
Phosphorescent Cu(I) complexes based on bis(pyrazol-1-yl-methyl)-pyridine derivatives for organic light-emitting diodes†
Fengshou
Wu
ab,
Jie
Li
c,
Hongbo
Tong
d,
Zaoying
Li
*a,
Chihaya
Adachi
c,
Adam
Langlois
e,
Pierre D.
Harvey
e,
Li
Liu
f,
Wai-Yeung
Wong
*b,
Wai-Kwok
Wong
*b and
Xunjin
Zhu
*b
aCollege of Chemistry and Molecular Sciences, Wuhan University, Wuhan, P. R. China. E-mail: zyliwuc@whu.edu.cn
bInstitute of Molecular Functional Materials and Department of Chemistry and Institute of Advanced Materials, Hong Kong Baptist University, Hong Kong, P. R. China. E-mail: xjzhu@hkbu.edu.hk; wkwong@hkbu.edu.hk; rwywong@hkbu.edu.hk
cCenter for Organic Photonics and Electronics Research (OPERA), Kyushu University, Japan
dInstitute of Applied Chemistry, Shanxi University, Taiyuan, P. R. China
eDépartement de Chimie, Université de Sherbrooke, Sherbrooke, PQ J1N 2X8, Canada
fHubei Collaborative Innovation Center for Advanced Organic Chemical Materials, Ministry of Education Key Laboratory for the Synthesis and Application of Organic Functional Molecules, School of Chemistry and Chemical Engineering, Hubei University, Wuhan 430062, P. R. China
First published on 23rd October 2014
Abstract
Mononuclear Cu(I) complexes based on bis(pyrazol-1-yl-methyl)-pyridine derivatives and ancillary triphenylphosphine have been prepared and characterized by 1H NMR, mass spectroscopy and single-crystal X-ray analysis. The thermogravimetric analysis shows that the complexes exhibit high thermal stability. The electronic absorption spectra display two features in the regions of 230–260 and 290–350 nm attributable to mixed ligand-to-ligand (LLCT) and metal-to-ligand-charge-transfer (MLCT) excited states, which is supported by the results of density functional theory (DFT) and time-dependent DFT (TDDFT) calculations on these Cu(I) complexes. These complexes are strongly emissive in the solid state at ambient temperature. Intense blue or green emission in the poly(methyl methacrylate) film is observed in the region of 475–518 nm for these complexes with the emission lifetimes in the microsecond time scale (12–20 μs), indicating that the emission may be phosphorescence emission. Increasing the steric hindrance of the substituents on the pyrazole unit results in a blue-shift of the emission bands and enhanced emission quantum efficiency in PMMA films. The two most emissive complexes have been used for the fabrication of phosphorescent organic light-emitting diodes (POLEDs).
Introduction
Most organic electrophosphorescent materials are organometallic phosphors with noble heavy metal ions as the core components, e.g. Pt(II),1–5 Ir(III),6–8 Os(II),9,10 Ru(II) (ref. 11) and Re(I).12–19 Owing to the efficient spin–orbit coupling (consequence of the heavy atom effect), a high internal quantum efficiency (IQE) beyond the statistical limit of 25% has been obtained from both singlet and triplet excitons. Despite their applications as the most efficient and versatile class of phosphorescent emitters produced to date, the synthesis of such compounds requires complex, multistep and time-consuming reactions. In addition to the high synthetic complexity, only moderate to poor yields are frequently achieved. Also, the natural abundance of this latter heavy element (Ir) is very limited and the risk of a drastic shortage of this metal is unquestionably a shortcoming. There is a need to develop cheap phosphorescent materials for commercialization of the phosphorescent organic light-emitting device (PhOLED). Cu(I) complexes have long been known as potential alternatives, with much lower cost than that of iridium phosphors.20–26 Cu(I) complexes of a tripodal ligand 2,6-bis(1-pyrazole)pyridine were ever prepared for ligand dynamic studies.27,28 Recently much attention has been paid to the alternative architecture bearing one diimine or analogues plus an ancillary bis-phosphine chelate, generally leading to compounds with greatly enhanced emission performance imposed by the highly rigid chelate and stronger metal–phosphine bonding.29–43 For example, a high external quantum efficiency (EQE) of 16% was achieved with heteroleptic Cu(I) complexes containing phenanthroline and triphenylphosphine as the ligands.17,44 Moreover, Wang and co-workers increased the efficiency and tuned the emission color to the orange-red region by extending the π-conjugation and augmenting the rigidity of the ligands in Cu(I) complexes.45 Chou's group also designed and synthesized a series of emissive, group 11 d10 transition metal complexes bearing various 2-pyridyl-pyrrolide chromophores.46 Recently, Osawa and co-workers demonstrated that the three-coordinate structure of Cu(I) complexes is another direction for high EQE.47 However, compared to those Cu(I) complexes emitting in the “green” and “red” region, the preparation of efficient and stable blue phosphorescent materials remains a challenge. Therefore, the fine-tuning of the phosphorescence wavelength to the deep blue region and enhancement of phosphorescent quantum yields of Cu(I) complexes are essential for blue PhOLEDs.We now report the synthesis, crystal structures, photoluminescence and electroluminescence properties of a new class of Cu(I) complexes P1–P5 based on a sterically congested 2,6-bis(1-pyrazole)pyridine ligand and its analogues (L). The use of the sterically congested ligand can afford a rigid four-coordinate structure around the copper center accompanied by the auxiliary triphenylphosphine ligand and counter anion ion BF4−, which are beneficial for high EQE because it hardly distorts in the excited state. Moreover, through the addition of bulky substituents to the pyrazole ring, fine tuning of the phosphorescence wavelength from the orange to blue region has been realized. Noteworthy, the ligands and complexes can be readily synthesized under mild conditions and separated in quantitative yields by facile precipitation and washing procedures. The two most emissive complexes have been used for the fabrication and characterization of organic light-emitting diodes (OLEDs).
Results and discussion
Synthesis and characterization
First, pyrazole derivatives, as a class of important precursors, were prepared in almost quantitative yields from volatile 1,3-diones and hydrazine hydrate according to the literature methods.48 Then a phase-transfer catalyzed reaction of 2,6-bis(bromomethyl)pyridine with various substituted pyrazoles afforded the corresponding 2,6-bis(1-pyrazole)pyridine ligand and its analogues (L) with yields of around 70%.49 As shown in Scheme 1, subsequent complexation upon stirring L with an equivalent amount of [Cu(CH3CN)4]BF4 salt, and then with the ancillary triphenylphosphine ligand, followed by purification through precipitation and washing steps, afforded Cu(I) complexes P1–P5 in almost quantitative yields.28,50 The Cu(I) complexes are air-stable in the solid state but will be oxidized by several days' air exposure in solution inducing the formation of more stable water coordinated Cu(II) complexes.51 The Cu(I) complexes were characterized by 1H NMR, elemental analysis and MALDI-TOF spectroscopy. Four of them were also analysed by single crystal X-ray diffraction. | ||
| Scheme 1 The synthetic routes for the Cu(I) complexes P1–P5. | ||
Single crystals of P1–P4 were obtained by slow evaporation of the respective saturated solutions of dichloromethane–hexane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, v/v). Perspectives views of P1–P4 are shown in Fig. 1. Pertinent crystallographic data and other experimental details are summarized in Table 1, and selected bond lengths and angles are given in Table 2. The single crystal structures reveal that the metal ions exhibit a highly distorted tetragonal coordination geometry. The N3–Cu1–N5 angle in P1–P4 is 110.84(15)°, 110.0(5)°, 120.5(2)° and 110.9(3)°, respectively. Specifically, the N1–Cu1–P1 angles, ranging from 113.5(14)° to 133.2(3)° in P1–P4, strongly deviate from the ideal tetrahedral value of 109.5°, which reflects the specific steric hindrance of the tripodal ligands. The Cu–N bond lengths of the complexes localize in a region of 2.049–2.229 Å, which are comparable to the literature values.52,53 The Cu–P bond lengths are similar to each other, 2.198(14) Å for P1, 2.161(4) Å for P2, 2.237(2) Å for P3 and 2.266(4) Å for P4.
