Polypyrrole doped epoxy resin nanocomposites with enhanced mechanical properties and reduced flammability†
Received
4th September 2014
, Accepted 17th October 2014
First published on 17th October 2014
Abstract
For the liquid epoxy nanosuspensions with both fibril and spherical polypyrrole (PPy) nanostructures, a stronger PPy nanofibers/epoxy interaction and more temperature stable behavior with a lower flow activation energy of nanosuspensions with nanofibers (54.34 kJ mol−1) than that with nanospheres (71.15 kJ mol−1) were revealed by rheological studies. As well as the common enhancing mechanism of limiting crack propagation in the polymer matrix, the nanofibers further initiated the shear bands in the epoxy resin to give a higher tensile strength (90.36 MPa) than that of pure epoxy (70.03 MPa) and even that of the epoxy nanocomposites with nanospheres (84.53 MPa). With a larger specific surface area, the nanofibers rather than nanospheres were observed to reduce the flammability of epoxy more efficiently by assisting more char formation of the epoxy resin. The hydroxyl groups formed between the protons of the doped acid in the PPy nanofillers and the epoxy broke the conjugate structure of PPy, leading to a higher bandgap in the nanocomposites (Eg1 = 3.08 eV for 1.0 wt% PPy nanofibers) than that of pure nanofillers (1.8 eV for PPy nanofibers and 1.2 eV for PPy nanospheres). Due to the high aspect ratio, the PPy nanofibers could form the conductive path more easily than the PPy nanospheres to provide a lower percolation threshold value. The real permittivity was observed to increase with increasing the PPy nanofiller loading, and the enhanced permittivity was interpreted by the interfacial polarization.
Introduction
The development of polymer nanocomposites (PNCs) has attracted more interest due to the introduced versatile physicochemical properties to the otherwise inert hosting polymers such as thermal stability,1–3 magnetic,4–7 optical8–10 and dielectric properties.11–14 As one of the most important engineered polymers, epoxy resin has drawn attention due to its wide applications including structural materials, tissue substitutes15 and anticorrosive coatings.16 However, the applications of epoxy resin are largely limited by the problem of flammability and the increasing industrial deployments demanded miniaturization of the multifunctional nanocomposites. For example, the dielectric properties are an important aspect for the PNCs due to their potential application in lightweight capacitor field. By introducing inorganic fillers including ceramic17 and metal18 particles, epoxy PNCs with high dielectric constant can be used as embedded capacitors.17 However, the addition of inorganic fillers could reduce their mechanical properties19–22 due to the poor interfacial adhesion between the inorganic fillers and the hosting polymer.23 The decreased mechanical properties also appear when traditional phosphorus-based flame retardants are applied to improve the fire retardancy performance of the epoxy PNCs.24,25 The properties of epoxy PNCs were summarized in Table 1. Carbon nanotubes (CNTs) are the most used nanofillers in enhancing the mechanical properties of epoxy resin; as summarized in Table 1, the PNCs show high tensile strength and Young's modulus. However, it is worth noting that the CNTs used to fabricate the PNCs were surface pre-treated, which will raise the complexity of manufacturing epoxy PNCs. The introduced functional groups such as NH2– and COOH– during the surface pre-treatment26–28 can react with epoxy resin and in turn improve the interaction between the CNTs and epoxy resin.
Table 1 The properties of epoxy PNCs
| Composites |
Dielectric constant |
Char residue (%) |
Young's modulus (GPa) |
Tensile strength (MPa) |
Elongation (%) |
Ref. |
| 3% polyhedral oligomeric silsesquioxane |
3.3 |
Not reported |
Not reported |
71.0 |
Not reported |
18
|
| 5% water-treated Al2O3 |
11–12 |
Not reported |
2.29 |
61.2 |
Not reported |
19
|
| 15% barium strontium titanate (BST) |
5.0 |
Not reported |
2.2 |
15–16 |
Not reported |
20
|
| 0.2% MWCNTs/44% piezoelectric lead zirconate titanate |
11 |
Not reported |
Not reported |
35 |
Not reported |
21
|
| 7.79% bis-phenoxy (3-hydroxy) phenyl phosphine oxide |
Not reported |
52.1 |
3.4 |
14.6 |
0.6 |
23
|
| 2.5% poly(DOPO substituted dihydroxyl phenyl pentaerythritol diphosphonate) (PFR) |
Not reported |
3.6 |
Not reported |
19.75 |
1.6 |
24
|
| Polyamidoamine generation-0 (PAMAM-0) dendrimer functionalized SWCNTs |
Not reported |
Not reported |
3.49 |
74.1 |
3.32 |
25
|
| Pristine SWCNTs |
Not reported |
Not reported |
3.27 |
74.1 |
4.05 |
25
|
| 1% F–SWNT–COOH (fluorinated acid-treated) |
Not reported |
Not reported |
2.63 |
95.0 |
Not reported |
26
|
| 1% SWNTs–R–NH2 (amino-terminated amide derivative) |
Not reported |
Not reported |
2.65 |
104 |
8.5 |
27
|
Among all the conjugated polymer nanofillers, PPy is more attractive owing to its outstanding physicochemical properties such as high electrical conductivity,29 good environmental stability and controllable doping levels,30 and its wide applications including cell adhesion,31 biosensors, counter electrodes,32 Cr(VI) removal,33 electromagnetic interference shielding materials34 and membranes for gas separation.35 PPy is one of the most widely used conductive nanofillers for fabricating conductive epoxy polymer nanocomposites (PNCs). Different dispersion methods including wing mixer, turax and ultrasonic stirring have been reported to increase the conductivity of PPy–epoxy composites and the highest conductivity value (2 × 10−5 S cm−1) was observed with 11.0 wt% PPy performed by turax mixing.36 Besides the increased conductivity, the composites exhibited other unique properties. For instance, the PPy/diglycidyl ether of bisphenol A (DGEBA) composites were used as an anticorrosion coating for mild steel; the minimum corrosion rate of PPy/DGEBA could reach 3 × 10−3 g m2 h when exposed to 5.0% HCl and 3.5% NaCl.37
For epoxy resin, three types of flame retardants including halogenated flame retardant, phosphorus-based flame retardants and layer double hydroxide flame retardants were mostly used. The utilization of halogenated flame retardant caused problems by releasing poisonous and corrosive smoke.25 Due to poor compatibility, the phosphorus-based flame retardants and layer double hydroxide flame retardants38 led to decreased mechanical properties, such as tensile strength and elongation. PPy was used as an anti-flammable coating for PET. For samples coated with PPy, no hole was observed in the surface of the samples after direct contact with fire, indicating that the PPy coating prevented the transferring of flame and heat to the polymer matrix.39 PPy contains a nitrogen component and it is known that the nitrogen-based flame retardants can enhance the char formation and promote the generation of carbonaceous char in the condensed phase and further slow the degradation of the matrix.40 Compared with CNTs, PPy nanofillers with amino groups can directly react with epoxy resin to enhance the mechanical properties, and the morphology (surface area) of PPy nanofillers becomes a dominant factor that affects the interaction between the nanofillers and epoxy matrix. However, most studies of the PPy–epoxy PNCs focus on their applications in conductive adhesive41 and anticorrosive coating42 fields; the mechanical properties, dielectric properties and flammability of the PNCs with the aid of pure PPy nanostructures are rarely reported.
In this paper, two different lab-synthesized PPy nanostructures, nanofiber and nanosphere, were applied as nanofillers in preparing epoxy resin nanocomposites to enhance the mechanical and dielectric properties and the fire retardancy performance. The effects of PPy loading and morphology on the rheological behavior of the uncured suspension, electrical conductivity, thermal stability, and thermo-mechanical properties, including storage and loss moduli as well as glass transition temperature, for the cured solid samples were systematically studied. In addition, the influence of PPy nanofillers on the properties of the PNCs was theoretically studied considering the interactions between the amine groups of the PPy nanofillers and the epoxy group of the polymer resin.
Experimental
Materials
The epoxy resin Epon 862 (bisphenol F epoxy) and EpiCure W curing agent were purchased from Miller-Stephenson Chemical Company Inc. The molecular structures of these chemicals are shown in Scheme 1, in the ESI.† Pyrrole (C4H5N), ammonium persulfate (APS, 98 wt%, (NH4)2S2O8) and p-toluene sulfonic acid (PTSA, 98.5 wt%, C7H8O3S) were purchased from Sigma Aldrich. The hydrochloric acid (HCl 36.5–38.0 wt%), ammonium metavanadate (NH4VO3, 99.9 wt%), Dowex® resin 50WX8 50-100 and acetone (CH3COCH3, 99.5 wt%) were purchased from VWR International LLC. All the chemicals were used as-received without any further treatment.
Preparation of PPy nanospheres (NSs)
The molar ratio used in this method was pyrrole![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :APS:PTSA = 6:3:5. For solution one, PTSA (60 mmol) and APS (36 mmol) were dissolved in deionized water (400 mL) in a beaker and ultrasonicated in an ice-water bath for one hour. Solution two was pyrrole (72 mmol) dissolved in deionized water (100 mL). Then solution two was added into solution one and the mixture was sonicated for an additional one hour in an ice-water bath for polymerization. Finally, the product was vacuum filtered and washed with ethanol and deionized water to remove any possible oligomers, excess acid and organic solvent. The obtained powders were dried at 50 °C overnight.
:APS:PTSA = 6:3:5. For solution one, PTSA (60 mmol) and APS (36 mmol) were dissolved in deionized water (400 mL) in a beaker and ultrasonicated in an ice-water bath for one hour. Solution two was pyrrole (72 mmol) dissolved in deionized water (100 mL). Then solution two was added into solution one and the mixture was sonicated for an additional one hour in an ice-water bath for polymerization. Finally, the product was vacuum filtered and washed with ethanol and deionized water to remove any possible oligomers, excess acid and organic solvent. The obtained powders were dried at 50 °C overnight.
