
Enhanced photochromic efficiency of transparent and flexible nanocomposite films based on PEO–PPO–PEO and tungstate hybridization
C.
Wang
,
B. P.
Zhou
,
X. P.
Zeng
,
Y. Y.
Hong
,
Y. B.
Gao
and
W. J.
Wen
*
Nano Science and Nano Technology Program and Department of Physics, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong. E-mail: phwen@ust.hk; Web: http://www.phys.ust.hk/phwen/ Fax: +852-23581652; Tel: +852-23587979
First published on 16th October 2014
Abstract
Nanocomposite hybrid films were prepared by depositing tungstate on PEO–PPO–PEO (EPE) templates under acidic conditions by a one-pot self-assembly sol–gel method. Uniform and transparent films based on this precursor were fabricated and easily manipulated using a facile casting method. Upon irradiation by sunlight, the film exhibits a rapid photochromic response that was reversible at room temperature. UV-Vis analysis revealed that folding of the –CH2–O– chains in the polymer and the larger tungstate clusters increased the red shift for the adsorption of visible light. The mechanism underlying the effect of hybridization of polyethylene oxide (PEO) on the enhanced photochromic effect was characterized by NMR, Raman and FTIR. The degree of folding of the –CH2–O– polymer chains was influenced by the presence of acid, which increased the oxygen coordination of WO6 and WO4 in the tungstate clusters. The characterized W–O bonding peaks shifted upon EPE incorporation, consistent with changes in oxygen coordination within the material. This weak coordination between tungstate and –CH2–O– improved the intervalence charge transfer (IVCT). Oxygen from EPE may enter the oxygen sites in the O–W–O clusters to produce an unbalanced electron state, resulting in oxygen defects, which has a critical effect on the enhancement of the photochromic response. In addition, this method provides protons via this weak coordination, thereby dramatically enhancing the efficiency of the hybrid film.
1. Introduction
The photochromism of composite inorganic–organic materials has been the subject of intense investigation because of its potential application in optical devices, optical switches, and chemical sensors.1 Polytungstates (PT) are particularly promising photochromic materials because of their rich topological, chemical, and physical properties and because they can be easily and inexpensively synthesized.2 Crystalline tungstate trioxides usually exhibit good chemical and thermal stabilities with slow switching speeds. By contrast, conjugated polymers are useful for multicoloration and improving coloration efficiency and switching speed but suffer from poor stabilities.3 To overcome the problems associated with using WO3 and conjugated polymers as photochromic materials, nanocomposite hybrid PT polymers have been combined to form polyoxometalates (POMs), and their photoactivity,4 water splitting capability5 and use in energy-efficient windows6 have been studied extensively.Many studies have examined the optical density and reversibility of POM hybrids using various organic molecules and solvents,7–9 but photochromic POMs remain time-consuming to produce, unstable, and difficult to fabricate on a large-scale. The optical contrast of such hybrids primarily arises from the switching of one component between different redox states, but the redox reactions that occur within the hybrid tend to be destructive to the polymer. Therefore, it is very important to choose organic molecules, which not only serve as proton donors for the coloration and stabilization of hybrids, but can also act as electron donors for the formation of a coordination bond. Moreover, color bleaching may occur due to the high temperatures necessary to revert the color of the films.10,11 To dynamically control solar radiation transmittance through nanocomposite materials, various “smart windows” have been developed and studied extensively. However, some smart windows require a relatively complex external power supply device to alter their transparency electrochromically,12,13 which is incompatible with the goal of decreasing energy consumption and slowing down heat propagation in commercial buildings.14–16
This article addresses the aforementioned issues by presenting hybrid systems based on the amphiphilic surfactant EPE and sodium tungstate to enhance photochromic efficiency. These two components are widely used chemically stable structural materials. Using these materials, transparent gels can be easily fabricated on large areas of the film by a one-pot casting method. Supporting PTs in or on uniform matrices with large surface areas can effectively increase the utility of the composites by facilitating the formation of functional tungstate species. Compared with previous studies on hybridization, EPE molecules are only used to create defect sites for PT, not provide protons. Herein, we demonstrate that this combination contributes to the unbalanced electron state of the W–O framework, which is beneficial for enhancing IVCT. The film offers a rapid solar response without additional energy consumption and has the unique ability of recovering full transparency simply by consuming O2 without heating, thereby exhibiting great potential for use in photochromic smart windows.
