-tryptase and 5(C)3 is 33 kJ mol−1 more stable than its 5(D)3 congener (Fig. 4). Furthermore, in their unbound states 5(D)3 is ca. 36 kJ mol−1 lower in energy than 5(C)3 (see ESI, Fig. S2†), due to more stabilizing intramolecular interactions. This might suggest that the improved inhibitory activity of 5(C)3 is not only the result of stronger binding to the protein, but also of a less stable ground state conformation of the inhibitor itself. This also explains why even though the two hydrazides D and C have similar activities (ca. 25 μM, cf.Table 1), the three-armed ligands 5(D)3 and 5(C)3 differ quite significantly in their inhibitory effects on the enzyme.
Further studies on the mode of inhibition
Inhibitory activities for the two other archetypal structurally similar serine proteases, trypsin and α-chymotrypsin, were also determined in order to determine the selectivities of the inhibitors. However, none of the inhibitors showed any significant inhibition of these two related enzymes at concentrations of up to 100 μM (Table 1). This again is in good agreement with the idea that the compounds inhibit β-tryptase by protein surface binding, as both enzymes are structurally rather similar to the β-tryptase monomer but do not possess the central pore of β-tryptase with the surrounding negative charges and the monomer–monomer interface. Furthermore, the active site of trypsin is nearly identical to the active site of tryptase. Hence, if binding to the active site, or for example some areas on one of the protein monomers, occurred, then the compounds would have to inhibit trypsin and chymotrypsin equally well. The modelling data confirm that these inhibitors can bind to the entrance as well as to specific regions inside the central pore of β-tryptase. Therefore, interactions with two different anionic binding sites at the monomer–monomer interface and around the active site (both specific to β-tryptase)18b would explain the selectivity observed relative to the two other serine proteases, trypsin and chymotrypsin, which lack these binding sites.Further studies were performed to test the reversibility of β-tryptase inhibition by using a dialysis experiment. For example, enzyme activity was restored in a time dependent manner (see Fig. S3†) by 14% upon dialysis with a buffer solution over a period of 48 h. This indicated that the inhibition of β-tryptase by these acyl hydrazones is in principle reversible. This result was confirmed by a second test in which excess heparin was used to compete with the cationic inhibitor for interaction with β-tryptase. In this experiment, 16% recovery of enzyme activity was observed, thus again indicating that the inhibition is reversible (for more details, see ESI†). It is well known that the binding of multivalent ligands is characterized by a significantly low “off-rate”, so that the kinetics of such reversibly binding inhibitors often resembles irreversible binding.25 Hence, it is not surprising that the enzymatic activity is only partially restored in these experiments. This slow off-rate is one of the major contributors to the high binding affinity of multivalent ligands.
The mode of inhibition was then obtained from analysis of the Dixon plot (plots of reciprocal velocities against inhibitor concentrations at different substrate concentrations) shown in Fig. 5. The lines intersect on the x axis which clearly indicates non-competitive inhibition.26 The inhibitory effect of the ligand is thus not affected by increasing the substrate concentration, which means that the substrate and the inhibitor do not compete for the same binding site. Otherwise, at sufficiently high concentrations the substrate should be able to displace the inhibitor from the common binding site, restoring enzymatic activity. This, however, is not the case here. Thus, the inhibitors evidently do not bind to the active sites of the enzyme. The most likely binding mode, in agreement with our earlier results18 and also confirmed by the modelling studies presented herein, is protein surface binding. The inhibitors bind to the anionic residues around the entrance to the central pore (Fig. 4) and thus prevent the substrate from reaching the active sites.