:3, v/v). Perspectives views of P1–P4 are shown in Fig. 1. Pertinent crystallographic data and other experimental details are summarized in Table 1, and selected bond lengths and angles are given in Table 2. The single crystal structures reveal that the metal ions exhibit a highly distorted tetragonal coordination geometry. The N3–Cu1–N5 angle in P1–P4 is 110.84(15)°, 110.0(5)°, 120.5(2)° and 110.9(3)°, respectively. Specifically, the N1–Cu1–P1 angles, ranging from 113.5(14)° to 133.2(3)° in P1–P4, strongly deviate from the ideal tetrahedral value of 109.5°, which reflects the specific steric hindrance of the tripodal ligands. The Cu–N bond lengths of the complexes localize in a region of 2.049–2.229 Å, which are comparable to the literature values.52,53 The Cu–P bond lengths are similar to each other, 2.198(14) Å for P1, 2.161(4) Å for P2, 2.237(2) Å for P3 and 2.266(4) Å for P4.
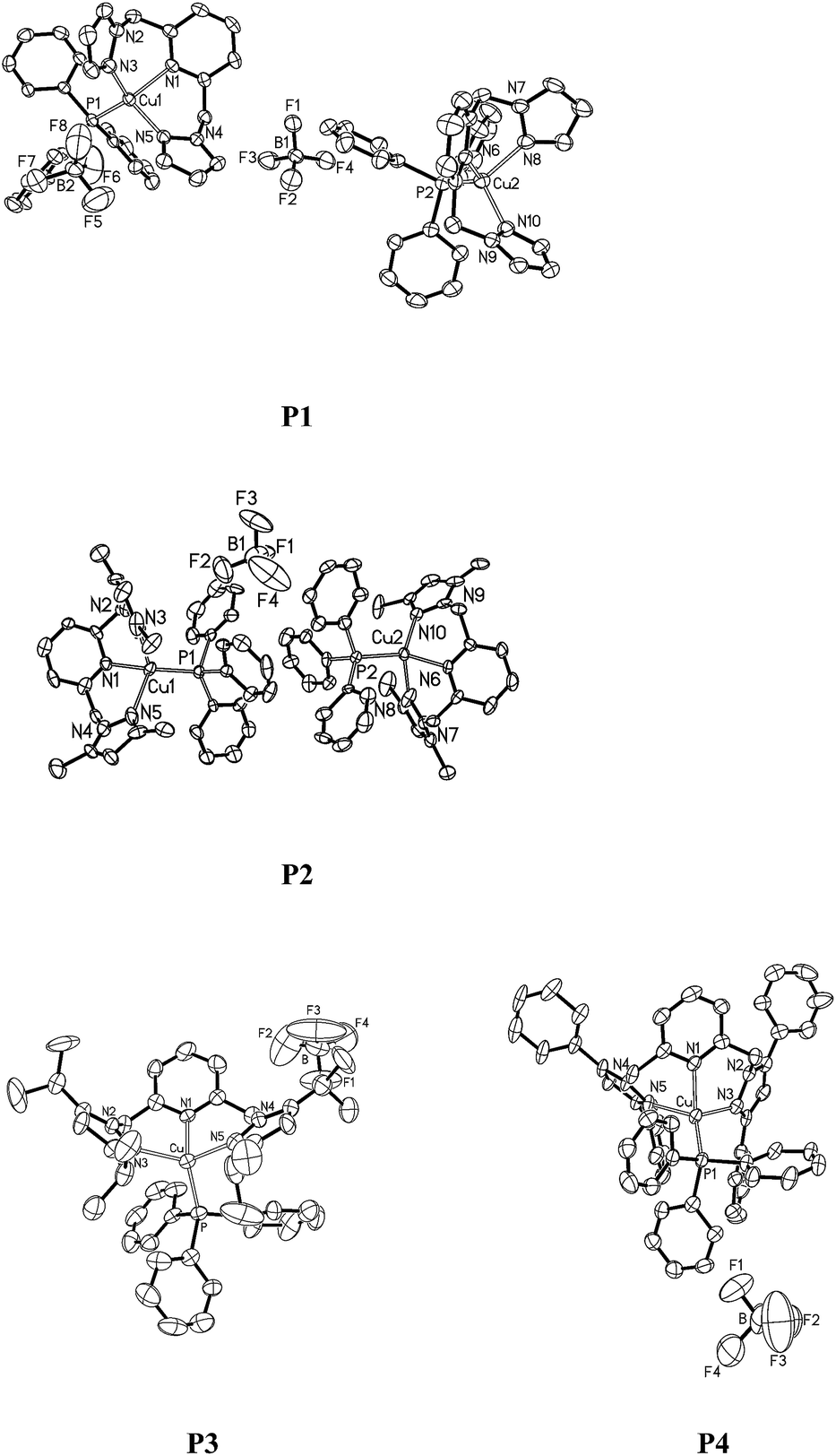 | ||
| Fig. 1 Perspective views of the structures of P1–P4. Thermal ellipsoids are drawn at the 30% probability level. H atoms in P1–P4, one solvent molecule (CH2Cl2) in P1 and one remote anion ion (BF4−) in P2 are omitted for clarity. | ||
| Complex | (P1)2·(CH2Cl2) | P2 | P3 | P4 |
|---|---|---|---|---|
| a R 1 = Σ‖Fo| − |Fc‖/Σ|Fo|. wR2 = [Σw(|Fo2| − |Fc2|)2/Σw|Fo2|2]1/2. | ||||
| Formula | C63H58B2Cl2Cu2F8N10P2 | C35H36BCuF4N5P | C43H52BCuF4N5P | C55H44BCuF4N5P |
| FW | 1388.73 | 708.02 | 820.22 | 956.27 |
| T (K) | 296(2) | 296(2) | 296(2) | 296(2) |
| Crystal system | Monoclinic | Orthorhombic | Monoclinic | Triclinic |
| Space group | P21/c | P21 | P21/n |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a (Å) | 10.470(3) | 20.5475(13) | 16.775(6) | 13.96(4) |
| b (Å) | 15.697(4) | 10.2304(6) | 16.685(6) | 14.57(2) |
| c (Å) | 39.879(11) | 32.811(2) | 18.348(7) | 14.67(2) |
| α (deg) | 90 | 90 | 90 | 117.544(19) |
| β (deg) | 91.893(4) | 90 | 112.177(9) | 99.86(3) |
| γ (deg) | 90 | 90 | 90 | 95.08(3) |
| V (Å3) | 6551(3) | 6897.3(7) | 4756(3) | 2559(9) |
| Z | 4 | 4 | 4 | 2 |
| D calcd (g cm−3) | 1.400 | 1.365 | 2.167 | 1.255 |
| μ(Mo-Kα) [mm−1] | 0.848 | 0.734 | 4.897 | 0.513 |
| F(000) | 2824 | 2932 | 2952 | 998 |
| Crystal size (mm3) | 0.51 × 0.50 × 0.49 | 0.30 × 0.20 × 0.20 | 0.31 × 0.25 × 0.35 | 0.35 × 0.32 × 0.32 |
| θ min, θmax (deg) | 1.39, 25.01 | 1.24, 25.00 | 1.71, 25.05 | 1.51, 25.00 |
| Reflections collected | 68238 |
70486 |
26332 |
26111 |
| Independent reflections | 11565 |
12132 |
8419 | 9012 |
| R int | 0.0583 | 0.0839 | 0.1147 | 0.2091 |
| GOF on F2 | 1.074 | 1.072 | 0.932 | 0.831 |
| R 1, wR2 [I > σ(I)]a | 0.0592, 0.1356 | 0.1001, 0.2609 | 0.0761, 0.1734 | 0.0964, 0.2268 |
| R 1, wR2 (all data) | 0.0841, 0.1495 | 0.1386, 0.2945 | 0.1779, 0.2021 | 0.1926, 0.2854 |
| Complex | P1 | P2 | P3 | P4 |
|---|---|---|---|---|
| Cu1–N1 | 2.104(3) | 2.137(11) | 2.137(5) | 2.173(6) |
| Cu1–N3 | 2.049(4) | 2.083(12) | 2.109(5) | 2.140(7) |
| Cu1–N5 | 2.063(3) | 2.229(15) | 2.093(6) | 2.166(7) |
| Cu1–P1 | 2.198(14) | 2.161(4) | 2.237(2) | 2.266(4) |
| N3–Cu1–N5 | 110.84(15) | 110.0(5) | 120.5(2) | 110.9(3) |
| N3–Cu1–N1 | 91.83(14) | 93.3(4) | 92.5(2) | 94.6(3) |
| N5–Cu1–N1 | 90.68(13) | 89.2(4) | 92.1(2) | 85.9(3) |
| N3–Cu1–P1 | 118.27(11) | 115.4(3) | 120.8(15) | 117.5(2) |
| N5–Cu1–P1 | 113.79(11) | 111.4(4) | 110.9(15) | 123.8(18) |
| N1–Cu1–P1 | 127.06(9) | 133.2(3) | 113.5(14) | 115.72(18) |
Thermogravimetric analysis (TGA) was carried out on the complexes P1–P5 (Table 3 and Fig. S1 in the ESI†). These Cu(I) complexes exhibit high decomposition temperatures (Tdec) at around 270 °C. Moreover, only one evaporation step was observed for all the complexes in the TGA traces, indicating the important role of an ancillary phosphine ligand in stabilizing the complexes.