2. Experimental section
Photochromic material preparation
The photochromic material was labelled in the order of surfactant (F127) + tungstate source + acid source (e.g. F127 + ST + H). F127STH precursor solution was synthesized by dissolving 3.3% w/v sodium tungstate (Na2WO4) into 10% w/v Pluronic F127 DI water solution under strong magnetic stirring at room temperature for 12 h. After being completely dissolved, the pH value was adjusted to 2 by using diluted sulfuric acid (5 mol L−1). F127WC precursor solution was also prepared following this procedure. 6% w/v ammonium paratungstate was added into 10% w/v F127 solution and citric acid was used to adjust the pH value to 2. The final film or block of the composite material was fabricated by a sol–gel method using the two clear precursor solutions described above. The two solutions were separately subjected to a vacuum defoaming process before being cast onto substrates, which were allowed to gelate by drying at room temperature overnight. These fabricated film samples are represented in black-bold text as F127STH and F127WC. The film for the X-ray diffraction (XRD) test was based on the F127WC precursor, whose tungstate weight ranged from 1% w/v to 6% w/v (samples 1–6). Samples a–f contained the same concentration of 6% w/v tungstate but 1% w/v, 2% w/v, 3% w/v, 5% w/v, 8% w/v and 10% w/v F127 concentration, respectively. The formaldehyde-containing sample was fabricated, in which 35% formaldehyde–water solution was used to replace the EPE solution. The fast-response reversible gel was fabricated by facile casting technology on commercial glass or PET substrates at room temperature. After evaporation, the films were covered with a single layer of a polyacrylamide polymer framework to enhance their mechanical properties. The resulting self-supporting nanocomposite films were easily peeled from the substrates due to the surface wettability of the PET plates. Uniform film thickness was achieved by casting the same concentration and amount of film-forming solution on substrates with the same surface area. The procedure of the dip-coating method was that the film was first exposed to plasma for 2 minutes; then it was dip-coated in an aqueous precursor for 30 seconds and then exposed to plasma for 2 minutes again after being dried in air, this process was repeated 10 times. The post-sintering of film samples was performed in air at 500 °C for 2 h and the temperature was increased by 10 °C per minute to 500 °C. All chemicals were purchased from Sigma-Aldrich and used without further purification.Characterization
The chemical structures and compositions of the thin films were characterized by Fourier transform infrared (FTIR) spectroscopy (Perkin Elmer Spectrum GX), X-ray photoelectron spectroscopy (XPS, Physical Electronics PHI) and Raman spectroscopy (Renishaw Raman spectroscopy with an excitation wavelength of 540 nm). The FTIR scans were conducted on a silicon wafer from 4000 to 600 cm−1, with 32 scans collected for signal averaging. To compensate for surface-charging effects in the XPS scans, the binding energy level of C1s was set at 285 eV for further data analysis. The structure of the hybrid thin films was characterized by XRD analysis using a Rigaku Dmax diffraction system using a Cu Kα source (λ = 1.54187 Å). UV-Vis transmittance spectra were recorded over a wavelength range of 200 to 1100 nm. The film coloration and bleaching data were recorded under mercury lamp irradiation with an average irradiance of 50 mW cm−2; transmittance data of coloration and bleaching processes were recorded at 640 nm and 600 nm. Scanning electron microscopy (SEM) images were obtained with a JOEL 6390 scanning electron microscope. The SEM samples were prepared by sintering sample powder onto a Cu substrate. High-resolution transmission electron microscopy (HRTEM) images were obtained with a JEOL-2010 transmission electron microscope at 200 kV.3. Results and discussion
3.1 One-pot synthesis of the hybrid film