Preparation of PPy nanofibers (NFs)
1 mL V2O5 sol–gel (V2O5 sol–gel was prepared by 0.35–0.5 M ammonium metavanadate solution run through an ion-exchange column containing Dowex 50W-X8 resin, which had been thoroughly washed with HCl and deionized water.) was introduced into 60 mL 1 M HCl at room temperature, under magnetic stirring. One mL pyrrole monomer was added into the mixture, 1.15 g APS was then added as the oxidant to initiate the formation of PPy. After 2 hours of polymerization under ambient conditions with magnetic stirring, bulk PPy nanofibers were finally vacuum filtered and washed with 1 M HCl and acetone, respectively. The V2O5 used in the experiment could be slowly converted to soluble VO2+ salts in aq. 1 M HCl and was also washed away. Finally, the obtained black powder precipitates were dried at 80 °C overnight.
Preparation of PPy/epoxy nanocomposites
Epoxy resin suspensions with 1.0, 5.0, 10.0 and 20.0 wt% of PPy NFs and NSs were prepared. Firstly PPy nanostructures were immersed in epoxy resin (the total weight of epoxy resin and curing agent was fixed to 40.0 g and the PPy loading in epoxy resin was controlled by varying the weight of PPy nanostructures) without any disturbance overnight so that the resin could wet the nanostructures completely. The mixture was then mechanically stirred (Heidolph, RZR 2041) at a speed of 600 rpm for one hour at room temperature. Curing agent W was added into the above suspension with a weight ratio of monomer/curing agent: 100/26.5 as recommended by the company and the solution was stirred at high-speed (600 rpm) for another one hour at room temperature. In order to remove bubbles in the solution and to prevent the settlement of PANI nanofillers during the curing process, low-speed (200 rpm) mechanical stirring was conducted at 70 °C for 3–4 hours in a water bath. Finally, the solution was poured into silicon rubber molds, cured at 120 °C for 5 hours, and then cooled down naturally to room temperature.
Characterization
Specific surface area characterization of PPy nanostructures.
Brunauer–Emmett–Teller (BET) theory was used to measure the specific surface area of the PPy nanofibers and nanospheres. BET adsorption and desorption isotherms were obtained using a surface area analyzer (NOVA 2200e, Quantachrome). The solid sample to be analyzed was weighed and placed inside the sample holder cell of a known volume. Prior to each measurement, samples were degassed at 60 °C for 24 h under high vacuum (<0.01 mbar). The refrigerant used was liquid nitrogen placed in a vacuum Dewar at about 77.4 K and the carrier gas was N2 (ultra high purity grade, Airgas).
Rheological behaviors of liquid epoxy resin nanosuspensions.
The rheological behaviors of the epoxy resin nanocomposite suspensions were investigated with a rheometer (AR 2000ex, TA Instrumental Company) at shear rates ranging from 1 to 1000 s−1 at 25, 50 and 70 °C (the samples were mixtures of only epoxy monomers and nanofillers). A series of measurements were performed in a cone-and-plate geometry with a diameter of 40 mm and a truncation of 64 μm.
Optical properties of PPy nanostructures and the PNCs.
The UV-vis-NIR diffuse reflectance spectra (DRS) of the PPy nanostructures and the PPy/epoxy PNCs were recorded on a JASCO spectrophotometer (Model V-670) equipped with a Jasco ISN-723 diffuse reflectance accessory. The PPy nanofibers and nanospheres used for the test were pressed into the form of disk pellets with a diameter of 25 mm by applying a pressure of 95 MPa in a hydraulic presser, and the average thickness was about 0.5 mm. The PNC samples used for the tests were disc pellets with a diameter of 40 mm and the average thickness was about 2 mm.
Doping level study of the PPy nanofillers.
Hitachi S-3400 scanning electron microscopy (SEM) combined with energy dispersive X-ray spectrometry (EDX) was applied to determine the elemental composition of the PPy nanofillers.
Thermal properties of PPy/epoxy nanocomposites.
The thermal stability of the cured PPy/epoxy PNCs was studied by thermogravimetric analysis (TGA, Q-500, TA instruments). All the samples were heated from 30 to 700 °C with an air flow rate of 60 mL min−1 and a heating rate of 10 °C min−1. The samples were tested under nitrogen with the same flow rate and heating rate to compare the thermal stability of PNCs in different conditions. Differential scanning calorimetry (DSC, TA Instruments Q2000) measurements were implemented under a nitrogen flow rate of approximately 20 mL min−1 at a heating rate of 10 °C min−1 from 0 to 300 °C.
Flame retardancy of cured epoxy nanocomposites.
The flame retardancy performance was evaluated by using a micro-scale combustion calorimeter (MCC, model “MCC-2”, Govmark, Farmingdale, New York) according to the American Society for Testing and Materials (ASTM D7309 – Method A). The sample (∼3 mg) was heated to a specified temperature using a linear heating rate (1 °C s−1) in a stream of nitrogen at a flow rate of 80 mL min−1. The thermal degradation products of the sample in nitrogen were mixed with a 20 mL min−1 stream of oxygen prior to entering the 900 °C combustion furnace. The reported MCC parameters were the averages of three measurements.
Mechanical properties of cured epoxy nanocomposites.
Dynamic mechanical analysis (DMA) measurements were carried out in the torsion rectangular mode using an AR 2000ex (TA Instrumental Company) with a strain of 0.05%, a constant frequency of 1 Hz and a heating rate of 2 °C min−1 in the temperature range of 30–200 °C. The sample dimensions were 12 × 3 × 40 mm3.
Tensile tests were carried out following ASTM (Standard D 412-98a, 2002) in a unidirectional tensile testing machine (ADMET tensile strength testing system). The parameters (displacement and load) were controlled by a digital controller (MTEST Quattro) with MTEST Quattro Materials Testing Software. The samples were prepared as described for the nanocomposite fabrication in silicon rubber molds, which were designed according to the standard ASTM requirement. A crosshead speed of 1.00 mm min−1 was used and the strain (mm mm−1) was calculated by dividing the crosshead displacement by the original gauge length.
Morphology of PPy/epoxy nanocomposites and PPy nanostructures.
After the tensile test, the broken samples of the PPy/epoxy nanocomposites were collected. The morphology of the fracture surfaces and the morphology of PPy nanostructures were characterized with a field emission scanning electron microscope (SEM, JEOL JSM-6700F). Before testing, the samples were first coated with a thin gold layer for better imaging.
Resistivity and permittivity of cured PPy/epoxy nanocomposites.
The samples of PPy nanostructures were pressed into the form of disc pellets with a diameter of 25 mm by applying a pressure of 95 MPa in a hydraulic presser and the average thickness was about 0.5 mm. The electrical conductivity was measured following a standard four probe method at room temperature. The volume resistivity was determined by measuring the DC resistance along disc samples with diameters of approximately 60 mm. An Agilent 4339B high resistance meter was used to measure the samples. This equipment allows resistivity measurement up to 1016 Ω. The reported values represent the mean value of eight measurements with a deviation less than 10%.
The dielectric permittivity was measured by a LCR meter (Agilent, E 4980A) equipped with a dielectric test fixture (Agilent, 16451B) in the frequency range of 20 to 2 × 106 Hz. The samples used for the tests were disc pellets with a diameter of 40 mm and the average thickness was about 2 mm. A piece of rectangular standard Teflon sample with a permittivity of 2.1–2.4 was used for calibration before each test.
Results and discussion
PPy nanofibers and nanospheres
Two different kinds of PPy nanostructures synthesized by two different methods were used as nanofillers to enhance the mechanical and dielectric properties of epoxy. PPy nanofibers were fabricated by a seeding approach, while PPy nanospheres were obtained by ultrasonication, Fig. 1. Compared with PPy nanospheres with an average diameter of 450 nm, PPy nanofibers have an average diameter of 77.17 nm and length of 0.73 μm. In addition, due to the higher aspect ratio, the PPy nanofibers exhibit larger specific surface area (18.97 cm2 g−1) than the PPy nanospheres (2.66 cm2 g−1). From the UV-vis absorbance spectra, Fig. 2(B & C), the photonic energy band gap (Eg) value of the PPy nanofillers can be obtained from the Tauc plot,43 which describes the αhν value as a function of photon energy (hν), eqn (1):where α, h and ν are the absorbance coefficient, Planck constant and photon frequency, respectively. The Tauc plot was converted from the diffuse reflectance UV-vis spectra. The parameter n is a pure number associated with the type of electronic transitions. n is 1/2 for the direct-allowed (characterized by the minimum energy level of the lowest conduction band positioned in k space directly under the maximum of the highest valence band), and 2 for the indirect-allowed (instead of directly under the maximum of the highest valence band, the minimum energy level of the lowest conduction band is shifted relative to the maximum of the highest valence band in k space).44,45 The value of n is reported to be 0.5 for PPy.46 The Eg value is obtained by extrapolation of the linear portion of the curve to the energy axis.