| Complex | λ abs (nm) | λ em (nm) | Φ | λ em (nm) | τ (μs) | Φ | E ox1/2 (V) | HOMO (eV) | LUMO (eV) | T dec (°C) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Measurements were done in 1 × 10−6 mol L−1 CH2Cl2 solution at 298 K. b In oxygen-free solution; [Ru(bpy)3]2+ as a standard in air-equilibrated water (Φ = 0.028). c Measured at the excitation of 320 nm in the PMMA film. d Quantum yield was measured in the PMMA film using a Hamamatsu C9920 system equipped with a calibrated integrating sphere. e Not emissive. f Defined as the temperature of 5% weight loss. | ||||||||||
| P1 | 256, 290, 348 | 518 | 12 | 0.05 | 0.80 | −5.60 | −2.34 | 272 | ||
| P2 | 258, 302, 349 | 563 | 0.0054 | 511 | 2 | 0.20 | 0.83 | −5.63 | −2.45 | 245 |
| P3 | 267, 350 | 562 | 0.0087 | 508 | 19 | 0.22 | 0.94 | −5.74 | −2.39 | 237 |
| P4 | 254, 348 | 576 | 0.0015 | 504 | 18 | 0.06 | 0.98 | −5.80 | −2.41 | 227 |
| P5 | 258, 350 | 512 | 0.0032 | 475 | 16 | 0.15 | 0.78 | −5.58 | −2.19 | 238 |
Photophysical properties
The absorption and emission spectra of the Cu(I) complexes in CH2Cl2 solution are shown in Fig. 2a. The Cu(I) complexes display intense absorption bands with maxima at 228 and 258 nm, which can be assigned to spin-allowed intraligand π–π* transitions of the bis(pyrazol-1-yl)-pyridine moiety, as compared to that of the free ligands (Fig. S1†). In addition, these complexes display additional bands between 290 and 350 nm, which are not present either in PPh3 or in the bis(pyrazol-1-yl)pyridine derivative. The low energy absorptions can be assigned to the low-lying metal-to-ligand charge-transfer (3MLCT) bands involving mainly the 3d orbitals of Cu(I) and the π* orbitals of the bis(pyrazol-1-yl)-pyridine ligands, mixed with some phosphine-to-L charge transfer (1LLCT).54 The conclusion was also supported by the results of density functional theory (DFT) and time-dependent DFT (TDDFT) calculations on the Cu(I) complexes, which indicate that the highest occupied molecular orbital (HOMO) resides on the metal 3d and the ancillary triphenylphosphine, while the lowest unoccupied orbital is mainly contributed by the π*-system of the pyridine ligand (vide infra).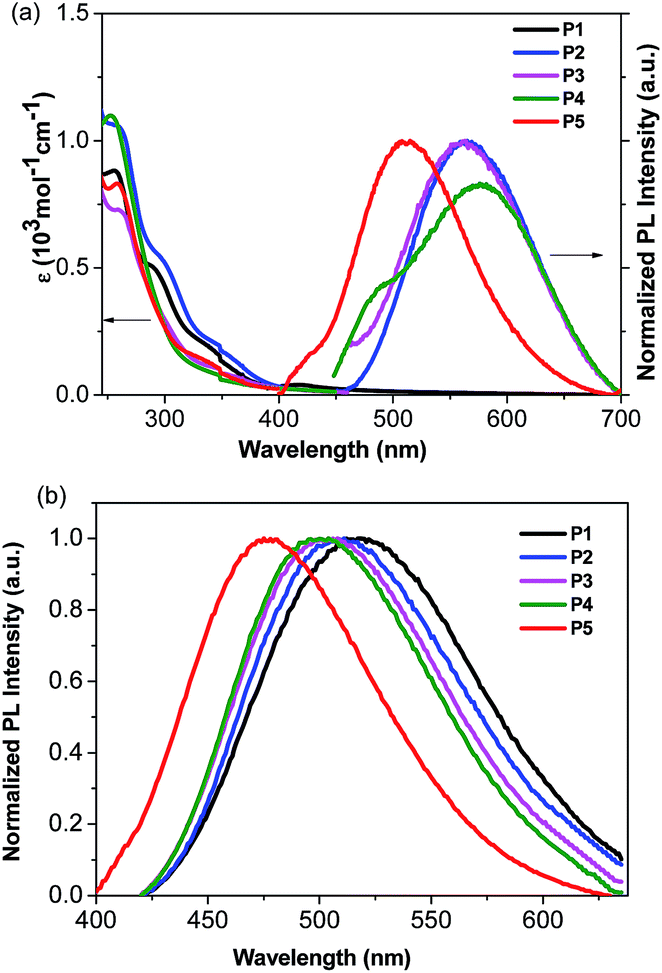 | ||
| Fig. 2 (a): Absorption and emission spectra of the Cu(I) complexes in CH2Cl2 solution at room temperature; (b): emission spectra of the Cu(I) complexes in 20 wt% PMMA films at room temperature. | ||
The emission maxima of P2–P5 in degassed CH2Cl2 range from 512 to 576 nm (Fig. 2a and Table 3). A possible assignment of the broad band is from the metal-to-ligand charge-transfer (MLCT), mixed with some ligand-to-ligand charge transfer (LLCT). In contrast, no emission peak appears in P1, probably due to the lack of a bulky group around the copper center, therefore leading to efficient non-radiative relaxation.20 Besides, P5 exhibits a higher energy emission relative to that of P2, indicating a larger HOMO–LUMO gap arising from the electron-withdrawing character of the trifluoromethyl group. This is consistent with the electrochemical and DFT calculations below (vide infra). The emission spectra of the Cu(I) complexes were also recorded in acetonitrile and methanol solutions, as these solvents have very different dielectric constants and coordination abilities from that of dichloromethane. Interestingly, the emission spectra of the complexes in acetonitrile and methanol are very similar, in which the emission bands in the visible region almost disappeared (Fig. S2†). The phenomenon may be the consequence of the higher dielectric constant of acetonitrile and methanol relative to that of dichloromethane, which stabilizes the charge transfer state, and their stronger coordinating ability can increase exciplex quenching by pentacoordination of Cu(II).55
It is known that the luminescence properties of the Cu(I) complexes are strongly dependent on the environment.7 Thus, thin films for optical measurements were prepared by spin coating 20 wt% poly(methyl methacrylate) (PMMA) solution in dichloromethane. As depicted in Fig. 2b, the emission spectra of the complexes in PMMA films are blue-shifted by 37–72 nm (1521–2480 cm−1) relative to that in dichloromethane solution. For instance, P2 shows a green emission (λmax = 511 nm) in the PMMA film and a yellow one (λmax = 563 nm) in dichloromethane solution. Similar trends were also observed for other complexes. Specifically, the emission maxima of these complexes are blue-shifted with the increasing steric hindrance effect on the pyrazole unit for P1–P4. It can be shown that incorporation of a bulky substituent group can sterically prevent structural relaxation of the Cu(I) complexes in the MLCT state, which may narrow the energy gap between excited and ground states.56 As suggested by the density