In this work, the synergistic effect of EPE, acid and tungstate is mainly discussed. The self-assembled PT is prepared in the aqueous phase. Under aqueous conditions, EPE has a high propensity for partitioning and favors a hexagonal structure as its concentration approaches saturation.17 The spherical structure shown in Scheme 1 is formed by many EPE molecules. One EPE molecule contains two PEO folding tails, which tend to form a double helical cylindrical (DNA-like) shape in water.19 The cylindrical structures formed by the two folding C–O structures efficiently form a space that is occupied by tungstate anions. These structures not only provide an interspace for PT, but also contribute to the formation of the defect sites. The photochromic mechanism is described in previous work.1,18 Light induces tungstate oxide to generate one hole–electron pair, with the electron trapped in the PT defect site. A proton is then inserted into the PT network. Our method exploits the role of acid ions (tridentate carboxylate or sulfate) via weak coordination to contribute to separate the function of the organic molecule for hybridization. Acidic ions also play a crucial role in the formation of interactions between EPE and tungstate in the high-yield coloration-bleaching process, which is dependent on the provision of protons to form a crown-ether structure.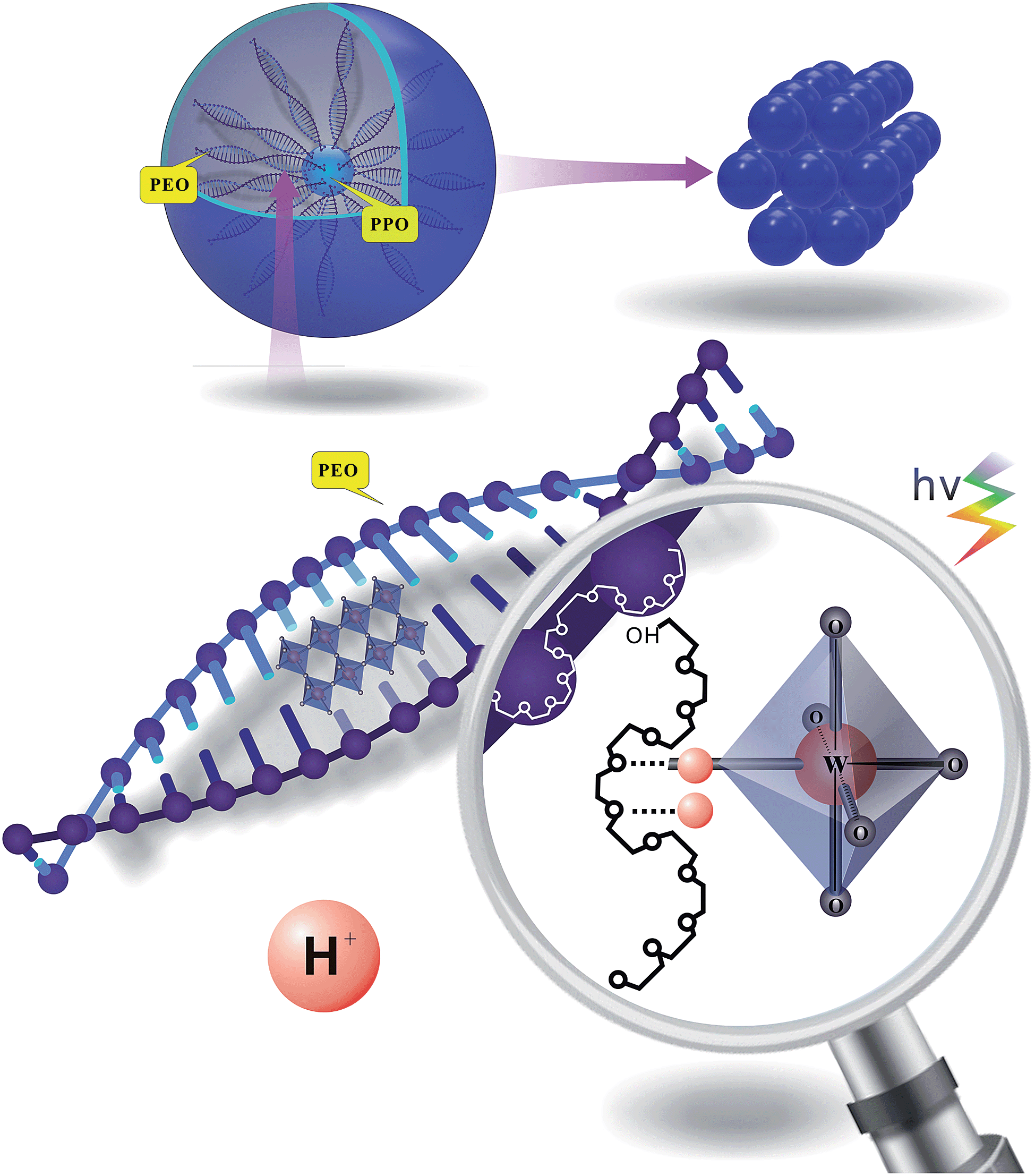 | ||
| Scheme 1 Synthesis of the EPE/WO3 hybrid film from aqueous precursors. | ||
To determine how the citrate and sulfate anions influence the EPE molecule differently at the same hydrogen ion concentration (pH value), the interaction between the acid and the triblock template was measured by NMR (Table 1). Integration of the peaks in the 1H-NMR spectra (Fig. 1), obtained in different chemical buffers demonstrated that the specific values of the functional groups of different molecules were influenced by their correlated action in aqueous solution. Resonances were observed for –CH3 groups at 1.16 ppm, –CH– groups as a broad peak at 3.5 ppm, –CH2– groups at 3.627 ppm, and –CH2–CH2– sequences at 3.694 ppm. The hyperfine structure of the EPE molecule contained broad peaks corresponding to the –CH2–CH– and –CH3 units of the PO groups that partially overlapped with the large peak of the (CH2)2 units of the EO groups; thus, it can be concluded that the block copolymer molecules dissolve as monomers and that the segments of the chains can consequently move freely. The values presented in Table 1 indicate that the PPO groups tend to dissociate in the core because [WO4]2− exhibited only minor changes in its specific value in systems containing the surfactant only and systems containing the surfactant and acid. However, the specific values for both the EO and PO groups decreased after the addition of citrate and sulfate to the solution from initial average values of 1.41/1 and 1.35/1 to 1.13/1 and 0.95/1, respectively. It can be concluded that the symmetric –CH2–O– molecules were locked into the interspace of tungstate upon acidification, without retaining their former symmetric structure. The presence of the acidic anions provided more weak coordination opportunities for the oxygen and alkane functionalities of the PEO units interacting with metal oxide anions due to the double helical structure of the alkali metal–PEO complexes. Moreover, the decrease in the PEO integrated value upon the addition of sulfuric acid (0.95/1) is larger than that observed upon the addition of citric acid (1.1/1). According to the Hofmeister series, the sulfate anion contributes more to the destabilization and destruction of EPE's former asymmetrical state than the citrate anion does.
| EPEc | C | EPE + W | EPE + C | EPE + WC | EPE + H | |
|---|---|---|---|---|---|---|
| a Specific value ratios of the hydrophobic groups –CH3, –CH–, and –CH2– and the hydrophilic group –CH2–CH2–. b Specific value ratios of carboxylic and hydroxylic groups. c EPE: Pluronic F127; C: citrate; W: tungstate; and H: sulfate. | ||||||
| 1.41/1 | — | 1.35/1 | 1.13/1 | 1.05/1 | 0.95/1 | |
| — | 1.55 | — | 1.68 | 1.66 | — | |
By contrast, when citric acid interacts with EPE, the integrated peak intensity of the carboxyl groups of the citrate molecule increased from 1.55 to 1.67 relative to the hydroxyl groups. This indicates that the carboxyl groups favor the asymmetric structure of the PEO–PPO–PEO molecules. The two-sided terminal –COOH also plays a role in forming mixed-metal complexes. Under certain pH conditions, a stable complex is irreversibly formed between PEO and carboxylic groups. During the process of complex formation, the dissociated carboxylic groups were influenced by neighboring undissociated PEO groups by taking up protons from the solution into the domain of the polymer chains through hydrogen bond formation. Moreover, esterification reactions occur between the alcohol group at the end of PEO and citric acid. Although this process was not dominant here, it may affect the established edge of the citrate–tungsten clusters.