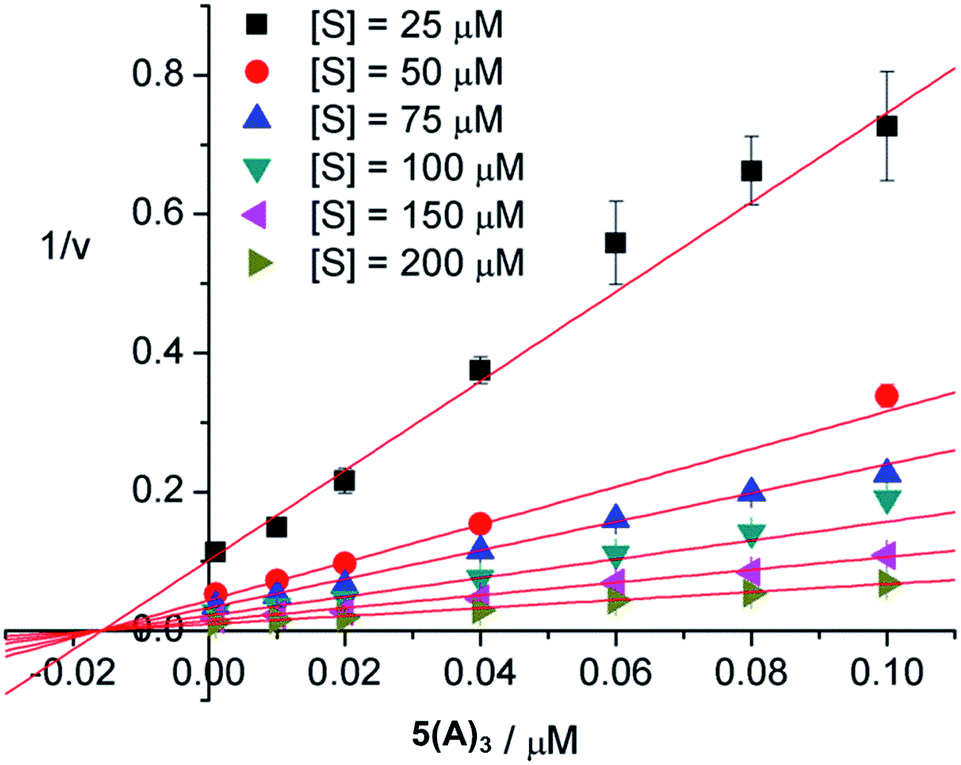 | ||
| Fig. 5 Dixon plot of reciprocal velocities (1/v) against inhibitor concentrations at different substrate concentrations: [S] = 25, 50, 75, 100, 150 and 200 μM. The error bars represent the standard deviations of triplicate measurements. | ||
Conclusions
In conclusion, we have shown that screening of a pre-equilibrated dynamic combinatorial library is an efficient way to identify high affinity inhibitors of β-tryptase. Two- and three-armed peptide hydrazones with rigid cores bind to (most likely the surface of) the protein with low nanomolar affinities and shut-down the enzymatic activity in a reversible and non-competitive way. Furthermore, we found that additional tailor-made binding motifs, such as the artificial arginine analog GCP, can significantly improve the activities of such inhibitors relative to inhibitors which only contain the corresponding proteinogenic amino acids. Work to further extend this approach of combining the rational design of tailor-made binding motifs for a given biological target with a combinatorial structural optimization is currently underway.Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft (DFG, SFB 1093) is gratefully acknowledged. Q.-Q. J. thanks the China Scholarship Council (CSC) for a PhD fellowship. We thank Dr Christoph Hirschhäuser for helpful suggestions for the preparation of the manuscript. We thank Mao Li for doing the library scrambling experiment.Notes and references
- For reviews on this topic, see: (a) O. Ramström and J.-M. Lehn, Nat. Rev. Drug Discovery, 2002, 1, 26–36 CrossRef PubMed; (b) P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J. L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed; (c) J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC; (d) S. Ladame, Org. Biomol. Chem., 2008, 6, 219–226 RSC; (e) R. A. R. Hunt and S. Otto, Chem. Commun., 2011, 47, 847–858 RSC; (f) E. Moulin, G. Cormos and N. Giuseppone, Chem. Soc. Rev., 2012, 41, 1031–1049 RSC; (g) F. B. L. Cougnon and J. K. Sanders, Acc. Chem. Res., 2012, 45, 2211–2221 CrossRef CAS PubMed.
- (a) P. A. Brady, R. P. Bonar-Low, S. J. Rowan, C. J. Suckling and J. K. M. Sanders, Chem. Commun., 1996, 319–320 RSC; (b) I. Huc and J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 2106–2110 CrossRef CAS; (c) P. A. Brady and J. K. M. Sanders, J. Chem. Soc., Perkin Trans. 1, 1997, 3237–3253 RSC; (d) B. Hasenknopf, J.-M. Lehn, N. Boumediene, A. Dupont-Gervais, A. Van Dorsserlaer, B. Kneisel and D. Fenske, J. Am. Chem. Soc., 1997, 119, 10956–10962 CrossRef CAS; (e) J.-M. Lehn, Chem.–Eur. J., 1999, 5, 2455–2463 CrossRef CAS.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef.