functional theory (DFT) studies (vide infra), the electron-withdrawing character of the trifluoromethyl substituents on the pyrazole unit for P5 can significantly lift the LUMO level, resulting in a blue emission with the highest energy. Obviously, the Cu(I) complexes exhibit a high energy CT absorption but a relatively low energy emission maximum, as shown in Fig. 2, which is attributed to the tetragonal flattening due to the geometric orbital vacancy.57–59 The quantum yield of photoluminescence was measured in the 20% doped PMMA film using an integrating sphere, following the method published by Porres et al.60 As shown in Table 3, P2 exhibits an enhanced quantum yield relative to P1, indicating that the methyl group on the pyrazole unit in P2 prevents the rearrangement of the excited state to a certain degree. In addition, the microsecond life-time scale of the emitting species (12–21 μs) at room temperature indicates that the emission may stem from the triplet state, which was further supported by temperature-dependent photoluminescence and lifetimes of the complexes. At an enough low temperature such as 77 K, the population is predominantly frozen in the triplet state and thus the emitting state can be assigned as a pure triplet state. To give an example, the photoluminescence for P3 at 77 K shifted less than 10 nm as compared with that at room temperature. And the corresponding life time at 77 K is 20 μs, which is very close to that of 19 μs at room temperature. The results indicate that photoluminescence was populated from the triplet state at both 77 K and room temperature. Moreover, the quantum yields of all complexes significantly increase in the polymer films, relative to those in solutions. The blue shifts, the increasing quantum yields, and the lengthening decayed times are associated with the increase of the matrix rigidity. With an increase of the matrix rigidity in PMMA, the freedom for changes of the molecular geometries upon MLCT excitation is decreased, and the distortions from tetrahedral-like ground state (d10) to flattened excited state (d9) can be suppressed. Thus, for the title complexes, higher emission quantum yields and long lifetimes are observed in solid states.45,61,62
DFT and TDDFT computations
The photophysical properties of P1–P5 were addressed by DFT and TDDFT in order to provide an assignment for the lowest energy absorption and emissive bands. For the sake of simplicity, only P1 is described as the observations are the same in all cases. All data for the four other compounds (P2–P5) are placed in the ESI† for convenience. The frontier MOs for P1 are presented in Fig. 3 and the relative atomic contributions are placed in Table 4. The two key features arising from these computations are firstly that the HOMO and LUMO levels are relatively well isolated from the other nearest MOs indicating that the mixing of the HOMO → LUMO process with other electronic transition is minimal. Second, the HOMOs of the complexes are mainly composed of Cu and PPh3, while LUMOs are mainly located on the central pyridine unit. The lowest energy electronic transition, HOMO → LUMO, is MLCT as the major component (>85%), and a minor contribution of LLCT (∼10–11%) is also computed, consistent with previously reported literature assignments. And the spin density distribution for P1 also shows the mixed MLCT and LLCT (Fig. 4).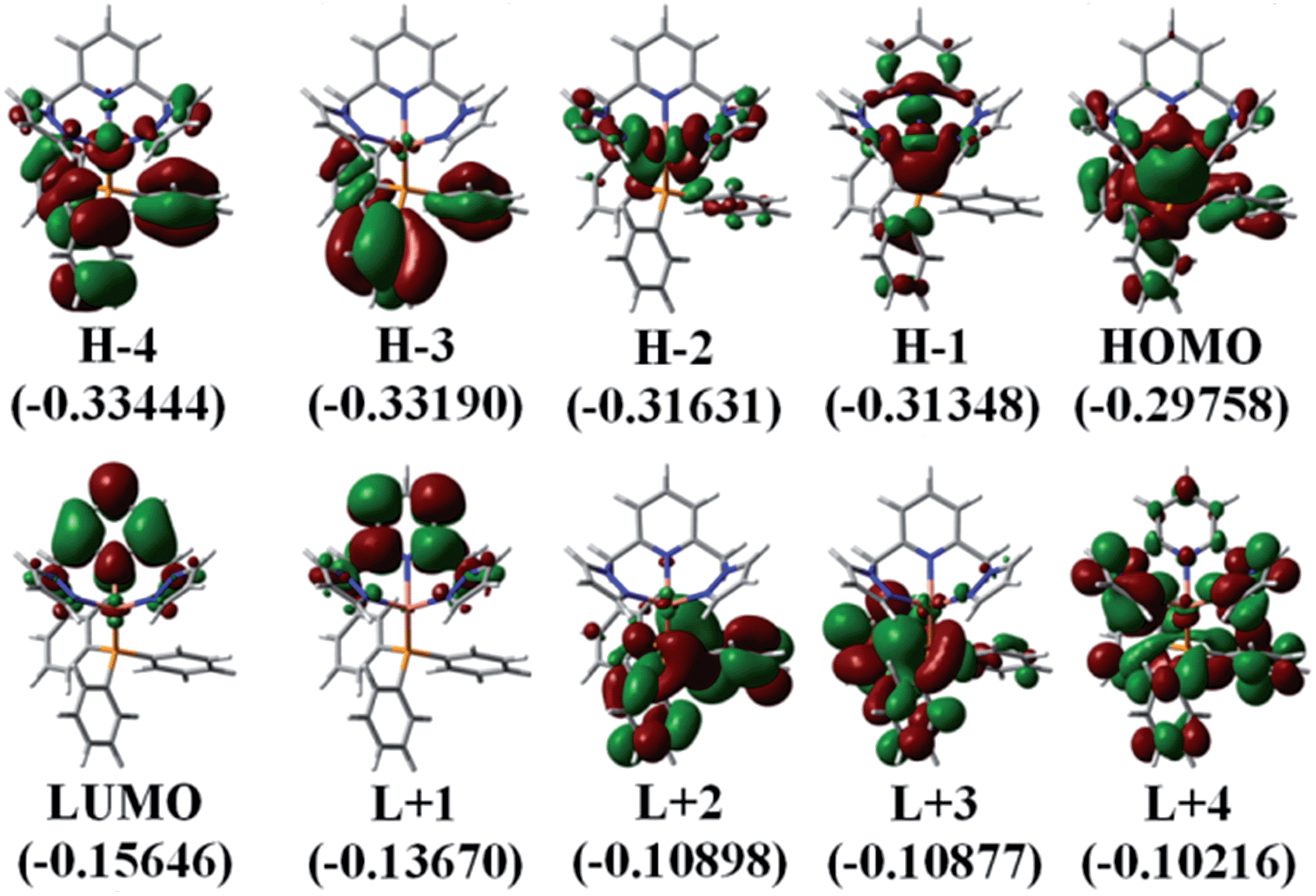 | ||
| Fig. 3 Representations of the frontier MOs for P1. The MO energies are in a.u. | ||
| Molecular fragment | H-4 | H-3 | H-2 | H-1 | HOMO | LUMO | L+1 | L+2 | L+3 | L+4 |
|---|---|---|---|---|---|---|---|---|---|---|
| Pyridine | 1.37 | 0.34 | 0.49 | 18.19 | 1.33 | 84.95 | 85.72 | 1.51 | 2.1 | 9.68 |
| Pyrazole groups | 7.46 | 0.43 | 23.15 | 7.54 | 10.33 | 11.43 | 13.1 | 0.95 | 1.74 | 36.96 |
| Copper | 19.59 | 1.18 | 71.44 | 69.13 | 42.2 | 1.75 | 0.62 | 0.98 | 1.51 | 6.42 |
| Triphenylphosphine | 71.57 | 98.04 | 4.92 | 5.14 | 46.15 | 1.87 | 0.56 | 96.56 | 94.65 | 46.95 |
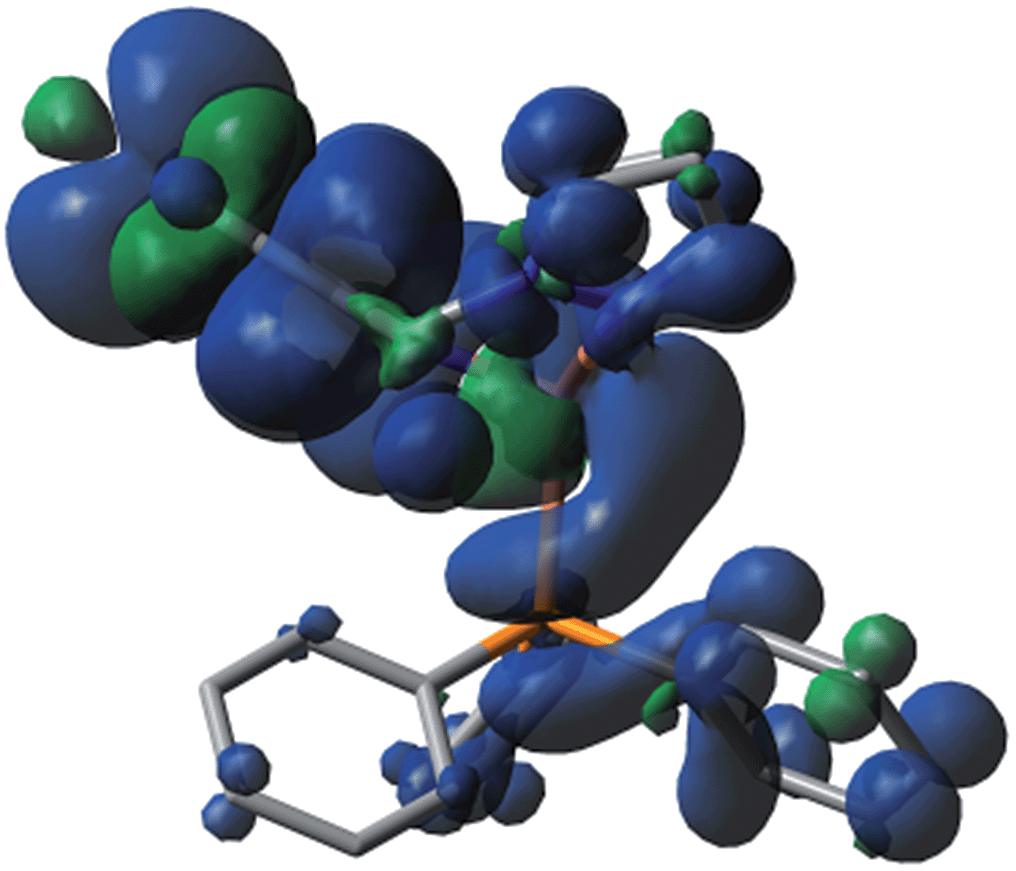 | ||
| Fig. 4 Side view image of the spin density distribution for P1. | ||
Using TDDFT computations, the position of the first 100 electronic transitions was obtained (where the first 10 are placed in Table 5), and used to generate a bar graph and a calculated spectrum by applying a thickness of 1000 cm−1 to each bar (Fig. 5). The calculated position of the pure electronic transition is 391.5 nm, which falls at the end of the long absorption tail of compound P1, exactly where the 0–0 peak would be expected. The two most intense calculated transitions are found in the vicinity of 275 and 280 nm, which match a strong shoulder at ∼270 nm in the experimental spectrum. The lowest energy transition is in all cases HOMO → LUMO almost exclusively.