Based on the results of dynamic light scattering experiments, the F127 micelle size increased upon the addition of citric or sulfuric acid to average diameters of 19 nm and 20 nm from their original size of 17 nm. Acid modification does not only affect the micelle surface but also appreciably changes the size of the micelles as well. PEO–polypropylene oxide has been shown to be very surface active in aqueous solution. When the pH of the solution decreases significantly, the resulting flattened conformation of PEO in water results in a majority of the molecular segments being in contact with the water interface. In this conformation, the tungstate anion and a suitable acid may occupy the folded portion of PEO. As indicated by the NMR results, the sulfate anion contributes more to the destabilization of PEO than citric acid does. Thus, the crown-ether-like structure of F127 in sulfuric acid is less asymmetrical than that in citric acid. Therefore, the relatively short, small tungstate anion can more easily occupy the defects.
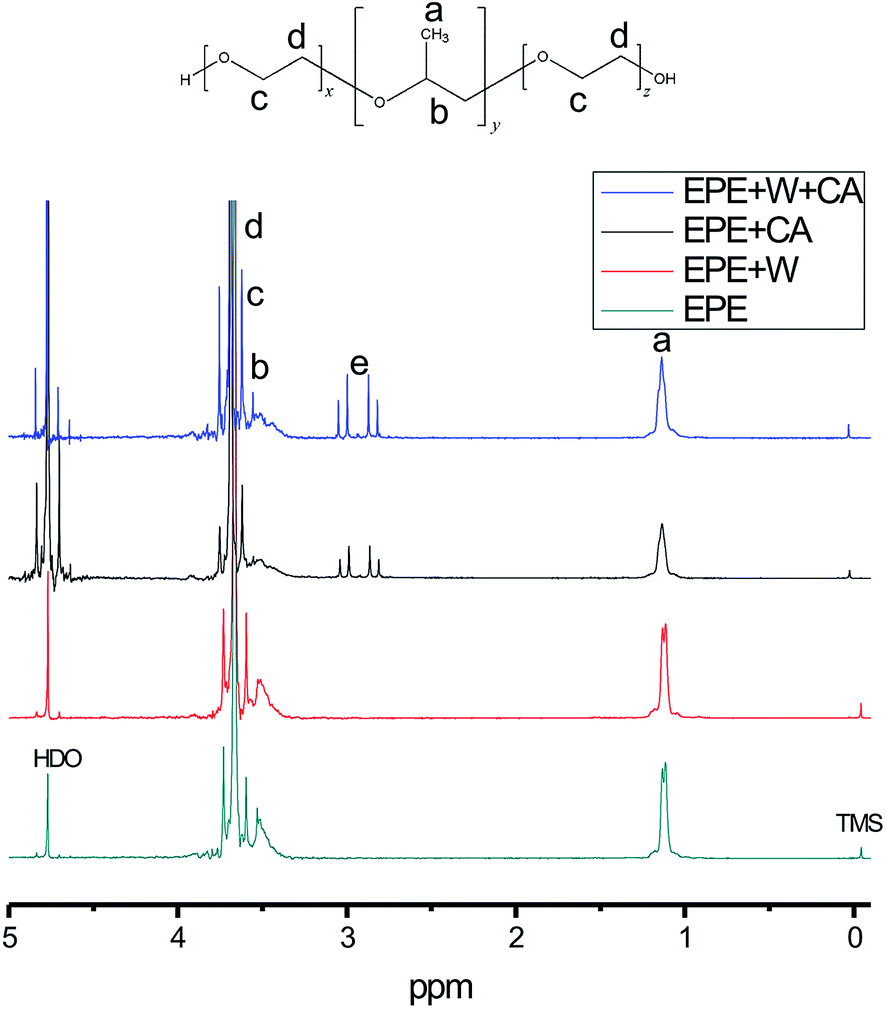 | ||
| Fig. 1 NMR analysis of tungstate acid with PEO–PPO–PEO. | ||
3.2 Mechanism of formation of the hybrid film
The chemical structures of the hybrid films were verified by FTIR and Raman spectroscopies. These two analytical methods are powerful tools for characterizing the structural alteration that occurs upon the introduction of the surfactant and acid. Fig. 2(A)–(C) describe tungstate, EPE and acid molecule peak shifts due to hybridization in the FTIR and Raman spectra, respectively. Fig. 2(A), tungstate (a) and (b), shows the FTIR spectra of different films composed of F127STH, F127WC, and F127 with each tungstate source and tungstate alone. For instance, as shown in the FTIR spectra of ST series one (a), the three characteristic peaks of tungstate were present in the sodium tungstate crystal but were lost upon the addition of sulfate. However, when measured in systems containing EPE, the intensities of these peaks all increase when acid was added (785 cm−1 ascribed to Oc–W–Oc; 837 cm−1 ascribed to Ob–W–Ob; and 962 cm−1 ascribed to W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ot, where Ot is the terminal oxygen, Ob is the bridged oxygen of two octahedra sharing a corner, and Oc is the bridged oxygen sharing an edge). It should be mentioned that the strong absorption band at 837 cm−1 was related to shared bonding in the O–W corner, which was lost when the pH changes. Meanwhile, the peak at 619 cm−1 for W–O increases significantly, which may be attributed to weak coordination arising from hydrogen bonding. These results also demonstrate that –CH2–O– was incorporated into the nanocomposite PT cluster film. Similar FTIR results were obtained for the citrate and paratungstate systems. Henceforth, the discussion of Fig. 2(A), tungstate (c) and (d), will focus mainly on the sulfate and sodium tungstate system. Based on the Raman spectra shown in Fig. 2(A), tungstate (c), the Raman bands of aqueous tetrahedral WO42− (ST) were assigned to a (WO) symmetric stretching at 931 cm−1 and a broad (W–O) asymmetric stretching at 834 cm−1. When the pH value reached 2.3 after acidifying the solution, the band at 931 cm−1 disappeared, and a wide band at 960–969 cm−1 appeared in the Raman spectra. These changes occurred because aqueous octahedral W12O396− and W6O192− polyanions displayed a (W–O) stretching mode at 970–960 cm−1. An additional band at higher wavenumber (999 cm−1) appears after sulfate addition; this band was attributable to the stretching modes of polyanionic species W10O324− and W6O192−.21 Upon the addition of EPE, the bands at 999 cm−1 attributed to the symmetric and asymmetric stretching vibrations of the WO terminal group decreased in intensity and shifted to 995 and 1005 cm−1, respectively, due