- For recent examples, see: (a) S. Otto, R. L. E. Furlan and J. K. M. Sanders, Science, 2002, 297, 590–593 CrossRef CAS PubMed; (b) P. T. Corbett, L. H. Tong, J. K. M. Sanders and S. Otto, J. Am. Chem. Soc., 2005, 127, 8902–8903 CrossRef CAS PubMed; (c) S. R. Beeren and J. K. M. Sanders, J. Am. Chem. Soc., 2011, 133, 3804–3807 CrossRef CAS PubMed; (d) J. M. Klein, V. Saggiomo, L. Reck, U. Lüning and J. K. M. Sanders, Org. Biomol. Chem., 2012, 10, 60–66 RSC.
- (a) S. Sando, A. Narita and Y. Aoyama, Bioorg. Med. Chem. Lett., 2004, 14, 2835–2838 CrossRef CAS PubMed; (b) A. M. Whitney, S. Ladame and S. Balasubramanian, Angew. Chem., Int. Ed., 2004, 43, 1143–1146 CrossRef CAS PubMed; (c) S. Ladame, A. M. Whitney and S. Balasubramanian, Angew. Chem., Int. Ed., 2005, 44, 5736–5739 CrossRef CAS PubMed; (d) M. Rauschenberg, S. Bomke, U. Karst and B. J. Ravoo, Angew. Chem., Int. Ed., 2010, 49, 7340–7373 CrossRef CAS PubMed.
- (a) M. Hochgürtel, H. Kroth, D. Piecha, M. W. Hofmann, C. Nicolau, S. Krause, O. Schaaf, G. Sonnenmoser and A. V. Eliseev, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 3382–3387 CrossRef PubMed; (b) A. Bugaut, J.-J. Toulmé and B. Rayner, Angew. Chem., Int. Ed., 2004, 43, 3144–3147 CrossRef CAS PubMed; (c) M. Hochgürtel, R. Biesinger, H. Kroth, D. Piecha, M. W. Hofmann, S. Krause, O. Schaaf, C. Nicolau and A. V. Eliseev, J. Med. Chem., 2003, 46, 356–358 CrossRef PubMed; (d) S. Zameo, B. Vauzeilles and J.-M. Beau, Angew. Chem., Int. Ed., 2005, 44, 965–969 CrossRef CAS PubMed; (e) S. Gerber-Lemaire, F. Popowycz, E. Rodríguez-García, A. T. C. Asenjo, I. Robina and P. Vogel, ChemBioChem, 2002, 5, 466–470 CrossRef.
- (a) T. Bunyapaiboonsri, O. Ramström, S. Lohmann, J.-M. Lehn, L. Peng and M. Goeldner, ChemBioChem, 2001, 2, 438–444 CrossRef CAS; (b) T. Bunyapaiboonsri, H. Ramström, O. Ramström, J. Haiech and J.-M. Lehn, J. Med. Chem., 2003, 46, 5803–5811 CrossRef CAS PubMed; (c) O. Ramström, S. Lohmann, T. Bunyapaiboonsri and J.-M. Lehn, Chem.–Eur. J., 2004, 10, 1711–1715 CrossRef PubMed.
- Recent studies have shown that the equilibration of hydrazones can be accelerated and generated at weak acidic conditions close to physiological pH by using aniline as a nucleophilic catalyst, which may open a window to the application of this DCC tool to biological targets in the future: (a) A. Dirksen, S. Dirksen, T. M. Hackeng and P. E. Dawson, J. Am. Chem. Soc., 2006, 128, 15602–15603 CrossRef CAS PubMed; (b) V. T. Bhat, A. M. Caniard, T. Luksch, R. Brenk, D. J. Campopiano and M. F. Greaney, Nat. Chem., 2010, 2, 490–497 CrossRef CAS PubMed; (c) A. J. Clipson, V. T. Bhat, I. McNae, A. M. Caniard, D. J. Campopiano and M. F. Greaney, Chem.–Eur. J., 2012, 18, 10562–10570 CrossRef CAS PubMed.