| Wavelength (nm) | Osc. Strength | Major contributors (%) |
|---|---|---|
| 391.5 | 0.0368 | HOMO → LUMO (97%) |
| 371.0 | 0.0045 | H-1 → LUMO (97%) |
| 356.6 | 0.0024 | H-2 → LUMO (99%) |
| 333.9 | 0.0055 | HOMO → L+1 (98%) |
| 312.4 | 0.0001 | H-1 → L+1 (98%) |
| 308.0 | 0.0003 | H-6 → LUMO (96%) |
| 302.9 | 0.0091 | H-9 → LUMO (10%), H-7 → LUMO (39%), H-5 → LUMO (27%), H-4 → LUMO (13%) |
| 299.6 | 0.0004 | H-2 → L+1 (97%) |
| 284.9 | 0.0079 | H-9 → LUMO (10%), H-7 → LUMO (14%), H-4 → LUMO (39%), H-3 → LUMO (23%) |
| 282.7 | 0.0007 | H-4 → LUMO (11%), H-3 → LUMO (73%) |
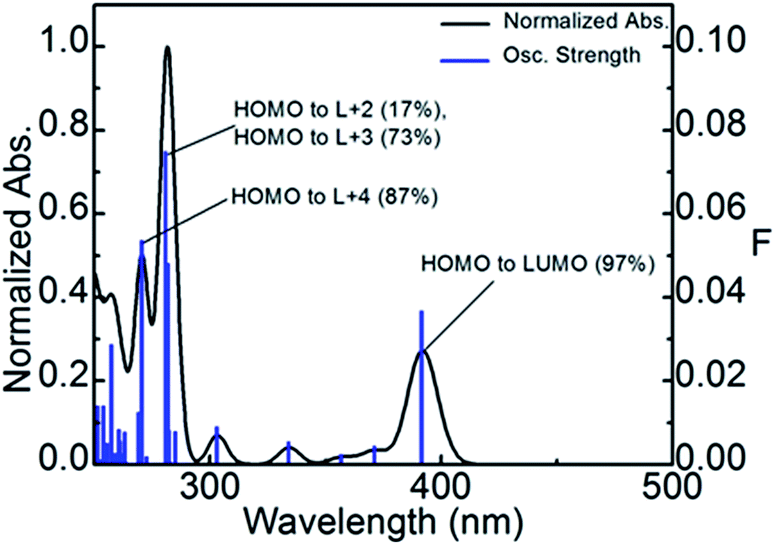 | ||
| Fig. 5 Bar graph representing the positions of the first hundred electronic transitions (blue). Calculated spectrum by applying a thickness of 1000 cm−1 to each bar (black). There is no vibronic component in these transitions. The assignments of the most intense transitions are provided. | ||
In order to strengthen the assignment of the MLCT, the geometric parameters about the Cu atom are listed in Table 6 for compound P1 in both the ground and the lowest energy triplet states. The excited state distortion is the most pronounced for the Cu–N bond length which shortens in the T1 state. This is fully consistent with the loss in Cu–N anti-bonding character of the HOMO when removing an electron from this MO to promote it into an MO where the Cu–N interactions are very weak. The large change in the P–Cu–N (pyridine) angle going from 122.0 to 105.5° (a change of 16.5°) indicates that the Cu environment is trying to adopt a flattened geometry, which is consistent with the Cu(II) nature of the metal in the excited state (according to the crystal field theory). Consequently, the N–Cu–N (pyridine) angles should become larger, as noted in Table 7. These computations support the assignment of these emissions to 3MLCT.
|
|
||||
|---|---|---|---|---|
| Geometric parameter | Singlet state | Triplet state | Difference | |
| Bond length (Å) | P–Cu | 2.2664 | 2.3626 | 0.0962 |
| N1–Cu | 2.1193 | 2.0786 | 0.0407 | |
| N2–Cu | 2.2237 | 1.9324 | 0.2913 | |
| N3–Cu | 2.116 | 2.0968 | 0.0192 | |
| Bond angle (°) | P–Cu–N1 | 120.8705 | 122.1726 | 1.3021 |
| P–Cu–N2 | 122.0231 | 105.5704 | 16.4527 | |
| P–Cu–N3 | 120.9005 | 117.9565 | 2.944 | |
| N1–Cu–N2 | 88.0958 | 97.1508 | 9.055 | |
| N2–Cu–N3 | 88.3386 | 97.7792 | 9.4406 | |
| N3–Cu–N1 | 108.1363 | 110.324 | 2.1877 | |
| Compound | T1–S0 gap | Predicted phosphorescence wavelength (nm) | |
|---|---|---|---|
| Hartree | EV | ||
| P1 | 0.10357 | 2.82 | 440 |
| P2 | 0.09678 | 2.63 | 471 |
| P3 | 0.09742 | 2.65 | 468 |
| P4 | 0.10065 | 2.74 | 453 |
| P5 | 0.10333 | 2.81 | 441 |
The position of the triplet state transition (S0 → T1) has been calculated by calculating the total energy of the optimized geometry of compound P1 in both the ground and T1 states. This energy difference is then translated into the nm scale and placed in Table 7. For all five compounds, the pure electronic transition is calculated to occur between 440 and 471 nm. Again this position falls pretty much at the beginning of the emission band. We find that the experimental emission maximum does not follow the calculated position of the 0–0 peaks. This is to be expected as the full-width-at-half-maximum (a feature associated with the excited state distortion) and the vibronic components associated with hot bands (for example: v′ = 1 → v = 0, where v and v′are the vibrational quantum numbers) are not necessarily the same for all five compounds. The important point is that the TDDFT computations provide information that is reasonable with the experimental observation.
Electrochemical properties
Cyclic voltammetry measurements were performed on complexes P1–P5 in CH2Cl2 solution, using the redox couple Cp2Fe/Cp2Fe+ as the internal reference (Fig. S3†). All the complexes show an irreversible oxidation peak with Epa values of ∼0.80, 0.83, 0.94, 0.98 and 0.78, respectively (Table 3), which can be assigned to the metal oxidation from Cu(I) to Cu(II). Oxidation on the metal is likely to result in a significant geometrical rearrangement, as divalent copper ions prefer a flattened geometry. The HOMO and the LUMO energy levels as listed in Table 3 were calculated from the onset potentials for oxidation together with their absorption spectra.Electroluminescent characteristics
Considering the highest photoluminescence quantum yields (PLQY) of P2 and P3 among all five Cu(I) complexes investigated (Table 3), the two complexes were selected for OLED fabrications with a structure of ITO/PEDOT:PSS (40 nm)/PYD2:Cu(I) complex (10 wt%, 30 nm)/DPEPO (50 nm)/LiF/Al. By doping P2 or P3 in a high triplet energy host, 2,6-dicarbazolo-1,5-pyridine (PYD2), with a concentration of 10 wt%, a 30 nm thick emitting layer was spin-coated on a PEDOT-treated ITO substrate. After the film was dried under vacuum, a 50 nm thick bis(2-(diphenylphosphino)phenyl)ether oxide (DPEPO) layer and a LiF/Al cathode were deposited in succession. The high triplet energy DPEPO layer was used here to transfer electrons and block excitons.63The EL spectra of both P2 and P3 are similar to their PL ones in PMMA films (Fig. 6a), revealing that EL emissions in this region come directly from the Cu(I) complexes. Although, the PL properties of these two complexes are very close, the P2-based OLED shows a better EL performance as compared to the P3-based OLED (Fig. 6b). The P2-based OLED turns on at 7 V, and achieves a maximum brightness of 850 cd m−2 and a maximum EQE of 1.0%. While the P3-based OLED turns on at 10 V, and achieves a maximum brightness of 210 cd m−2 and a maximum EQE of 0.24%. The relatively poor performance of P3-based OLEDs can be ascribed to the more sterically congested ligand of P3, which inhibits the redistribution of the counter ions in the EML under a bias. It is known that the accumulation of BF4− ions near the anode can significantly decrease the hole injection barrier, and therefore enhance the hole current density and even the charge balance of the device.44 This finding also indicates that the performance of these Cu(I) complex based PhOLEDs can be further improved by employing a hole injection layer with a better-matched HOMO level.