to changes in the charge of the tungstate cluster. The W–O corner-sharing band at 960–970 cm−1 shifted to 980 cm−1, characteristic of tetrahedral WO4. The 983 cm−1 peak in Fig. 2(C), acid (b), was also consistent with formation of a tetrahedral structure tungstate cluster. The W–O–W band also clearly shifted, as indicated by the dotted line in Fig. 2(A), tungstate (a). These changes indicate that the vibrations of tungstate were disturbed in the nanocomposite film due to hydrogen-bonding interactions between the tungstate molecule and EO groups and weak coordination between the tungstate and sulfate groups. The oxygen in EPE may also contribute to bonding in the WO6 and WO4 frameworks. The special bonding observed in this study resulted in an unbalanced electron state, which is likely to manifest as a bond for one tungstate unit. In this manner, the formation of the composite increases the number of defects in the tungstate and enhances its photochromic character. Similar phenomena were observed in the paratungstate and citrate systems shown in Fig. 2(A), tungstate (b) and (d).
Ot, where Ot is the terminal oxygen, Ob is the bridged oxygen of two octahedra sharing a corner, and Oc is the bridged oxygen sharing an edge). It should be mentioned that the strong absorption band at 837 cm−1 was related to shared bonding in the O–W corner, which was lost when the pH changes. Meanwhile, the peak at 619 cm−1 for W–O increases significantly, which may be attributed to weak coordination arising from hydrogen bonding. These results also demonstrate that –CH2–O– was incorporated into the nanocomposite PT cluster film. Similar FTIR results were obtained for the citrate and paratungstate systems. Henceforth, the discussion of Fig. 2(A), tungstate (c) and (d), will focus mainly on the sulfate and sodium tungstate system. Based on the Raman spectra shown in Fig. 2(A), tungstate (c), the Raman bands of aqueous tetrahedral WO42− (ST) were assigned to a (WO) symmetric stretching at 931 cm−1 and a broad (W–O) asymmetric stretching at 834 cm−1. When the pH value reached 2.3 after acidifying the solution, the band at 931 cm−1 disappeared, and a wide band at 960–969 cm−1 appeared in the Raman spectra. These changes occurred because aqueous octahedral W12O396− and W6O192− polyanions displayed a (W–O) stretching mode at 970–960 cm−1. An additional band at higher wavenumber (999 cm−1) appears after sulfate addition; this band was attributable to the stretching modes of polyanionic species W10O324− and W6O192−.21 Upon the addition of EPE, the bands at 999 cm−1 attributed to the symmetric and asymmetric stretching vibrations of the WO terminal group decreased in intensity and shifted to 995 and 1005 cm−1, respectively, due to changes in the charge of the tungstate cluster. The W–O corner-sharing band at 960–970 cm−1 shifted to 980 cm−1, characteristic of tetrahedral WO4. The 983 cm−1 peak in Fig. 2(C), acid (b), was also consistent with formation of a tetrahedral structure tungstate cluster. The W–O–W band also clearly shifted, as indicated by the dotted line in Fig. 2(A), tungstate (a). These changes indicate that the vibrations of tungstate were disturbed in the nanocomposite film due to hydrogen-bonding interactions between the tungstate molecule and EO groups and weak coordination between the tungstate and sulfate groups. The oxygen in EPE may also contribute to bonding in the WO6 and WO4 frameworks. The special bonding observed in this study resulted in an unbalanced electron state, which is likely to manifest as a bond for one tungstate unit. In this manner, the formation of the composite increases the number of defects in the tungstate and enhances its photochromic character. Similar phenomena were observed in the paratungstate and citrate systems shown in Fig. 2(A), tungstate (b) and (d).