 |
| | Fig. 1 SEM microstructure of the PPy nanofibers and nanospheres. | |
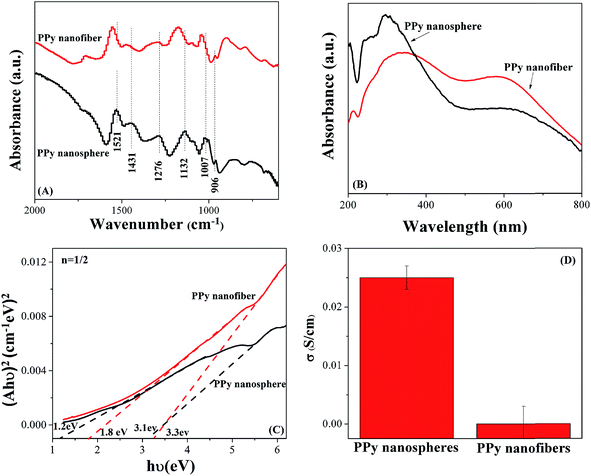 |
| | Fig. 2 (A) FT-IR spectra, (B) UV-vis absorbance spectra, (C) Tauc plot of (αhν)2vs. hν and (D) electrical conductivity of PPy nanofibers and nanospheres. | |
For each PPy nanostructure, two band gaps were observed. It is known that when two polarons on the PPy chain begin an interaction, they can attract each other and form a bipolaron, which introduces two states (bonding and antibonding bipolaron states) in the gap between the valence band and conductive band.47,48 Here, the observed Eg values of 1.8 and 3.3 eV for PPy nanofibers and 1.2 and 3.1 eV for PPy nanospheres are quite close to the reported 1.0 eV for the transition from the valence band to the bonding bipolaron state and 2.7 eV for the transition from the valence band to the antibonding bipolaron state of PPy.47 It is worth noting that the Eg value of PPy nanospheres was lower than that of PPy nanofibers; different Eg values could be related to different doping levels. In addition, the Fourier transform infrared (FT-IR) spectroscopy result shows that compared with PPy nanospheres, the peak of PPy nanofibers shifts to a higher wavenumber range, Fig. 2(A); the bathochromic shift (wavelength shift to higher number and Eg shift to lower value) was also observed in the UV-vis spectra of PPy(SO3H)–DEHS (DEHS = di(2-ethylhexyl) sulfosuccinic acid) and was attributed to the greater doping of PPy by anionically charged sulfonyl groups.49 Here, the doping level of the PPy nanofillers was studied with the EDX of the N:S atomic ratio and N:Cl atomic ratio in the PPy nanospheres and nanofilers, respectively. The N is from the NH-group of PPy, S is from the doping acid PTSA for PPy nanospheres and Cl is from the doping acid HCl for PPy nanofibers. The higher N:S atomic ratio of PPy nanospheres (0.20) than the N:Cl atomic ratio of PPy nanofibers (<0.01) observed in the EDX indicates a higher doping level of PPy nanospheres than that of the PPy nanofibers, leading to a lower Eg of PPy nanospheres than that of PPy nanofibers. Meanwhile, the electrical conductivity study of the PPy nanostructures shows a similar phenomenon: higher electrical conductivity of the pure PPy nanospheres (0.025 S cm−1) than that of the PPy nanofibers (1.65 × 10−5 S cm−1), Fig. 2(D), which is also caused by the higher doping level.50
Rheological behaviors of PPy–epoxy resin nanosuspensions
The rheological behaviors were tested for pure epoxy resin and its nanosuspensions containing PPy nanofillers. Fig. 3(A) shows the curve of shear stress (σ) vs. shear rate (γ). The σ is observed to increase with increasing the γ. The viscosity (η, Pa s) is observed to decrease with increasing the shear rate, Fig. 3(B). The power law model is employed to correlate σ and γ, eqn (2):51where K is the flow consistency index (corresponds to η for a Newtonian fluid52 and will increase with increasing the solid content53) and m is the flow behavior index. For a Newtonian fluid, m is equal to 1 and m is less than 1 for a pseudoplastic fluid. The value of m is summarized in Table 2. For pure epoxy resin, the m value is almost equal to 1, indicating its Newtonian fluid nature. However, for the epoxy nanosuspension with PPy nanofillers, the m value is observed to decrease with increasing the PPy nanofiller loading. A reduced m indicates that the nanofillers favor the pseudoplastic nature (η decreases with increasing γ54) of the nanosuspensions.
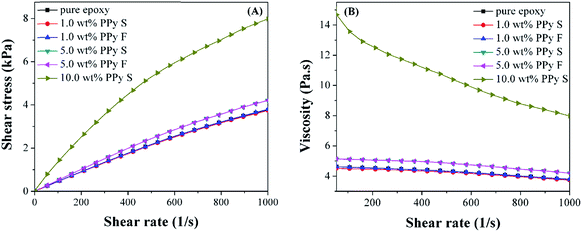 |
| | Fig. 3 (A) Shear stress (σ) and (B) viscosity (η) vs. shear rate (γ) of pure epoxy resin and its nanosuspensions with different loading levels of PPy nanofibers (F) and nanospheres (S). | |
Table 2 The rheological data of liquid pure epoxy and its nanosuspensions
|
|
m
|
K (Pa s) |
| Pure epoxy |
0.9601 |
6.394 |
| 1.0 wt% PPy S |
0.8792 |
8.871 |
| 1.0 wt% PPy F |
0.8770 |
9.128 |
| 5.0 wt% PPy S |
0.8699 |
11.39 |
| 5.0 wt% PPy F |
0.7794 |
27.55 |
| 10.0 wt% PPy S |
0.7081 |
62.29 |
The rheological behavior of the nanosuspensions was further studied with the zero shear viscosity (limiting viscosity at zero shear rate,55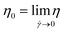 ), which was obtained by employing the Carreau model to correlate η and
), which was obtained by employing the Carreau model to correlate η and ![[small gamma, Greek, dot above]](https://www.rsc.org/images/entities/i_char_e0a2.gif) , eqn (3):56
, eqn (3):56
| | 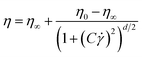 | (3) |
where
η∞ is the infinite shear viscosity (limiting viscosity at infinite shear rate,
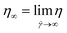
),
C is the cross time constant, which represents the transition to a constant viscosity in the limit of zero shear rate
57 and
d is the rate index. The value of
η0, summarized in
Table 3, is observed to increase with increasing PPy nanostructure loading. At the same loading, the
η0 of the nanosuspensions with PPy nanofibers is higher than that of the nanosuspensions with PPy nanospheres. Higher
η0 can be attributed to the confined polymer chains, in which case the flow activation energy (
Eη, kJ mol
−1) should also be larger than that of the bulk polymer.
58 Here, the
Eη for pure epoxy resin and its nanosuspensions containing PPy nanofillers was studied with a plot of viscosity as a function of temperature (
Fig. 4).
Fig. 4(A & B) show the shear stress (
σ) and viscosity (
η)
vs. shear rate (
γ) at 25, 50 and 70 °C, respectively. The gradual increase of
σ with increasing
γ indicates the reliability of the measurements. The
η of the nanosuspensions with 1.0 wt% PPy nanofibers decreases with increasing the temperature (
Fig. 4(B)). The
σ and
η as a function of temperature for the nanosuspensions with 10.0 wt% PPy nanospheres, 10.0 wt% and 20.0 wt% PPy nanofibers are shown in Fig. S1–S3 in the ESI,
† respectively. The relationship between temperature and
η is shown in
eqn (4):
59| | 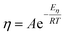 | (4) |
where
R is the gas constant (8.314 J K
−1 mol
−1),
T is the temperature (K) and
A is a pre-exponential factor.
60 The
Eη values of pure epoxy and its nanosuspension at the
γ of 1 s
−1 are summarized in
Table 3. The value of
Eη represents the sensitivity of
η to the temperature and is known to be related to the molecular weight, chain rigidity, intermolecular force of attraction and the chain morphology.
61 Here, in the nanosuspensions with lower loading of PPy nanofillers, for example, 1.0 wt%, the
Eη shows a higher value than that of pure epoxy, indicating that the increased
η0 is associated with the larger flow barrier for the confined polymer chains.
58 However, for the nanosuspensions with higher loading of PPy such as 10.0 and 20.0 wt%, the
Eη values (54.34, 71.15 and 86.42 kJ mol
−1 for 10.0 wt% PPy nanofibers, 10.0 wt% PPy nanospheres and 20.0 wt% PPy nanofibers, respectively) are observed to be lower than that of pure epoxy (98.21 kJ mol
−1). At the same loading, the nanosuspensions with PPy nanofibers exhibit lower
Eη (54.34 kJ mol
−1) than the nanosuspensions with PPy nanospheres (71.15 kJ mol
−1), implying that the increased
η0 is not attributed to the confined polymer chains.
58 Meanwhile, the
η0 is known to be proportional to molecular weight
62 and
Eη decreases with increasing the polymer molecular weight.
63 Thus, the increased
η0 value in the nanosuspensions indicates a larger molecular weight. In the epoxy nanosuspensions with PPy nanofillers, the PPy nanofillers containing amine groups
64 can react with epoxy resin
7,65 to form longer polymer chains and cause larger molecular weight. In addition, at the same loading, the interaction of PPy nanofibers having higher specific surface area with epoxy will be stronger than that of PPy nanospheres with epoxy, and thus higher
η0 and lower
Eη are observed in the nanosuspensions with PPy nanofibers.