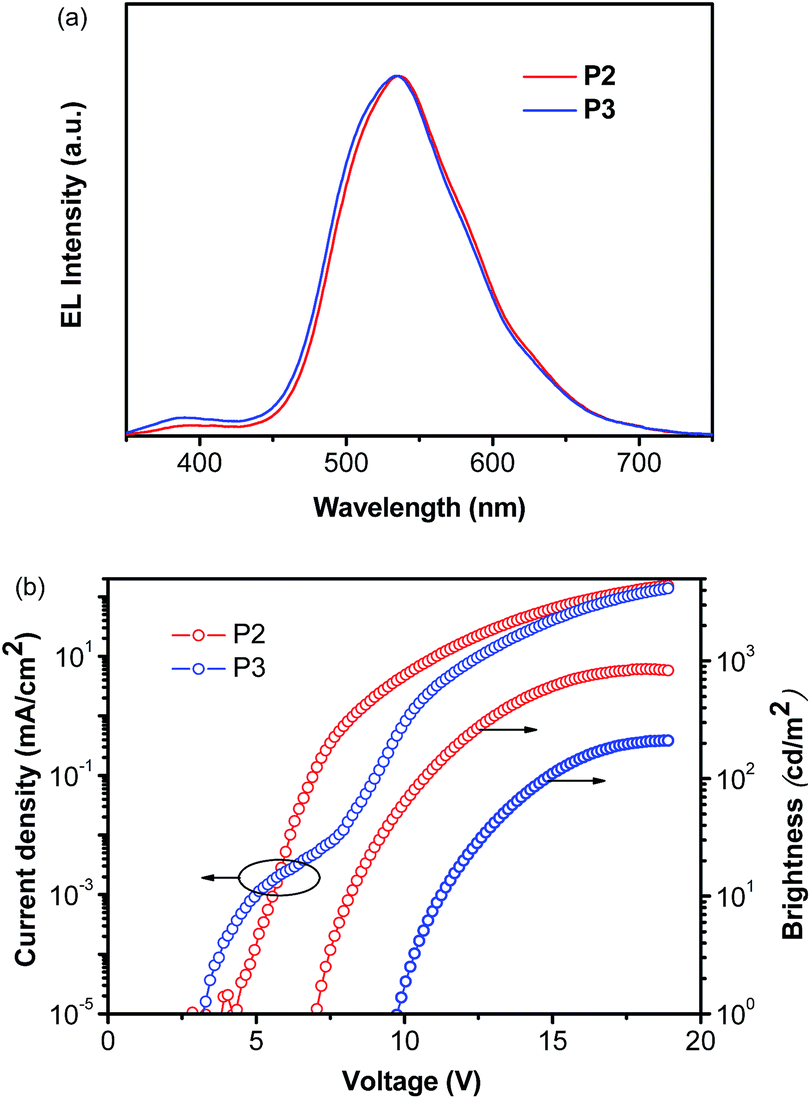 | ||
| Fig. 6 EL characteristics of P2 and P3-based OLEDs: (a) EL spectra measured at 10 mA cm−2; (b) current density–voltage–brightness characteristics. | ||
Conclusions
A series of mononuclear Cu(I) complexes P1–P5 have been prepared using different substituted pyridine–pyrazole ligands. Crystallographic analyses reveal that the Cu(I) complexes maintain the distorted tetragonal coordination geometry. With changes of the substituted groups on the pyrazole unit, the emissions arising from triplet excited states exhibit maxima that vary over the visible region from orange to blue. The combined effects of a sterically hindered dipyrazole ligand and a rigid environment result in an exceptional stability and a good emission quantum yield of the complexes in the solid state. The complexes exhibit long lived excited state emission and these excited states are assigned to metal-to-ligand charge transfer (3MLCT) states. Easy preparation and facile tunability of the emission energy of the Cu(I) complexes render them potentially useful light-emitting materials. Bright electroluminescence was observed from these Cu(I) complexes doped into a simple double-layer OLED, indicating that they are promising materials for EL applications.Experimental sections
General procedures
All reagents and solvents were of commercial reagent grade and used without further purification except where noted. Dichloromethane was distilled from calcium hydride. Toluene and tetrahydrofuran (THF) were distilled under nitrogen in the presence of sodium chips by using benzophenone ketyl as an indicator. The freshly distilled solvents for use in reactions were bubbled with nitrogen for at least 10 min to remove residual oxygen. The ligands (L1–L5) of bis(pyrazol-1-yl-methyl)-pyridine derivatives were prepared according to the literature method34 and the characterization data are given in ESI.† The copper complex [Cu(CH3CN)4]BF4 was synthesized by dropwise addition of aqueous HBF4 (50%) into a suspension of Cu2O in acetonitrile according to the literature procedures.64 The details of the instrumentation are provided in the ESI.†Synthesis of the Cu(I) complexes [CuL(PPh3)]BF4
To a dry and degassed CH2Cl2 (10 mL) solution of L (1 mmol) was added [Cu(CH3CN)4]BF4 (314 mg, 1 mmol). The mixture was kept stirring under nitrogen at room temperature. After 2 h, PPh3 (262 mg, 1 mmol) was added and the yellow solution was changed to colorless, which was then allowed to stir for another 2 h. A white solid was collected after the addition of dry and degassed hexane (45 mL), washed with dry diethyl ether and recrystallized from dichloromethane–hexane (1:3, v/v) solution as white crystals.
P1: (yield 90%). 1H NMR (400 MHz, CDCl3): δ 5.45 (m, 4H, CH2), 6.28–6.32 (t, 2H, J(HH) = 2.2 Hz, H4-pz), 7.18–7.23 (m, 6H, o-Ph), 7.27–7.31 (m, 6H, m-Ph), 7.38–7.42 (m, 3H, p-Ph), 7.56 (d, 2H, J(HH) = 8.0 Hz, H3-pz), 7.63 (d, 2H, J(HH) = 2.0 Hz, H5-pz) and 7.92–7.96 (m, 3H, H-py) ppm. 31P NMR (400 MHz, CDCl3) δ −1.39 ppm. Anal. calcd (found) for C31H28BCuF4N5P (%): C 57.11 (56.73), H 4.33 (4.08), N 10.74 (10.60). MALDI-TOF MS: calcd for [M-PPh3]+, 302.0536; found: 302.2547.
P2: (yield 90%). 1H NMR (400 MHz, CDCl3): δ 2.17 (s, 6H, Me5-pz), 2.36 (s, 6H, Me3-pz), 5.01 (s, 4H, CH2), 5.96 (s, 2H, H4-pz), 7.27–7.36 (m, 12H, o,m-Ph), 7.43–7.46 (m, 3H, p-Ph), 7.64 (d, 2H, J(HH) = 8.0 Hz, H3-py), 8.03 (t, 1H, J(HH) = 8.0 Hz, H4-py) ppm. 31P NMR (400 MHz, CDCl3) δ −5.90 ppm. Anal. calcd (found) for C35H36BCuF4N5P (%): C 59.37 (59.55), H 5.12 (5.12), N 9.89 (10.09). MALDI-TOF MS: calcd for [M-PPh3]+, 358.1162; found: 358.6413.
P3: (yield 90%). 1H NMR (400 MHz, CDCl3): δ 1.12 (d, 12H, J = 6.8 Hz, Me5-pz), 1.28 (d, 12H, J = 6.8 Hz, Me3-pz), 2.80–2.84 (m, 2H, CH-5-pz), 2.98–3.02 (m, 2H, CH-3-pz), 4.97 (s, 4H, CH2), 5.97 (s, 2H, H4-pz), 7.18–7.22 (m, 6H, o-ph), 7.27–7.31 (m, 6H, m-Ph), 7.37–7.40 (m, 3H, p-Ph), 7.75 (d, 2H, J = 8.0 Hz, H3-py) and 8.02 (t, 1H, J = 8.0 Hz, H4-py) ppm. 31P NMR (400 MHz, CDCl3) δ −6.83 ppm. Anal. calcd (found) for C43H52BCuF4N5P (%): C 62.97 (61.68), H 6.39 (6.33), N 8.54 (7.96). MALDI-TOF MS: calcd for [M-PPh3]+, 470.2312; found: 470.3458.