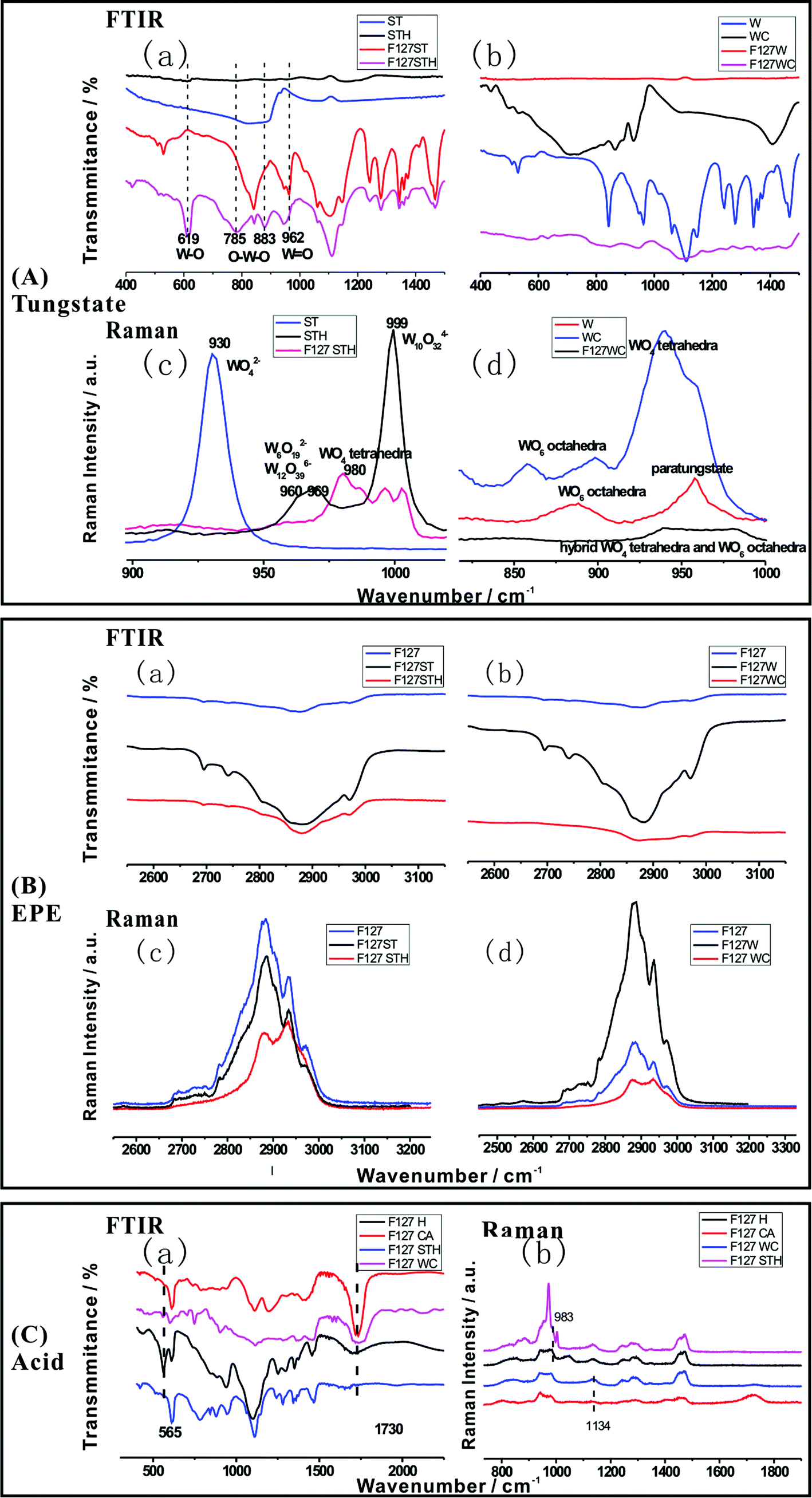 | ||
| Fig. 2 (A) FTIR spectral changes (a), (b) and Raman spectral changes (c), (d) of tungstate; (B) FTIR spectral changes (a), (b) and Raman spectral changes (c), (d) of EPE; (C) FTIR spectral changes (a) and Raman spectral changes (b) of acid as a function of wavenumber in the hybrid. | ||
For the EPE molecule, shown in Fig. 2(B), EPE (a) and (b), the strong characteristic FTIR bands for the –CH2–O– group, which were typically found at 2800–3000 cm−1, were initially larger than usual owing to the enhancement caused by resonance between the transition metal oxide (W–O) and EPE structure.20 However, the bands were lost when the pH was reduced to 2 by the addition of sulfate or citrate, indicating that a portion of the groups in the EPE molecule was intercalated into the hybrid. From this point, the EO molecule is also affected by the tungstate anion and the acid, as shown in Fig. 2(B). The characteristic peaks of the EO chain in both the FTIR and Raman spectra (Fig. 2(B), EPE (c) and (d)) increased in intensity upon the addition of tungstate and decreased upon the addition of acid due to the domination of proton interactions over the weak coordination between –CH2–O– and tungstate.
The same conclusion is reached based on the changes in the acid peak shown in Fig. 2(C). The results shown in Fig. 2(C), acid (b), indicate that when tungstate was added, the spectral peaks associated with –COOH (1730 cm−1) and S–O (565 cm−1), shown in Fig. 2(C), acid (a), weakened. Therefore, acid can be incorporated into the hybrid structure to form a correlated hybridization composite system comprising three parts (tungstate, acid, and –CH2–O–).