Table 3 The flow activation energy (Eη) of liquid pure epoxy and its nanosuspensions at a shear rate (γ) of 1 s−1
|
|
E
η
(kJ mol−1) |
Zero viscosity (η0, Pa s) |
| Pure epoxy |
98.21 |
5.590 |
| 1.0 wt% PPy F |
109.71 |
9.13 |
| 10.0 wt% PPy F |
54.34 |
136.60 |
| 10.0 wt% PPy S |
71.15 |
62.34 |
| 20.0 wt% PPy F |
86.42 |
2814.00 |
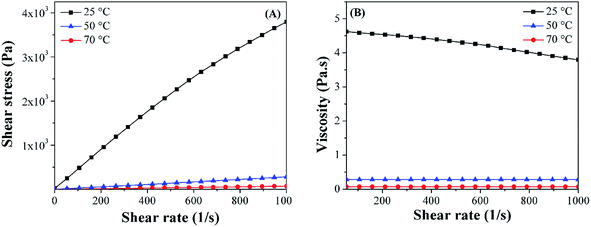 |
| | Fig. 4 (A) Shear stress (σ) and (B) viscosity (η) vs. shear rate (γ) of epoxy nanosuspensions with 1.0 wt% PPy nanofibers at different test temperatures. | |
Differential scanning calorimetry of cured epoxy resin and its nanocomposites
Generally, gelation, curing, vitrification, and devitrification events can be studied with DSC tests for thermosetting systems.66 The former two processes can be observed in the uncured samples, however, the exothermal peak, which represents the curing process, can also be observed in the cured PNCs (Fig. 5). It is known that the existence of nanoparticles would limit the curing extent (α) of epoxy resin, and cause the incompletely-formed networks in PNCs.67 On increasing the temperature higher than the glass transition temperature (Tg), the polymer segments can move again and tend to accomplish the curing process, thus an additional curing process was observed in the DSC curve. The value of residual heat of curing can be used to calculate the α of PNCs based on eqn (5):68| | 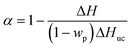 | (5) |
where ΔH is the residual heat of the reaction in PNCs (J g−1), wp is the particle weight percentage in the composites and ΔHuc is the residual heat of the uncured pure resin (319.3 J g−1, obtained by running the DSC test on the uncured pure epoxy). The value of α for PNCs was calculated and summarized in Table 4. For pure epoxy and PNCs with lower loading of PPy nanostructures (1.0 wt%), no additional exothermal peak was observed, indicating that the sample was well cured, however, for PNCs with higher loading of PPy nanostructures, α first decreased and then increased with increasing PPy nanostructure loading. As aforementioned, the existence of nanofillers has a detrimental effect on the curing process of the matrix resin, which causes a reduced α (0.76 and 0.78 for 10.0 wt% PPy nanofibers and nanospheres, respectively). However, the covalent bonds formed between PPy and epoxy resin favor the network formation in PNCs. Thus, an increased α (0.90 and 0.92 for 20.0 wt% PPy nanofibers and nanospheres, respectively) implies that the positive effect becomes a dominant factor in PNCs with higher loading of PPy. A similar tendency was also observed in the variation of Tg. With PPy nanostructure loading less than 20.0 wt%, the Tg of PNCs (78.40 and 63.23 °C for 1.0 wt% PPy nanofibers and nanospheres, respectively) was lower than that of pure epoxy (108.31 °C). The lower Tg was attributed to the enlarged free volume arising from the interface between fillers and epoxy resin,69 which provides more space for polymer chain segments to move even at a lower temperature. However, when the loading of PPy reached 20.0 wt%, the interaction between PPy nanostructures and epoxy resin limits the movement of polymer chains, and leads to an increment in Tg. For PNCs with 20.0 wt% PPy nanofibers, the Tg (115.18 and 99.05 °C for PPy nanofibers and nanospheres, respectively) becomes even larger than that of pure epoxy.
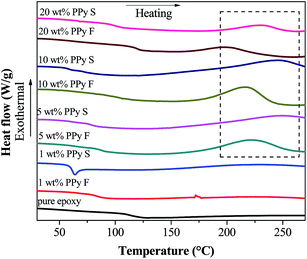 |
| | Fig. 5 DSC curves of cured pure epoxy and its PNCs with different loading levels of PPy nanofibers and nanospheres. | |
Table 4 Onset decomposition temperature (T1onset and T2onset), glass transition temperature (Tg), residual heat of curing (ΔH), and curing extent (α) of the cured pure epoxy and its PNCs
| Sample name |
T
1onset (°C) |
T
2onset (°C) |
T
g (°C) |
ΔH (J g−1) |
α
|
| Cured pure epoxy |
363.84 |
522.82 |
108.31 |
0.00 |
1.00 |
| PNCs with 1.0 wt% PPy nanofiber |
355.82 |
518.20 |
78.40 |
0.00 |
1.00 |
| PNCs with 1.0 wt% PPy nanosphere |
359.83 |
518.24 |
63.23 |
0.00 |
1.00 |
| PNCs with 5.0 wt% PPy nanofiber |
323.78 |
520.35 |
79.83 |
63.02 |
0.80 |
| PNCs with 5.0 wt% PPy nanosphere |
299.08 |
521.03 |
74.20 |
31.90 |
0.90 |
| PNCs with 10.0 wt% PPy nanofiber |
292.41 |
524.85 |
103.41 |
78.00 |
0.76 |
| PNCs with 10.0 wt% PPy nanosphere |
294.41 |
523.32 |
67.52 |
68.74 |
0.78 |
| PNCs with 20.0 wt% PPy nanofiber |
264.37 |
523.99 |
115.18 |
30.50 |
0.90 |
| PNCs with 20.0 wt% PPy nanosphere |
277.72 |
524.84 |
99.05 |
26.27 |
0.92 |
Thermomechanical properties of cured epoxy resin and its nanocomposites
The thermomechanical properties of pure epoxy and its PNCs were studied by the variation of shear storage modulus (G′), shear loss modulus (G′′) and tanδ as a function of temperature (Fig. 6). G′ and G′′ represent the elastic behavior (energy storage in the nanocomposites during the test) and the viscous behavior (energy dissipation in the nanocomposites during the test), respectively.70,71 The introduction of nanofillers also has two opposite effects on the thermo-mechanical properties of the polymer matrix, consistent with the aforementioned DSC analysis. On one hand, the nanofiller leads to an enlarged free volume between the interface of the nanofiller and polymer matrix, which can enhance the mobility of the polymer chains and cause a reduction of both moduli.72 On the other hand, the agglomeration of nanofillers and the interaction between nanofiller and polymer matrix would prohibit the movement of polymer chains and result in the increment of both moduli.72,73 The G′ and G′′ values for the cured pure epoxy resin and its PNCs with lower nanofiller loading (1.0 and 5.0 wt%) were almost the same in the glass plateau (below 60 °C) (Fig. 6(A & B)). However, with increasing the nanofillers loading to 10.0 wt%, the free volume was enlarged. Thus both G′ and G′′ were decreased to even lower than those of pure epoxy. With further increasing of the nanofiller loading to 20.0 wt%, both moduli depended on the morphology of PPy nanofiller. For the PNCs with nanospheres, the G′ and G′′ kept decreasing with increasing the loading of PPy nanospheres from 10.0 to 20.0 wt%. The change in G′ from decrease to increase was considered as an indicator to identify the percolation threshold of the nanofillers in epoxy resin.74 PNCs with PPy nanofibers are observed to reach the percolation threshold with a lower loading (10.0 wt%) than the PNCs with PPy nanospheres (above 20.0 wt%).75 This is due to the PPy nanofibers possessing a larger aspect ratio than PPy nanospheres.
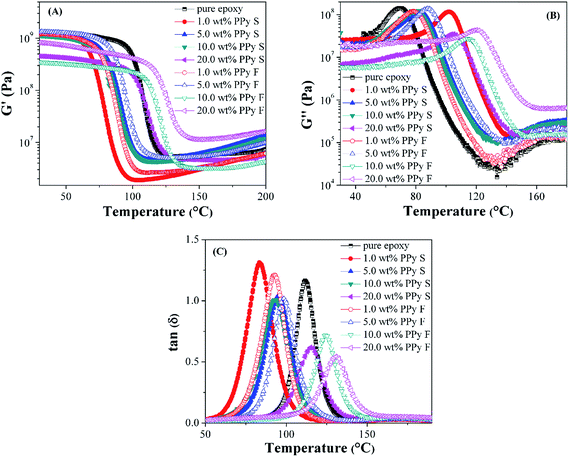 |
| | Fig. 6 (A) Storage modulus (G′), (B) loss modulus (G′′) and (C) tanδ vs. temperature of cured pure epoxy and its PNCs with different loading levels of PPy nanofibers and nanospheres. | |
The tanδ is the ratio of the G′′ to the G′, and the peak of tanδ is often used to determine the Tg. As shown in Fig. 6(C), for the PNCs with the PPy nanofillers, the Tg first decreased with increasing the loading of PPy nanofibers from 0 to 1.0 wt%, which is attributed to the enlarged free volume between the epoxy chains.69 With further increase in the loading of PPy nanofiller, the Tg increased and for PNCs with 20.0 wt% PPy nanofibers, the Tg value became higher than that of pure epoxy, which is consistent with the result obtained in the DSC study.
Tensile mechanical property and fracture surface analysis of cured nanocomposites
Fig. 7 shows the curve of tensile stress as a function of tensile strain for pure epoxy and its PNCs with different PPy nanostructures. An enhanced tensile strength was observed in the PNCs with 1.0 wt% PPy nanostructures. The highest tensile strength shown for 1.0 wt% PPy nanofibers PNCs (90.36 MPa), was 29% higher than that of pure epoxy (70.03 MPa). The mechanism of the increased tensile strength is studied from the morphology of fracture surfaces (Fig. 8). As shown in pure epoxy, Fig. 8(a & b), the fracture surface is observed to be very smooth and can be considered as a typical brittle fracture. However, in the PNCs with lower loading of PPy nanostructures, the fracture surface became rougher with more microcrazings, which can relieve the stress state and limit the void formation in the bulk polymer.76,77 In addition, the crack distribution was observed to depend on the morphology of the PPy; for the PNCs with 1.0 wt% PPy nanofibers, Fig. 8(c), more laminated microcrazings are observed on the fracture surface, and the microcrazings are homogeneously distributed in the PNC resin. In addition, shear yielding of epoxy resin, Fig. 8(d), indicates that the PPy nanofibers can initiate the shear bands in the epoxy resin.78 However, for the PNCs with 1.0 wt% PPy nanospheres, Fig. 8(e & f), the cracks are observed propagating in the same direction, which would lead to a lower tensile strength. The morphology variation of the cracks indicates that the PPy nanofibers were more efficient in obstructing the propagation of the cracks. Compared with the reference PNCs, as shown in Table 1 the lower tensile strength of the PPy/epoxy PNCs is associated with the different curing process.