P4: (yield 90%). 1H NMR (400 MHz, CDCl3): δ 5.19 (s, 4H, CH2), 6.80 (s, 2H, H4-pz), 7.21 (m, 15H, p-Ph), 7.40–7.58 (m, 20H, Ph-pz), 7.67 (d, 2H, J(HH) = 8.0 Hz, H3-py) and 8.36 (t, 1H, J(HH) = 8.0 Hz, H4-py) ppm. 31P NMR (400 MHz, CDCl3) δ −5.52 ppm. Anal. calcd (found) for C43H52BCuF4N5P (%): C 62.97 (61.66), H 6.39 (6.54), N 8.54 (8.44). Anal. calcd (found) for C55H44BCuF4N5P (%): C 69.08 (68.43), H 4.64 (4.83), N 7.32 (7.21). MALDI-TOF MS: calcd for [M-PPh3]+, 606.1702; found: 606.0880.
P5: (yield 90%). 1H NMR (400 MHz, CDCl3): δ 2.45 (s, 6H, Me3-pz), 5.30 (s, 4H, CH2), 6.40 (s, 2H, H4-pz), 7.31–7.44 (m, 15H, P-Ph), 7.85 (d, 2H, J(HH) = 8.0 Hz, H3-py) and 8.01 (t, 1H, J(HH) = 8.0 Hz, H4-py) ppm. 31P NMR (400 MHz, CDCl3) δ −3.14 ppm. Anal. calcd (found) for C35H30BCuF10N5P (%): C 51.52 (51.43), H 3.71 (3.63), N 8.58 (8.21). MALDI-TOF MS: calcd for [M-PPh3]+, 466.0542; found: 466.3701.
X-ray crystallography
Single crystals suitable for crystal structure analysis were obtained by slow evaporation of a dichloromethane–hexane solution of the complex at room temperature. Data were measured using Mo-Kα radiation on a Bruker SMART 1000 CCD diffractometer. Data collection at 296 K and reduction were performed using SMART and SAINT software.65 Absorption correction was applied using the multi-scan method (SADABS).66 The crystal structure of P1–P5 was solved by direct methods and refined by full matrix least-squares on F2 using the SHELXTL program package.67Acknowledgements
We thank the National Natural Science Foundation of China (NSFC) (Grant 91222201 and 51373145), Hong Kong Research Grants Council (Grant no. HKBU202210, HKBU202811, HKBU203011, HKBU203312 and HKUST2/CRF/10), and Hong Kong Baptist University (FRG2/12-13/050 and FRG2/11-12/156) for financial support. W.-K.W. and W.-Y.W. also acknowledge a grant from Areas of Excellence Scheme, University Grants Committee, Hong Kong (Project no. AoE/P-03/08). W.-Y.W. thanks the financial support from the National Basic Research Program of China (973 Program) (2013CB834702). PDH thanks the Natural Sciences and Engineering Research Council of Canada (NSERC) for support. L.L. thanks Natural Science Foundation of Hubei Province of China (2013CFA087). We also thank Dr Maggie M. Y. Chan at the University of Hong Kong, Prof. K.-W. Cheah and Dr H.-L. Tam at the Hong Kong Baptist University for their assistances in PL measurements.Notes and references
- E. Rossi, A. Colombo, C. Dragonetti, D. Roberto, F. Demartin, M. Cocchi, P. Brulatti, V. Fattori and J. A. G. Williams, Chem. Commun., 2012, 48, 3182 RSC.
- A. F. Rausch, L. Murphy, J. A. G. Williams and H. Yersin, Inorg. Chem., 2012, 51, 312 CrossRef CAS PubMed.
- W. Mroz, C. Botta, U. Giovanella, E. Rossi, A. Colombo, C. Dragonetti, D. Roberto, R. Ugo, A. Valore and J. A. G. Williams, J. Mater. Chem., 2011, 21, 8653 RSC.
- K. Li, X. G. Guan, C. W. Ma, W. Lu, Y. Chen and C. M. Che, Chem. Commun., 2011, 47, 9075 RSC.
- N. Komiya, M. Okada, K. Fukumoto, D. Jomori and T. Naota, J. Am. Chem. Soc., 2011, 133, 6493 CrossRef CAS PubMed.
- Y. C. Zhu, L. Zhou, H. Y. Li, Q. L. Xu, M. Y. Teng, Y. X. Zheng, J. L. Zuo, H. J. Zhang and X. Z. You, Adv. Mater., 2011, 23, 4041 CrossRef CAS PubMed.
- Q. Zhao, C. H. Huang and F. Y. Li, Chem. Soc. Rev., 2011, 40, 2508 RSC.
- R. J. Wang, D. Liu, H. C. Ren, T. Zhang, X. Z. Wang and J. Y. Li, J. Mater. Chem., 2011, 21, 15494 RSC.
- S. H. Chang, C. F. Chang, J. L. Liao, Y. Chi, D. Y. Zhou, L. S. Liao, T. Y. Jiang, T. P. Chou, E. Y. Li, G. H. Lee, T. Y. Kuo and P. T. Chou, Inorg. Chem., 2013, 52, 5867 CrossRef CAS PubMed.
- B. S. Du, J. L. Liao, M. H. Huang, C. H. Lin, H. W. Lin, Y. Chi, H. A. Pan, G. L. Fan, K. T. Wong, G. H. Lee and P. T. Chou, Adv. Funct. Mater., 2012, 22, 3491 CrossRef CAS.
- Y. L. Tung, S. W. Lee, Y. Chi, L. S. Chen, C. F. Shu, F. I. Wu, A. J. Carty, P. T. Chou, S. M. Peng and G. M. Lee, Adv. Mater., 2005, 17, 1059 CrossRef CAS.
- T. Yu, D. P. K. Tsang, V. K. M. Au, W. H. Lam, M. Y. Chan and V. W. W. Yam, Chem.–Eur. J., 2013, 19, 13418 CrossRef CAS PubMed.
- X. Li, D. Y. Zhang, G. H. Lu, G. Y. Xiao, H. J. Chi, Y. Dong, Z. Q. Zhang and Z. Z. Hu, J. Photochem. Photobiol., A, 2012, 241, 1 CrossRef CAS.
- M. Mauro, C. H. Yang, C. Y. Shin, M. Panigati, C. H. Chang, G. D'Alfonso and L. De Cola, Adv. Mater., 2012, 24, 2054 CrossRef CAS PubMed.
- T. T. Zhang, J. F. Jia, Y. Ren and H. S. Wu, J. Phys. Chem. A, 2011, 115, 3174 CrossRef CAS PubMed.
- X. Li, D. Y. Zhang, W. L. Li, B. Chu, L. L. Han, J. Z. Zhu, Z. S. Su, D. F. Bi, D. Wang, D. F. Yang and Y. R. Chen, Appl. Phys. Lett., 2008, 92, 083302 CrossRef.
- G. B. Che, Z. S. Su, W. L. Li, B. Chu, M. T. Li, Z. Z. Hu and Z. Q. Zhang, Appl. Phys. Lett., 2006, 89, 103511 CrossRef.
- Q. S. Zhang, Q. G. Zhou, Y. X. Cheng, L. X. Wang, D. G. Ma, X. B. Jing and F. S. Wang, Adv. Mater., 2004, 16, 432 CrossRef CAS.
- S. Ranjan, S. Y. Lin, K. C. Hwang, Y. Chi, W. L. Ching, C. S. Liu, Y. T. Tao, C. H. Chien, S. M. Peng and G. H. Lee, Inorg. Chem., 2003, 42, 1248 CrossRef CAS PubMed.
- D. R. McMillin and K. M. McNett, Chem. Rev., 1998, 98, 1201 CrossRef PubMed.
- M. Osawa, Chem. Commun., 2014, 50, 1801 RSC.
- K. A. Vinogradova, V. F. Plyusnin, A. S. Kupryakov, M. I. Rakhmanova, N. V. Pervukhina, D. Y. Naumov, L. A. Sheludyakova, E. B. Nikolaenkova, V. P. Krivopalovd and M. B. Bushuev, Dalton Trans., 2014, 43, 2953 RSC.