XPS was used to further confirm the chemical structures of the hybrid films and probe interfacial interactions. Fig. 3b shows photoelectron spectra obtained during hybridization, which reveal variations in W4f levels. The shape of the XPS curve before hybridization is convoluted into two doublets, of which the main doublet at 35.8 eV was due to the W4f7/2 orbital and the other at 37.8 eV was assigned to W4f5/2, corresponding to the W6+ oxidation state. As shown in Fig. 3a, a second doublet appeared after hybridization at a lower binding level with peaks at 34.7 eV and 36.7 eV, which were assigned to W4f7/2 and W4f5/2 of the W5+ oxidation state, respectively. The results indicated the formation of W5+ species after hybridization, in agreement with the electron state changes of tungstate. The percentage of W5+ after hybridization was 25.1%, corresponding to W5+/(W5+ + W6+) = 1/5. This result illustrates that the unbalanced oxygen number is induced by hybridization. The chemical formula of the hybrid gel component can be simplified to H0.2WO3.
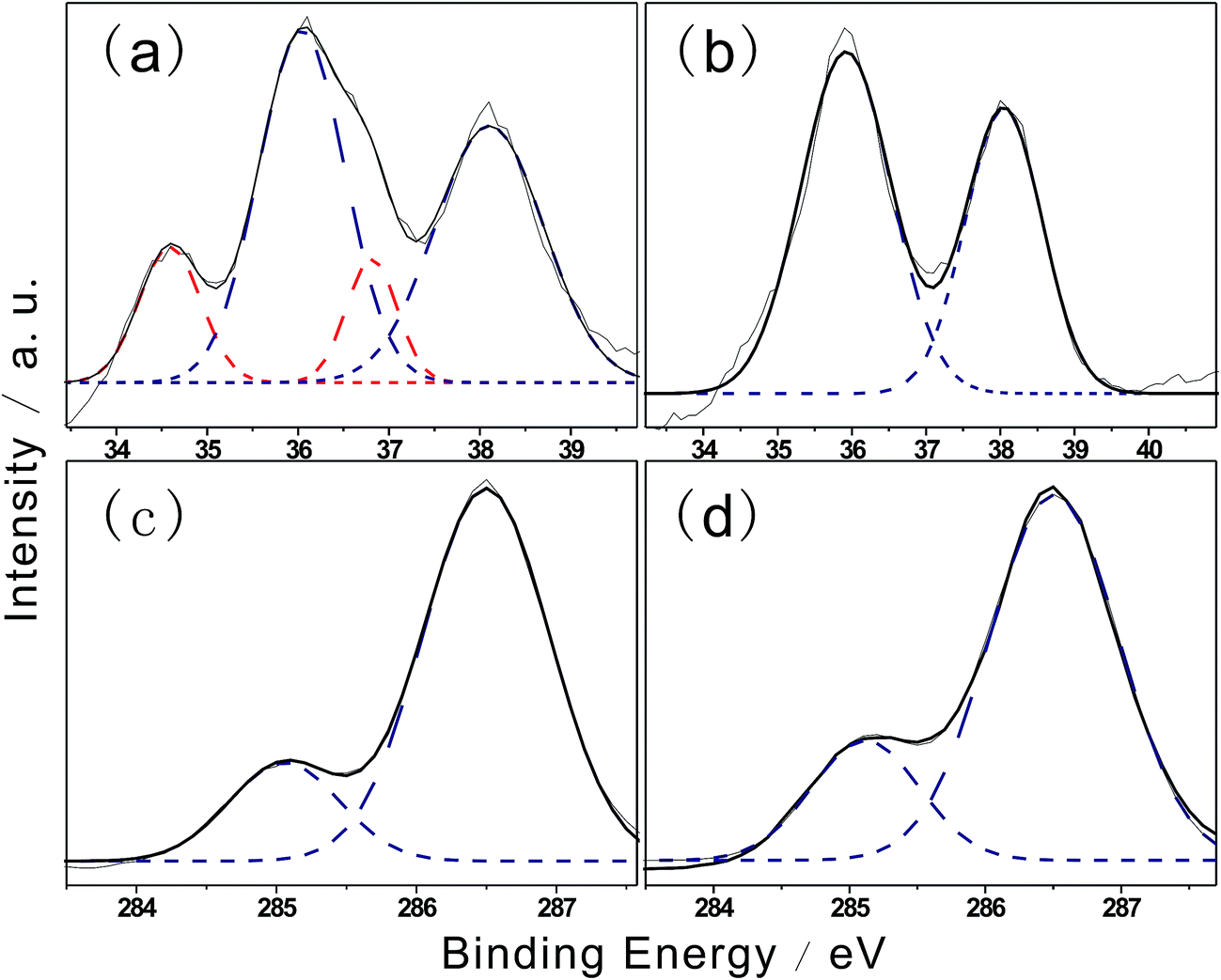 | ||
| Fig. 3 XPS analysis of the hybridization of the thin films F127STH (a and c) and F127WC (b and d). | ||
When tungstate was deposited with the EPE layer, the EPE/WO3 hybrid thin film exhibited two main characteristic peaks at 38.1 eV (W4f5/2) and 36.1 eV (W4f7/2); these peaks were 0.3 eV higher than those of the WO3 film.22,23 The increase in binding energy was likely due to the presence of a large amount of defects caused by the weak coordination.
C1s spectra of F127STH and F127WC are shown in Fig. 3c and d, respectively. The lower binding energy peak at 285.0 eV was assigned to carbon bonded to hydrogen. The appearance of a second peak at 286.5 eV was assigned to C–O bonding present in weak coordination. Compared with results reported in previous work,24 –CH2–O– bonding was greatly enhanced after hybridization.