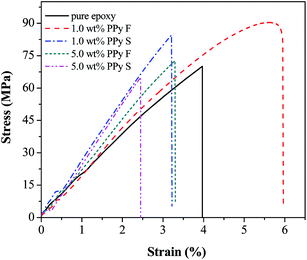 |
| | Fig. 7 Stress–strain curves of cured pure epoxy and its nanocomposites filled with different PPy nanofiller loading levels. | |
 |
| | Fig. 8 SEM microstructures of the fracture surface of (a & b) cured pure epoxy and its PNCs with (c & d) 1.0 wt% nanofibers, (e & f) 1.0 wt% nanospheres, (g) 5.0 wt% nanofibers, (h) 5.0 wt% nanospheres, (i) 20.0 wt% nanofibers and (j) 20.0 wt% nanospheres. | |
With increasing the loading of PPy (5.0 wt%) nanofillers, as aforementioned in the DSC section, the α was decreased, and it is known that tensile strength decreases with reducing α.79 In addition, although the PPy nanofiller led to a rougher fracture surface, the agglomeration of PPy nanofillers was also noticed in PNCs with 5.0 wt% PPy nanofillers, Fig. 8(g & h), and with further increasing the loading of PPy nanostructures to 20.0 wt%, Fig. 8(i & j), a large amount of the agglomerated nanofillers was obviously observed with voids in the fracture surface, which caused a decreased tensile strength, Fig. S4 in the ESI.†
Thermogravimetric analysis of cured epoxy resin and its nanocomposites
Fig. 9 shows the TGA curve of cured pure epoxy and its PNCs. Under nitrogen, Fig. 9(A), only one sharp weight loss stage is observed in all the samples, which is due to the chain break-down of the polymer structure. From the onset decomposition temperature (extrapolated onset temperature is obtained by software and the temperature range is set between 200 to 400 °C) summarized in Table 5, the addition of the PPy nanofillers leads to a decreased decomposition temperature in the PNCs, and the reduction is caused by a lower decomposition temperature of pure PPy nanofibers (200.78 °C) and PPy nanospheres (213.63 °C) than that of pure epoxy, as shown in Fig. S5 in the ESI.† In addition, due to the higher decomposition temperature of PPy nanospheres than that of PPy nanofibers, the PNCs with nanospheres exhibit higher onset decomposition temperature than that of the PNCs with nanofibers at the same filler loading (Table 5). The weight loss at 700 °C was also studied and summarized in Table 5; it is worth noting that for PNCs with 20.0 wt% PPy nanofibers (75.65%) and nanospheres (79.57%), the weight loss is 8 and 12% lower, respectively, than that of pure epoxy (87.85%). The lower weight loss indicates a higher char residue, which is further studied in the flammability analysis of the PNCs.
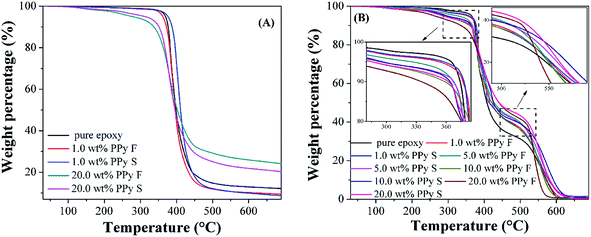 |
| | Fig. 9 TGA curves of cured pure epoxy and its PNCs with different loadings of PPy nanofibers and nanospheres in (A) nitrogen and (B) air. | |
Table 5 Onset decomposition temperature and weight loss at 700 °C under nitrogen of the cured pure epoxy, pure PPy nanostructure and its PNCs
| Sample name |
Onset decomposition temperature (°C) |
Weight loss (%) |
| Cured pure epoxy |
369.08 |
87.85 |
| PPy nanofibers |
200.78 |
88.52 |
| PPy nanospheres |
213.63 |
41.31 |
| PNCs with 1.0 wt% PPy nanofibers |
361.83 |
90.61 |
| PNCs with 1.0 wt% PPy nanospheres |
375.18 |
91.39 |
| PNCs with 20.0 wt% PPy nanofibers |
276.38 |
75.65 |
| PNCs with 20.0 wt% PPy nanospheres |
315.10 |
79.57 |
When the TGA test was run in air, Fig. 9(B), two weight loss stages are observed, different from the results obtained under nitrogen. Same as for the nitrogen case, the first degradation stage in air is due to the breaking of the crosslinked network in epoxy, and the onset decomposition temperature (T1onset) of PNCs (323.78 and 299.08 °C for 5.0 wt% PPy nanofibers and nanospheres, respectively) for the first weight loss stage is observed to be lower than that of pure epoxy (363.84 °C) (Table 4). However, the observed extra weight loss stage in the higher temperature range (450–600 °C) is attributed to the structure conversion or oxidation of aromatic rings in epoxy.80 And the onset decomposition temperature (T2onset) of the PNCs (523.99 and 524.84 °C for 20.0 wt% PPy nanofibers and nanospheres, respectively) for the second weight loss second stage is higher than that of pure epoxy (522.82 °C) (Table 4). The enhanced thermal stability is associated with the covalent bonds formed between PPy nanostructures and epoxy resin, which can postpone the break-down of the aromatic rings.81 A similar phenomenon has also been observed in the PANI/epoxy PNCs.82
Flammability analysis of cured epoxy nanocomposites
The flame retardancy behaviors of the cured PNCs with different PPy nanostructures were evaluated by studying the heat release capacity (HRC), peak heat release rate (PHRR), total heat release (THR), and char residue, which are summarized in Table 6. The increased char residue with increasing the PPy nanofiller loading indicates that the existence of PPy favors char formation of the polymer matrix and the same phenomenon was also observed in the PANI/epoxy PNCs.82 In addition, it is worth noting that the morphology of the char residue is completely different for the pure epoxy and its PNCs with PPy nanostructures (Fig. 10). For pure epoxy (Fig. 10(A)), only a thin layer of char residue was formed on the surface of the sample holder, however, for PNCs with 20.0 wt% PPy nanofibers and nanospheres Fig. 10(B & C), a block of char was formed, implying that the PPy nanostructures promoted the char yield of the epoxy resin; the same phenomenon can also be observed in the SEM image of the char layer, Fig. S5 in the ESI.† Char yield is considered as a characteristic of flame retardancy.83,84 The char formed on the surface of materials is known to prevent heat from being transferred from the heat source to the inner material85 and also to obstruct the distribution of combustible gases produced during the burning process.86 Thus, as shown in Fig. 11, the existence of PPy nanofillers reduced the heat release from the epoxy resin. In addition, the PHRR, HRC and THR of the PNCs are also decreased with increasing loading of the PPy nanofibers (Table 6). Moreover, due to the larger specific surface area of the PPy nanofibers than that of the PPy nanospheres, the nanofibers can act more efficiently in assisting the char formation of the epoxy resin. As shown in Table 6, the char residue of PNCs with PPy nanofibers (33% for 20.0 wt%) was higher than that of PNCs with PPy nanospheres (30% for 20.0 wt%), thus the PHRR value of PNCs with the PPy nanofibers (345.1 W g−1 for 20.0 wt%) was observed to be lower than that of the PNCs with the PPy nanospheres (360.3 W g−1 for 20.0 wt%) (Fig. 11).
Table 6 Heat release capacity (HRC), peak heat release rate (PHRR), total heat release (THR) and char residue for the cured pure epoxy and its PNCs
|
|
HRC (J g−1 K−1) |
PHRR (W g−1) |
THR (kJ g−1) |
Char residue (%) |
| Cured pure epoxy |
491 |
664.3 |
28 |
12 |
| PNCs with 1.0 wt% PPy nanofibers |
477 |
515.7 |
26.6 |
13 |
| PNCs with 5.0 wt% PPy nanofibers |
450 |
487.2 |
23.9 |
21 |
| PNCs with 10.0 wt% PPy nanofibers |
378 |
408.9 |
23.0 |
24 |
| PNCs with 20.0 wt% PPy nanofibers |
319 |
345.1 |
19.3 |
33 |
| PNCs with 20.0 wt% PPy nanospheres |
355 |
360.3 |
23.0 |
30 |
 |
| | Fig. 10 The optical images of residue for (A) cured pure epoxy and its PNCs with 20.0 wt% PPy (B) nanofibers and (C) nanospheres. | |
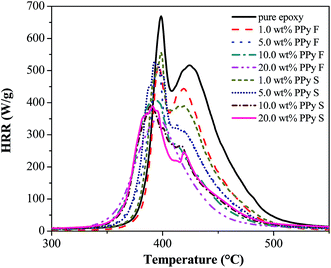 |
| | Fig. 11 The HRR vs. temperature curves of cured pure epoxy and its PNCs with nanofibers and nanospheres. | |
Bandgap of cured nanocomposites
The optical properties of cured pure epoxy resin and its PNCs were also studied by ultraviolet-visible (UV-vis) spectroscopy, and the Eg values are obtained from the Tauc plot43 (Fig. 12). Both 2 and 0.5 for pure number n have been reported for epoxy.87 Considering the Eg of PPy nanostructures with n of 0.5, 0.5 is selected to obtain the Eg of pure epoxy and its nanocomposites. For the epoxy PNCs, two absorption bands are also observed, Fig. 12, and the calculated Eg values are summarized in Table 7. The higher band gap (Eg2) at around 3.50 eV, attributed to the π–π* transition of the benzene group in the epoxy backbone, which is also shown in the pure epoxy sample, is very close to the reported 3.45 eV.87 On the other hand, the Eg1 with a lower value associated with the band transition of PPy nanofillers is observed to decrease with increasing the loading of PPy. The protons of the doped acid in the PPy nanofillers can react with epoxy more easily to form hydroxyl groups88 due to the more electronegative nature of oxygen than nitrogen. This reaction further broke the conjugate structure of PPy, leading to a higher Eg1 in the PNCs (3.08 eV for 1.0 wt% PPy nanofibers) than in pure nanofillers (1.8 eV for pure PPy nanofibers).89 The enlarged Eg1 of poly(m-phenylene) (PMP) arising from the broken conjugation was also observed with a weakened π-conjugation than that of poly(p-phenylene).90 In addition, due to the higher Eg of pure PPy nanofibers (1.8 eV) than that of nanospheres (1.2 eV), the Eg1 of PNCs with PPy nanofibers (2.20 eV for 20.0 wt%) is observed to be higher than those of PNCs with nanospheres (1.98 eV for 20.0 wt%). The changing trend of Eg1 of the PNCs is consistent with the electrical conductivity of the PNCs, i.e., the PNCs with higher conductivity exhibit a lower Eg1.