- (a) J. M. Clark, W. M. Abraham, C. E. Fishman, R. Forteza, A. Ahmed, A. Cortes, R. L. Warne, W. R. Moore and R. D. Tanaka, Am. J. Respir. Crit. Care Med., 1995, 152, 2076–2083 CrossRef CAS PubMed; (b) J. F. Molinari, M. Scuri, W. R. Moore, J. Clark, R. D. Tanaka and W. M. Abraham, Am. J. Respir. Crit. Care Med., 1996, 154, 649–653 CrossRef CAS PubMed; (c) J. M. Clark, W. R. Moore and R. D. Tanaka, Drugs Future, 1996, 21, 811–816 CAS.
- (a) P. J. B. Pereira, A. Bergner, S. Macedo-Ribeiro, R. Huber, G. Matschiner, H. Fritz, C. P. Sommerhoff and W. Bode, Nature, 1998, 392, 306–311 CrossRef CAS PubMed; (b) C. P. Sommerhoff, W. Bode, P. J. B. Pereira, M. T. Stubbs, J. Stürzebecher, G. P. Piechottka, G. Matschiner and A. Bergner, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 10984–10991 CrossRef CAS.
- C. P. Sommerhoff, C. Söllner, R. Mentele, G. P. Piechottka, E. A. Auerswald and H. Fritz, Biol. Chem. Hoppe-Seyler, 1994, 375, 685–694 CrossRef CAS.
- K. C. Elrod, W. R. Moore, W. M. Abraham and R. D. Tanaka, Am. J. Respir. Crit. Care Med., 1997, 156, 375–381 CrossRef CAS PubMed.
- L. Cregar, K. C. Elrod, D. Putnam and W. R. Moore, Arch. Biochem. Biophys., 1999, 366, 125–130 CrossRef CAS PubMed.
- G. C. Paesen, C. Siebold, K. Harlos, M. F. Peacey, P. A. Nuttall and D. I. Stuart, J. Mol. Biol., 2007, 368, 1172–1186 CrossRef CAS PubMed.
- (a) P. Thongyoo, C. Bonomelli, R. J. Leatherbarrow and E. W. Tate, J. Med. Chem., 2009, 52, 6197–6200 CrossRef CAS PubMed; (b) C. P. Sommerhoff, O. Avrutina, H.-U. Schmoldt, D. Gabrijelcic-Geiger, U. Diederichsen and H. Kolmar, J. Mol. Biol., 2010, 395, 167–175 CrossRef CAS PubMed.
- (a) M. J. Costanzo, S. C. Yabut, H. R. Almond, P. Andrade-Gordon, T. W. Corcoran, L. de Garavilla, J. A. Kauffman, W. M. Abraham, R. Recacha, D. Chattopadhyay and B. E. Maryanoff, J. Med. Chem., 2003, 46, 3865–3876 CrossRef CAS PubMed; (b) N. Schaschke, A. Dominik, G. Matschiner and C. P. Sommerhoff, Bioorg. Med. Chem. Lett., 2002, 12, 985–988 CrossRef CAS; (c) D. Scarpi, J. D. McBride and R. J. Leatherbarrow, Bioorg. Med. Chem., 2004, 12, 6045–6052 CrossRef CAS PubMed; (d) C.-S. Lee, W. Liu, P. A. Sprengeler, J. R. Somoza, J. W. Janc, D. Sperandio, J. R. Spencer, M. J. Green and M. E. McGrath, Bioorg. Med. Chem. Lett., 2006, 16, 4036–4040 CrossRef CAS PubMed; (e) M. Tetsuhashi, M. Ishikawa, M. Hashimoto, Y. Hashimoto and H. Aoyama, Bioorg. Med. Chem., 2010, 18, 5323–5338 CrossRef CAS PubMed.