- D. M. Zink, D. Volz, T. Baumann, M. Mydlak, H. Flügge, J. Friedrichs, M. Nieger and S. Bräse, Chem. Mater., 2013, 25, 4471 CrossRef CAS.
- M. Wallesch, D. Volz, D. M. Zink, U. Schepers, M. Nieger, T. Baumann and S. Bräse, Chem.–Eur. J., 2014, 20, 6578 CrossRef CAS PubMed.
- A. Kaeser, M. Mohankumar, J. Mohanraj, F. Monti, M. Holler, J. Cid, O. Moudam, I. Nierengarten, L. Karmazin-Brelot, C. Duhayon, B. Delavaux-Nicot, N. Armaroli and J. Nierengarten, Inorg. Chem., 2013, 52, 12140 CrossRef CAS PubMed.
- Q. S. Zhang, B. Li, S. P. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326 CrossRef CAS.
- P. Manikandan, B. Varghese and P. T. Manoharan, J. Chem. Soc., Dalton Trans., 1996, 371 RSC.
- P. Manikandan, M. Subramoni, B. Varghese and P. T. Manoharan, J. Chem. Soc., Dalton Trans., 1998, 3219 RSC.
- T. Aida, A. Kishimura and T. Yamashita, J. Am. Chem. Soc., 2005, 127, 179 CrossRef PubMed.
- W. F. Fu, Y. Chen, J. L. Li, G. S. M. Tong, W. Lu, S. W. Lai and C. M. Che, Chem. Sci., 2011, 2, 1509 RSC.
- Z. W. Mao, Q. H. Wei, G. Q. Yin, L. Y. Zhang, L. X. Shi and Z. N. Chen, Inorg. Chem., 2004, 43, 3484 CrossRef PubMed.
- M. A. Omary, M. A. Rawashdeh-Omary, M. W. A. Gonser, O. Elbjeirami, T. Grimes and T. R. Cundari, Inorg. Chem., 2005, 44, 8200 CrossRef CAS PubMed.
- S. Shinkai, M. Shirakawa, N. Fujita, T. Tani, K. Kaneko, M. Ojima, A. Fujii and M. Ozaki, Chem.–Eur. J., 2007, 13, 4155 CrossRef PubMed.
- M. E. Thompson, V. A. Krylova, P. I. Djurovich and M. T. Whited, Chem. Commun., 2010, 46, 6696 RSC.
- I. I. Vorontsov, A. Y. Kovalevsky, Y. S. Chen, T. Graber, M. Gembicky, I. V. Novozhilova, M. A. Omary and P. Coppens, Phys. Rev. Lett., 2005, 94, 193003 CrossRef PubMed.
- L. X. Wang, J. H. Min, Q. S. Zhang, W. Sun and Y. X. Cheng, Dalton Trans., 2011, 40, 686 RSC.
- S. N. Wang, W. L. Jia, T. McCormick, Y. Tao and J. P. Lu, Inorg. Chem., 2005, 44, 5706 CrossRef PubMed.
- S. N. Wang, T. McCormick and W. L. Jia, Inorg. Chem., 2006, 45, 147 CrossRef PubMed.
- D. T. Song, W. L. Jia, G. Wu and S. N. Wang, Dalton Trans., 2005, 3, 433 Search PubMed.
- I. Andres-Tome, J. Fyson, F. B. Dias, A. P. Monkman, G. Iacobellis and P. Coppo, Dalton Trans., 2012, 41, 8669 RSC.
- S. Igawa, M. Hashimoto, I. Kawata, M. Yashima, M. Hoshino and M. Osawa, J. Mater. Chem. C., 2013, 1, 542 RSC.
- X. L. Chen, R. M. Yu, Q. K. Zhang, L. J. Zhou, C. Y. Wu, Q. Zhang and C. Z. Lu, Chem. Mater., 2013, 25, 3910 CrossRef CAS.
- X. L. Xin, M. Chen, Y. B. Ai, F. L. Yang, X. L. Li and F. Y. Li, Inorg. Chem., 2014, 53, 2922 CrossRef CAS PubMed.
- Q. S. Zhang, Q. G. Zhou, Y. X. Cheng, L. X. Wang, D. G. Ma, X. B. Jing and F. S. Wang, Adv. Funct. Mater., 2006, 16, 1203 CrossRef CAS.
- Q. S. Zhang, J. Q. Ding, Y. X. Cheng, L. X. Wang, Z. Y. Xie, X. B. Jing and F. S. Wang, Adv. Funct. Mater., 2007, 17, 2983 CrossRef CAS.
- Y. Chi, C. W. Hsu, C. C. Lin, M. W. Chung, G. H. Lee, P. T. Chou, C. H. Chang and P. Y. Chen, J. Am. Chem. Soc., 2011, 133, 12085 CrossRef PubMed.
- M. Hashimoto, S. Igawa, M. Yashima, I. Kawata, M. Hoshino and M. Osawa, J. Am. Chem. Soc., 2011, 133, 10348 CrossRef CAS PubMed.
- Z. X. Wang and H. L. Qin, Green Chem., 2004, 6, 90 RSC.
- D. A. H. Watson and P. J. Steel, Inorg. Chim. Acta, 1987, 130, 167 CrossRef.
- S. M. Kuang, D. G. Cuttell, D. R. McMillin, P. E. Fanwick and R. A. Walton, Inorg. Chem., 2002, 41, 3313 CrossRef CAS PubMed.
- P. J. Burke, K. Henrick and D. R. McMillin, Inorg. Chem., 1982, 21, 1881 CrossRef CAS.
- T. McCormick, W. L. Jia and S. N. Wang, Inorg. Chem., 2006, 45, 147 CrossRef CAS PubMed.
- L. Yang, J. K. Feng, A. M. Ren, M. Zhang, Y. G. Ma and X. D. Liu, Eur. J. Inorg. Chem., 2005, 10, 1867 CrossRef.
- I. Andres-Tome, J. Fyson, F. B. Dias, A. P. Monkman, G. Iacobellis and P. Coppo, Dalton Trans., 2012, 41, 8669 RSC.
- Y. Pellegrin, M. Sandroni, E. Blart, A. Planchat, M. Evain, N. C. Bera, M. Kayanuma, M. Sliwa, M. Rebarz, O. Poizat, C. Daniel and F. Odobel, Inorg. Chem., 2011, 50, 11309 CrossRef CAS PubMed.
- M. T. Miller, P. K. Gantzel and T. B. Karpishin, J. Am. Chem. Soc., 1999, 121, 4292 CrossRef CAS.
- M. Z. Zgierski, J. Chem. Phys., 2003, 118, 4045 CrossRef CAS.
- Z. A. Siddique, Y. Yamamoto, T. Ohno and K. Nozaki, Inorg. Chem., 2003, 42, 6366 CrossRef CAS PubMed.
- L. X. Chen, G. B. Shaw, I. Novozhilova, T. Liu, G. Jennings, K. Attenkofer, G. J. Meyer and P. Coppens, J. Am. Chem. Soc., 2003, 125, 7022 CrossRef CAS PubMed.
- L. Porres, A. Holland, L. O. Palsson, A. P. Monkman, C. Kemp and A. Beeby, J. Fluoresc., 2006, 16, 267 CrossRef CAS PubMed.
- R. D. Costa, E. Orti, H. J. Bolink, S. Graber, S. Schaffner, M. Neuburger, C. E. Housecroft and E. C. Constable, Adv. Funct. Mater., 2009, 19, 3456 CrossRef CAS.
- L. Qin, Q. S. Zhang, W. Sun, J. Y. Wang, C. Z. Lu, Y. X. Cheng and L. X. Wang, Dalton Trans., 2009, 43, 9388 RSC.
- Q. S. Zhang, T. Komino, S. P. Huang, S. Matsunami, K. Goushi and C. Adachi, Adv. Funct. Mater., 2012, 22, 2327 CrossRef CAS.
- G. J. Gubas, Inorg. Synth., 1990, 28, 68 CrossRef.
- SMART 5.0 and SAINT 4.0 for Windows NT, Area Detector Control and Integration Software, Bruker Analytical X-Ray Systems Inc., Madison, WI, 1998 Search PubMed.
- G. M. Sheldrick, SADABS: Program for Empirical Absorption Correction of Area Detector Data, University of Göttingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, SHELXTL 5.1 for Program for Windows NT: Structure Determination Software Programs, Bruker Analytical X-Ray Systems Inc., Madison, WI, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details, complete results of density functional theory (DFT) and time-dependent DFT (TDDFT) calculations, figures and CIF files with crystallographic data for the structures of P1–P4. CCDC 985983–985986. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4tc01885k |
| This journal is © The Royal Society of Chemistry 2015 |