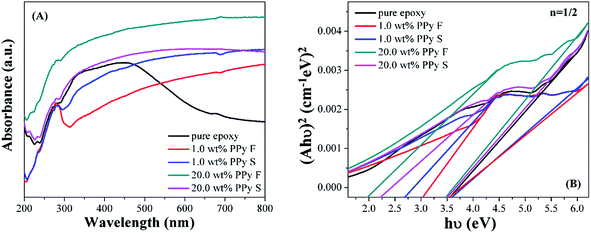 |
| | Fig. 12 (A) UV-vis absorbance spectra and (B) Tauc plot of (Ahν)2vs. hν for pure epoxy and its PNCs. | |
Table 7 Band gap value of the cured pure epoxy and its PNCs
| Loading of CNTs |
E
g1/eV |
E
g2/eV |
| Pure epoxy |
N/A |
3.62 |
| PNCs with 1.0 wt% PPy nanofibers |
3.08 |
3.62 |
| PNCs with 1.0 wt% PPy nanospheres |
2.70 |
3.63 |
| PNCs with 20.0 wt% PPy nanofibers |
2.20 |
3.49 |
| PNCs with 20.0 wt% PPy nanospheres |
1.98 |
3.50 |
Electrical properties of cured nanocomposites
Fig. 13 shows the volume resistivity of the PNCs as a function of the loading of different PPy nanostructures. For PNCs with both nanostructures, the resistivity was observed to decrease with increasing the loading of PPy nanostructures. The changing trend in resistivity value of the PNCs is consistent with the Eg1 of the PNCs: the PNCs with lower Eg1 exhibit lower resistivity. In addition, the resistivity of PNCs also depends on the morphology of the PPy nanofillers; at lower loading of PPy nanostructures (1.0, 5.0 and 10.0 wt%), the resistivity of PNCs with the nanospheres is higher than that of the PNCs with nanofibers, which is attributed to higher electrical conductivity of the pure PPy nanospheres (0.025 S cm−1) than that of the PPy nanofibers (1.65 × 10−5 S cm−1) (Fig. 2(D)). However, due to the higher aspect ratio, nanofibers can form the conductive network more easily with less contact resistance91 than the nanospheres, thus, with the loading of the nanostructure increased to 20.0 wt%, although the Eg1 value of PNCs with PPy nanospheres (1.98 eV) is lower than that of PNCs with PPy nanofibers (2.20 eV), Table 7, the PNCs with PPy nanofibers show lower volume resistivity than that of PNCs with PPy nanospheres (Fig. 13).
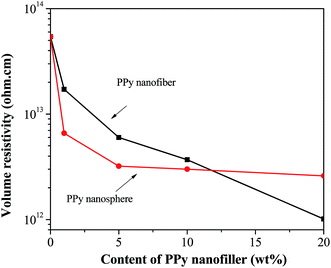 |
| | Fig. 13 Volume resistivity of cured pure epoxy and its PNCs with different loading levels of the nanofibers and nanospheres. | |
Dielectric permittivity
The dielectric property of pure epoxy and its PNCs was studied with the variation of real permittivity (ε′), imaginary permittivity (ε′′) and dielectric loss (tanδ) as a function of the frequency. As shown in Fig. 14, for all the samples, both ε′ and ε′′ decrease with increasing frequency, which is typical dielectric relaxation and can be attributed to the reorientation of the permanent molecular dipoles.92 The orientation of dipolar groups under an alternative electric field depends on the frequency value. At lower frequency, the dipolar functional groups can orient themselves and a high permittivity value is observed; however, at increased frequency, the dipolar groups can not orient at the same rate of the alternating electric field and thus cause a reduced permittivity.93 All the PNCs show higher ε′ and ε′′ than pure epoxy and the values of both permittivities increase with increasing the loading of the PPy nanofillers (Fig. 14(A & B)). The enhanced permittivity was widely observed in the nanocomposites and was attributed to the interfacial polarization,94 in which the charge carriers were blocked at the internal surface or at interfaces between the matrix and the fillers leading to an increased permittivity.95 In the PPy/epoxy PNCs, although the protons (hydrogen ions) provided by the doping acid can move along the PPy chains, these charge movements will be hindered by epoxy resin, thus, a large number of space charges will accumulate at the interface of PPy and epoxy and result in a larger interfacial polarization. The observed higher tanδ value of the PNCs than that of pure epoxy, Fig. 14(C), is associated with the free charge motion difference,96 indicating an interfacial polarization occurring in the PNCs.
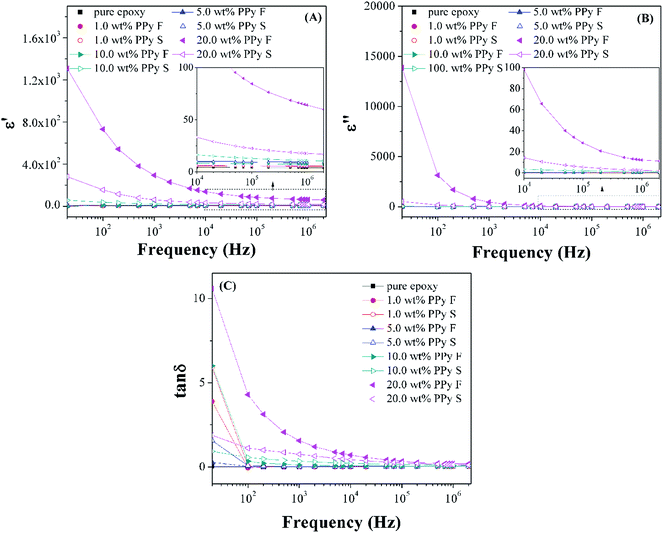 |
| | Fig. 14 (A) Real permittivity (ε′), (B) imaginary permittivity (ε′′), and (C) dielectric loss (tanδ) of cured pure epoxy and its PNCs with different loading levels of nanofibers and nanospheres. | |
In addition, the dielectric properties of the PNCs were observed to depend on the PPy nanofiller morphology. For the PNCs with a lower loading (1.0, 5.0 and 10.0 wt%) of PPy nanofibers and nanospheres, Fig. 14(A & B), the permittivity values are almost the same. However at higher loading (20 wt%), the PNCs with PPy nanofibers show much higher ε′ and ε′′ than the PNCs with PPy nanospheres. The variation of the permittivity is due to the larger specific surface area of PPy nanofibers, which would lead to a stronger interfacial polarization in the PNCs.97 Thus, at higher loading of PPy nanofillers, the permittivity of the PNCs with the PPy nanofibers is much larger than that of the PNCs with the PPy nanospheres.
Conclusions
The epoxy resin nanocomposites with both nanofibers and nanospheres at different loading levels were prepared and systematically studied for both solid and liquid phases. The rheological behavior revealed a reduced flow behavior index with increasing the PPy loading, indicating that the nanofillers favored the pseudoplastic nature of the nanosuspensions. For the cured solid samples, the nanocomposites with 1.0 wt% PPy nanofibers exhibited an enhanced tensile strength (90.36 MPa), 29% higher than that of pure epoxy (70.03 MPa). The fracture surface study indicates that the PPy nanofillers released the stress state. The crack distribution was observed to depend on the PPy morphology. The PPy nanofillers promoted char formation and reduced the heat release from the epoxy resin. With increasing the loading of PPy nanofillers, the PHRR, HRC and THR of nanocomposites were all decreased. The G′ of the nanocomposites was initially decreased due to the enlarged interface free volume between the nanofiller and polymer matrix. However, when the loading of PPy nanofiller was further increased, the agglomeration of nanofillers and the nanofillers–polymer interaction would prohibit the movement of polymer chains and result in the increment of both G′ and G′′. The minimum value of G′ was considered as a criterion to identify the percolation threshold in epoxy resin. PPy nanofibers with a higher aspect ratio reached the percolation threshold with a lower loading (10.0 wt%) than the PPy nanospheres (above 20.0 wt%). For the conductivity study, at the high loading level of 20.0 wt%, the observed lower volume resistivity in the nanocomposites with nanofibers than that in the nanocomposites with nanospheres was discussed considering the contact resistance difference. Compared with pure epoxy, the improved dielectric properties of the nanocomposites was attributed to the interfacial polarization. The nanocomposites with nanofibers showed stronger interfacial polarization than the nanocomposites with nanospheres due to the larger specific surface area of the PPy nanofibers than that of the PPy nanospheres. Epoxy PNCs with improved mechanical properties, dielectric properties and flame retardancy performance at the same time were reported.