- (a) M. del Fresno, D. Fernández-Forner, M. Miralpeix, V. Segarra, H. Ryder, M. Royo and F. Albericio, Bioorg. Med. Chem. Lett., 2005, 15, 1659–1664 CrossRef CAS PubMed; (b) M. García, X. del Río, S. Silvestre, M. Rubiralta, E. Lozoya, V. Segarra, D. Fernández, M. Miralpeix, M. Aparici and A. Diez, Org. Biomol. Chem., 2004, 2, 1633–1642 RSC; (c) J. T. Palmer, R. M. Rydzewski, R. V. Mendonca, D. Sperandio, J. R. Spencer, B. L. Hirschbein, J. Lohman, J. Beltman, M. Nguyen and L. Liu, Bioorg. Med. Chem. Lett., 2006, 16, 3434–3439 CrossRef CAS PubMed; (d) M. E. McGrath, P. A. Sprengeler, B. Hirschbein, J. R. Somoza, I. Lehoux, J. W. Janc, E. Gjerstad, M. Graupe, A. Estiarte, C. Venkataramani, Y. Liu, R. Yee, J. D. Ho, M. J. Green, C.-S. Lee, L. Liu, V. Tai, J. Spencer, D. Sperandio and B. A. Katz, Biochemistry, 2006, 45, 5964–5973 CrossRef CAS PubMed; (e) G. Liang, S. Aldous, G. Merriman, J. Levell, J. Pribish, J. Cairns, X. Chen, S. Maignan, M. Mathieu, J. Tsay, K. Sides, S. Rebello, B. Whitely, I. Morize and H. W. Pauls, Bioorg. Med. Chem. Lett., 2012, 22, 1049–1054 CrossRef CAS PubMed.
- (a) P. R. Wich and C. Schmuck, Angew. Chem., Int. Ed., 2010, 49, 4113–4116 CrossRef CAS PubMed; (b) Q.-Q. Jiang, L. Bartsch, W. Sicking, P. R. Wich, D. Heider, D. Hoffmann and C. Schmuck, Org. Biomol. Chem., 2013, 11, 1631–1639 RSC.
- (a) C. Schmuck and M. Schwegmann, J. Am. Chem. Soc., 2005, 127, 3373–3379 CrossRef CAS PubMed; (b) C. Schmuck and M. Schwegmann, Org. Lett., 2005, 7, 3517–3520 CrossRef CAS PubMed.
- (a) C. Schmuck, P. Frey and M. Heil, ChemBioChem, 2005, 6, 628–631 CrossRef CAS PubMed; (b) C. Schmuck and M. Heil, Chem.–Eur. J., 2006, 12, 1339–1348 CrossRef CAS PubMed.
- (a) H. Y. Kuchelmeister and C. Schmuck, Chem.–Eur. J., 2011, 17, 5311–5318 CrossRef CAS PubMed; (b) K. Klemm, M. R. Stojković, G. Horvat, V. Tomišić, I. Piantanida and C. Schmuck, Chem.–Eur. J., 2012, 18, 1352–1363 CrossRef CAS PubMed.
- H. Y. Kuchelmeister, A. Gutschmidt, S. Tillmann, S. Knauer and C. Schmuck, Chem. Sci., 2012, 3, 996–1002 RSC.
- (a) F. McTaggart, G. R. Brown, R. G. Davidson, S. Freeman, G. A. Holdgate, K. B. Mallion, D. J. Mirrlees, G. J. Smith and W. H. J. Ward, Biochem. Pharmacol., 1996, 51, 1477–1487 CrossRef CAS; (b) T.-C. Mou, N. Masada, D. M. F. Cooper and S. R. Sprang, Biochemistry, 2009, 48, 3387–3397 CrossRef CAS PubMed.
- Absolute interaction energies obtained from force field calculations are difficult to compare for different molecules due to the uncertainties and approximations in these calculations. For isomers only relative data are compared, which are significantly more reliable.
- (a) M. Mammen, C. Seok-Ki and G. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2754–2794 CrossRef; (b) S. Cha, Biochem. Pharmacol., 1975, 24, 2177–2185 CrossRef CAS; (c) J. F. Morrison, Biochim. Biophys. Acta, 1969, 185, 269–286 CrossRef CAS.
- (a) M. Dixon, Biochem. J., 1953, 55, 170–171 CAS; (b) A. Cornish-Bowden, Biochem. J., 1974, 137, 143–144 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the synthetic procedures and characterization data, as well as detailed preparation of DCLs, enzyme assay procedures, and description of the molecular modeling calculations. See DOI: 10.1039/c4sc02943g |
| This journal is © The Royal Society of Chemistry 2015 |