Acknowledgements
This project is supported by the National Science Foundation (CMMI 10-30755, CMMI 13-14486). S. Wei acknowledges the Welch foundation (V-0004).
References
- J. Zhu, Q. He, Z. Luo, A. Khasanov, Y. Li, L. Sun, Q. Wang, S. Wei and Z. Guo, J. Mater. Chem., 2012, 22, 15928–15938 RSC.
- Q. He, T. Yuan, J. Zhu, Z. Luo, N. Haldolaarachchige, L. Sun, A. Khasanov, Y. Li, D. P. Young, S. Wei and Z. Guo, Polymer, 2012, 53, 3642–3652 CrossRef CAS PubMed.
- J. Zhu, S. Wei, Y. Li, L. Sun, N. Haldolaarachchige, D. P. Young, C. Southworth, A. Khasanov, Z. Luo and Z. Guo, Macromolecules, 2011, 44, 4382–4391 CrossRef CAS.
- Q. He, T. Yuan, S. Wei, N. Haldolaarachchige, Z. Luo, D. P. Young, A. Khasanov and Z. Guo, Angew. Chem., Int. Ed., 2012, 51, 8842–8845 CrossRef CAS PubMed.
- J. Zhu, S. Wei, N. Haldolaarachchige, D. P. Young and Z. Guo, J. Phys. Chem. C, 2011, 115, 15304–15310 CAS.
- J. Zhu, S. Wei, X. Chen, A. B. Karki, D. Rutman, D. P. Young and Z. Guo, J. Phys. Chem. C, 2010, 114, 8844–8850 CAS.
- J. Guo, X. Zhang, H. Gu, Y. Wang, X. Yan, D. Ding, J. Long, S. Tadakamalla, Q. Wang, M. A. Khan, J. Liu, X. Zhang, B. L. Weeks, L. Sun, D. P. Young, S. Wei and Z. Guo, RSC Adv., 2014, 4, 36560–36572 RSC.
- J. Zhu, S. Wei, M. J. Alexander, T. D. Dang, T. C. Ho and Z. Guo, Adv. Funct. Mater., 2010, 20, 3076–3084 CrossRef CAS.
- H. Wei, X. Yan, Y. Li, S. Wu, A. Wang, S. Wei and Z. Guo, J. Phys. Chem. C, 2012, 116, 4500–4510 CAS.
- H. Wei, X. Yan, Y. Li, H. Gu, S. Wu, K. Ding, S. Wei and Z. Guo, J. Phys. Chem. C, 2012, 116, 16286–16293 CAS.
- X. Zhang, S. Wei, N. Haldolaarachchige, H. A. Colorado, Z. Luo, D. P. Young and Z. Guo, J. Phys. Chem. C, 2010, 116, 15731–15740 Search PubMed.
- J. Zhu, S. Wei, L. Zhang, Y. Mao, J. Ryu, A. B. Karki, D. P. Young and Z. Guo, J. Mater. Chem., 2012, 21, 342–348 RSC.
- J. Zhu, X. Zhang, N. Haldolaarachchige, Q. Wang, Z. Luo, J. Ryu, D. P. Young, S. Wei and Z. Guo, J. Mater. Chem., 2012, 22, 4996–5005 RSC.
- J. Zhu, S. Wei, J. Ryu and Z. Guo, J. Phys. Chem. C, 2011, 115, 13215–13222 CAS.
- M. Fini, G. Giavaresi, N. N. Aldini, P. Torricelli, R. Botter, D. Beruto and R. Giardino, Biomaterials, 2002, 23, 4523–4531 CrossRef CAS.
- V. B. Mišković-Stanković, M. R. Stanić and D. M. Dražić, Prog. Org. Coat., 1999, 36, 53–63 CrossRef.
- Y. Rao, S. Ogitani, P. Kohl and C. P. Wong, J. Appl. Polym. Sci., 2002, 83, 1084–1090 CrossRef CAS.
- L. Qi, B. I. Lee, S. Chen, W. D. Samuels and G. J. Exarhos, Adv. Mater., 2005, 17, 1777–1781 CrossRef CAS.
- M. Takala, M. Karttunen, J. Pelto, P. Salovaara, T. Munter, M. Honkanen, T. Auletta and K. Kannus, IEEE Trans. Dielectr. Electr. Insul., 2008, 15, 1224–1235 CrossRef CAS.
- H. Zhao and R. K. Y. Li, Composites, Part A, 2008, 39, 602–611 CrossRef PubMed.
- K. Sonoda, J. Juuti, Y. Moriya and H. Jantunen, Compos. Struct., 2010, 92, 1052–1058 CrossRef PubMed.
- S. Tian and X. Wang, J. Mater. Sci., 2008, 43, 4979–4987 CrossRef CAS PubMed.
- M. W. Noh and D. C. Lee, Polym. Bull., 1999, 42, 619–626 CrossRef CAS.
- H. Ren, J. Sun, B. Wu and Q. Zhou, Polym. Degrad. Stab., 2007, 92, 956–961 CrossRef CAS PubMed.
- X. Wang, Y. Hu, L. Song, W. Xing, H. Lu, P. Lv and G. Jie, Polymer, 2010, 51, 2435–2445 CrossRef CAS PubMed.
- L. Sun, G. L. Warren, J. Y. O'Reilly, W. N. Everett, S. M. Lee, D. Davis, D. Lagoudas and H. J. Sue, Carbon, 2008, 46, 320–328 CrossRef CAS PubMed.
- J. Zhu, J. Kim, H. Peng, J. L. Margrave, V. N. Khabashesku and E. V. Barrera, Nano Lett., 2003, 3, 1107–1113 CrossRef CAS.
- J. Zhu, H. Peng, F. Rodriguez-Macias, J. L. Margrave, V. N. Khabashesku, A. M. Imam, K. Lozano and E. V. Barrera, Adv. Funct. Mater., 2004, 14, 643–648 CrossRef CAS.
- P. M. Carrasco, H. J. Grande, M. Cortazar, J. M. Alberdi, J. Areizaga and J. A. Pomposo, Synth. Met., 2006, 156, 420–425 CrossRef CAS PubMed.
- D. S. Maddison, J. Unsworth and R. B. Roberts, Synth. Met., 1988, 26, 99–108 CrossRef CAS.
- B. Lakard, L. Ploux, K. Anselme, F. Lallemand, S. Lakard, M. Nardin and J. Y. Hihn, Bioelectrochemistry, 2009, 75, 148–157 CrossRef CAS PubMed.
- K. Ding, H. Jia, S. Wei and Z. Guo, Ind. Eng. Chem. Res., 2011, 50, 7077–7082 CrossRef CAS.
- R. Ansari and N. K. Fahim, React. Funct. Polym., 2007, 67, 367–374 CrossRef CAS PubMed.
- J. Guo, H. Gu, H. Wei, Q. Zhang, N. Haldolaarachchige, Y. Li, D. P. Young, S. Wei and Z. Guo, J. Phys. Chem. C, 2013, 117, 10191–10202 CAS.
- L. Shao, X. Cheng, Z. Wang, J. Ma and Z. Guo, J. Membr. Sci., 2014, 452, 82–89 CrossRef CAS PubMed.
- C. Cassignol, M. Cavarero, A. Boudet and A. Ricard, Polymer, 1999, 40, 1139–1151 CrossRef CAS.
- U. Riaz, S. M. Ashraf and S. Ahmad, Prog. Org. Coat., 2007, 59, 138–145 CrossRef CAS PubMed.
- C. M. Becker, A. D. Gabbardo, F. Wypych and S. C. Amico, Composites, Part A, 2011, 42, 196–202 CrossRef PubMed.
- A. Varesano, C. Tonin, F. Ferrero and M. Stringhetta, J. Therm. Anal. Calorim., 2008, 94, 559–565 CrossRef CAS.
- Y. Hu, S. Wang, Z. Ling, Y. Zhuang, Z. Chen and W. Fan, Macromol. Mater. Eng., 2003, 288, 272–276 CrossRef CAS.
- I. A. Mir and D. Kumar, J. Adhes., 2010, 86, 447–462 CrossRef CAS.
- E. Armelin, R. Pla, F. Liesa, X. Ramis, J. I. Iribarren and C. Alemán, Corros. Sci., 2008, 50, 721–728 CrossRef CAS PubMed.
- J. Tauc, Mater. Res. Bull., 1970, 5, 721–729 CrossRef CAS.
- A. Hjelm, C. G. Granqvist and J. M. Wills, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 2436–2445 CrossRef CAS.
- N. Serpone, D. Lawless and R. Khairutdinov, J. Phys. Chem., 1995, 99, 16646–16654 CrossRef CAS.
- Y. Ali, K. Sharma, V. Kumar, R. G. Sonkawade and A. S. Dhaliwal, Appl. Surf. Sci., 2013, 280, 950–956 CrossRef CAS PubMed.
- J. L. Brédas, J. C. Scott, K. Yakushi and G. B. Street, Phys. Rev. B: Condens. Matter Mater. Phys., 1984, 30, 1023–1025 CrossRef.
- Y. Hu, R. Yang, D. F. Evans and J. H. Weaver, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 13660–13665 CrossRef CAS.
- K. S. Jang, H. Lee and B. Moon, Synth. Met., 2004, 143, 289–294 CrossRef CAS PubMed.
- G. Zotti, Synth. Met., 1998, 97, 267–272 CrossRef CAS.
- R. M. Manglik and P. Fang, Int. J. Heat Mass Transfer, 2002, 45, 803–814 CrossRef.
- J. Floury, A. Desrumaux and J. Lardières, Innovative Food Sci. Emerging Technol., 2000, 1, 127–134 CrossRef CAS.
- S. D. Holdsworth, J. Texture Stud., 1971, 2, 393–418 CrossRef PubMed.
- D. V. Boger, Nature, 1977, 265, 126–128 CrossRef CAS.
- M. M. Cross, J. Colloid Sci., 1965, 20, 417–437 CrossRef CAS.
- Y. H. Hyun, S. T. Lim, H. J. Choi and M. S. Jhon, Macromolecules, 2001, 34, 8084–8093 CrossRef CAS.
- K. Khellaf and G. Lauriat, J. Non-Newtonian Fluid Mech., 2000, 89, 45–61 CrossRef CAS.
- G. Galgali, C. Ramesh and A. Lele, Macromolecules, 2001, 34, 852–858 CrossRef CAS.
- J. Saltiel and J. T. D'Agostino, J. Am. Chem. Soc., 1972, 94, 6445–6456 CrossRef CAS.
- B. Mossel, B. Bhandari, B. D'Arcy and N. Caffin, LWT–Food Sci. Technol., 2000, 33, 545–552 CrossRef CAS.
- L. Li, J. Yang, X. Chen and X. Hao, Synth. Met., 2009, 159, 2462–2468 CrossRef CAS PubMed.
- D. Nichetti and I. Manas-Zloczower, J. Rheol., 1998, 42, 951–969 CrossRef CAS.
- E. A. Collins and A. P. Metzger, Polym. Eng. Sci., 1970, 10, 57–65 CAS.
- M. Choi and J. Jang, J. Colloid Interface Sci., 2008, 325, 287–289 CrossRef CAS PubMed.
- H. Gu, S. Tadakamalla, X. Zhang, Y.-D. Huang, Y. Jiang, H. A. Colorado, Z. Luo, S. Wei and Z. Guo, J. Mater. Chem. C, 2013, 1, 729–743 RSC.
- G. Wisanrakkit, J. K. Gillham and J. B. Enns, J. Appl. Polym. Sci., 1990, 41, 1895–1912 CrossRef CAS.
-
K. Dušek, Epoxy Resins and Composites III, Springer, Berlin Heidelberg, 1986, vol. 78, pp. 1–59 Search PubMed.
- Z. Guo, H. W. Ng, G. L. Yee and H. T. Hahn, J. Nanosci. Nanotechnol., 2009, 9, 3278–3285 CrossRef CAS PubMed.
- Y. Sun, Z. Zhang, K.-S. Moon and C. P. Wong, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 3849–3858 CrossRef CAS.
- J. Zhu, S. Wei, J. Ryu, M. Budhathoki, G. Liang and Z. Guo, J. Mater. Chem., 2010, 20, 4937–4948 RSC.
- B. Fabry, G. N. Maksym, J. P. Butler, M. Glogauer, D. Navajas and J. J. Fredberg, Phys. Rev. Lett., 2001, 87, 148102 CrossRef CAS.
- E. Kontou and M. Niaounakis, Polymer, 2006, 47, 1267–1280 CrossRef CAS PubMed.
- N. Roy and A. K. Bhowmick, J. Phys. Chem. C, 2012, 116, 8763–8772 CAS.
- S. Barrau, P. Demont, C. Maraval, A. Bernes and C. Lacabanne, Macromol. Rapid Commun., 2005, 26, 390–394 CrossRef CAS.
- J. K. W. Sandler, J. E. Kirk, I. A. Kinloch, M. S. P. Shaffer and A. H. Windle, Polymer, 2003, 44, 5893–5899 CrossRef CAS.
- B. J. Ash, R. W. Siegel and L. S. Schadler, Macromolecules, 2004, 37, 1358–1369 CrossRef CAS.
- Y. Sha, C. Y. Hui, A. Ruina and E. J. Kramer, Macromolecules, 1995, 28, 2450–2459 CrossRef CAS.
- B. Akbari and R. Bagheri, Eur. Polym. J., 2007, 43, 782–788 CrossRef CAS PubMed.
- X. Zhang, V. Bitaraf, S. Wei, Z. Guo, X. Zhang, S. Wei and H. A. Colorado, AIChE J., 2014, 60, 266–274 CrossRef CAS.
- Y. Liu, Z. Du, C. Zhang and H. Li, Int. J. Polym. Anal. Charact., 2006, 11, 299–315 CrossRef CAS.
- J. Jang, J. Bae and K. Lee, Polymer, 2005, 46, 3677–3684 CrossRef CAS PubMed.
- X. Zhang, Q. He, H. Gu, H. A. Colorado, S. Wei and Z. Guo, ACS Appl. Mater. Interfaces, 2013, 5, 898–910 CAS.
- D. Wang, K. Zhou, W. Yang, W. Xing, Y. Hu and X. Gong, Ind. Eng. Chem. Res., 2013, 52, 17882–17890 CrossRef CAS.
- D. Wang, Q. Zhang, K. Zhou, W. Yang, Y. Hu and X. Gong, J. Hazard. Mater., 2014, 278, 391–400 CrossRef CAS PubMed.
- J. W. Gilman, Appl. Clay Sci., 1999, 15, 31–49 CrossRef CAS.
- Y. L. Liu, Polymer, 2001, 42, 3445–3454 CrossRef CAS.
- H. Durmus, H. Safak, H. Z. Akbas and G. Ahmetli, J. Appl. Polym. Sci., 2011, 120, 1490–1495 CrossRef CAS.
- W. Jia, R. Tchoudakov, E. Segal, R. Joseph, M. Narkis and A. Siegmann, Synth. Met., 2003, 132, 269–278 CrossRef CAS.
- S. Y. Hong, D. Y. Kim, C. Y. Kim and R. Hoffmann, Macromolecules, 2001, 34, 6474–6481 CrossRef CAS.
- S. Tsuzuki, K. Honda, T. Uchimaru, M. Mikami and K. Tanabe, J. Am. Chem. Soc., 2000, 122, 11450–11458 CrossRef CAS.
- X. Zhang, J. Zhu, N. Haldolaarachchige, J. Ryu, D. P. Young, S. Wei and Z. Guo, Polymer, 2012, 53, 2109–2120 CrossRef CAS PubMed.
- M. Rahman, C.-W. Hsieh, C.-T. Wang, B.-R. Jian and W. Lee, Dyes Pigm., 2010, 84, 128–133 CrossRef CAS PubMed.
- S. Singha and M. J. Thomas, IEEE Trans. Dielectr. Electr. Insul., 2008, 15, 12–23 CrossRef CAS PubMed.
- C.-D. Liu, S.-N. Lee, C.-H. Ho, J.-L. Han and K.-H. Hsieh, J. Phys. Chem. C, 2008, 112, 15956–15960 CAS.
- A. Schönhals, H. Goering, F. R. Costa, U. Wagenknecht and G. Heinrich, Macromolecules, 2009, 42, 4165–4174 CrossRef.
- J. Lu, K.-S. Moon, B.-K. Kim and C. P. Wong, Polymer, 2007, 48, 1510–1516 CrossRef CAS PubMed.
- J. B. Yin and X. P. Zhao, J. Phys. Chem. B, 2006, 110, 12916–12925 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4tc01978d |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 



 ), which was obtained by employing the Carreau model to correlate η and
), which was obtained by employing the Carreau model to correlate η and 
 ), C is the cross time constant, which represents the transition to a constant viscosity in the limit of zero shear rate57 and d is the rate index. The value of η0, summarized in Table 3, is observed to increase with increasing PPy nanostructure loading. At the same loading, the η0 of the nanosuspensions with PPy nanofibers is higher than that of the nanosuspensions with PPy nanospheres. Higher η0 can be attributed to the confined polymer chains, in which case the flow activation energy (Eη, kJ mol−1) should also be larger than that of the bulk polymer.58 Here, the Eη for pure epoxy resin and its nanosuspensions containing PPy nanofillers was studied with a plot of viscosity as a function of temperature (Fig. 4). Fig. 4(A & B) show the shear stress (σ) and viscosity (η) vs. shear rate (γ) at 25, 50 and 70 °C, respectively. The gradual increase of σ with increasing γ indicates the reliability of the measurements. The η of the nanosuspensions with 1.0 wt% PPy nanofibers decreases with increasing the temperature (Fig. 4(B)). The σ and η as a function of temperature for the nanosuspensions with 10.0 wt% PPy nanospheres, 10.0 wt% and 20.0 wt% PPy nanofibers are shown in Fig. S1–S3 in the ESI,† respectively. The relationship between temperature and η is shown in eqn (4):59
), C is the cross time constant, which represents the transition to a constant viscosity in the limit of zero shear rate57 and d is the rate index. The value of η0, summarized in Table 3, is observed to increase with increasing PPy nanostructure loading. At the same loading, the η0 of the nanosuspensions with PPy nanofibers is higher than that of the nanosuspensions with PPy nanospheres. Higher η0 can be attributed to the confined polymer chains, in which case the flow activation energy (Eη, kJ mol−1) should also be larger than that of the bulk polymer.58 Here, the Eη for pure epoxy resin and its nanosuspensions containing PPy nanofillers was studied with a plot of viscosity as a function of temperature (Fig. 4). Fig. 4(A & B) show the shear stress (σ) and viscosity (η) vs. shear rate (γ) at 25, 50 and 70 °C, respectively. The gradual increase of σ with increasing γ indicates the reliability of the measurements. The η of the nanosuspensions with 1.0 wt% PPy nanofibers decreases with increasing the temperature (Fig. 4(B)). The σ and η as a function of temperature for the nanosuspensions with 10.0 wt% PPy nanospheres, 10.0 wt% and 20.0 wt% PPy nanofibers are shown in Fig. S1–S3 in the ESI,† respectively. The relationship between temperature and η is shown in eqn (4):59