3.3 Structural verification
To investigate the structural changes that occur upon calcination of the films, F127WC was analyzed by XRD and TEM (Fig. 4). In this analysis, samples fabricated with ammonium paratungstate were compared to a control sample, labeled 0. Although these two tungstate oxides exhibited similar XRD patterns post-sintering (Fig. 4a and b), individual WO42− and the tungstate clusters formed by ammonium paratungstate underwent a different process from aqueous solution. It can be deduced that aqueous polytungstate loses its photochromic effect when the oxygen defects vanish during the sintering. PT fills space in the PEO folding framework, with multi-dentate acids also contributing to the formation of ordered nanostructures.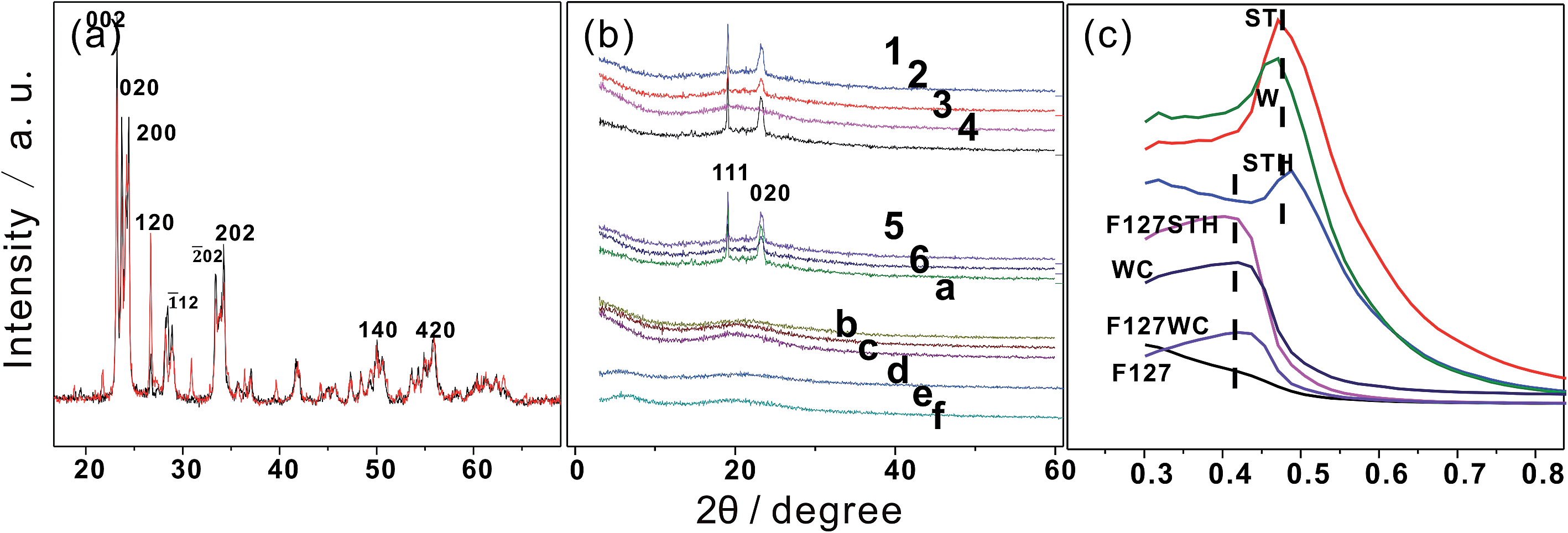 | ||
| Fig. 4 (a) X-ray diffraction pattern of WO3 prepared from sodium tungstate and ammonium paratungstate precursors; (b) XRD patterns of samples from 1–6 and a–g after gel drying. (c) SAXRD of different films. | ||
In addition, as shown in Fig. 4c, the evolution of layered structures can be observed in the SAXRD patterns. The inorganic moieties of the tungstate clusters exhibited a layered structure with d-spacings of 9.2 nm and 9.3 nm. For F127STH, the observed increase in the d-spacing was due to the insertion of additional –CH2–O– molecules into its inorganic layers with the continuous addition of EPE, which did not in itself exist as part of the ordered structure. By contrast, the carboxyl groups were incorporated into the hybrid film by adsorption onto an underlying organic layer to form an acid–anion complex.
After sintering the film at 500 °C for 2 h, WO3 crystallized from the gel and aggregated as 5 μm particles, as shown in the SEM images in Fig. 5. The uniform distribution of WO3 was the result of self-assembly of precursor clusters. In Fig. 5a and b, ball-shaped and flower-like structures were apparent due to the spherical aggregation behavior of the clusters. In addition, the nanoscale WO3 appeared as small dots on the surface of larger microparticles in Fig. 5c. These nanoscale WO3 particles grew by the Ostwald ripening process and aggregated at the surface of the WO3 microparticles. By adopting this sol–gel self-assembly approach, a uniform nanocrystal dispersion that incorporates both ligand interactions and polytungstate crystal formation can be achieved.
 | ||
| Fig. 5 SEM (a–c) and TEM (d–l) micrographs showing the morphology of WO3 after annealing. Image (c) shows an amplified section of image (b). Micrographs (d–f) show samples prepared without the addition of the surfactant or acid. The samples shown in (g–i) were prepared in the presence of acid. Micrographs (j–l) are images of samples containing both surfactant and acid. | ||
The addition of the surfactant or acid enhanced the nanocrystallization process, leading to a significant increase in the number of nanoparticles formed. TEM micrographs acquired after the sintering process (d–l) demonstrate that the formation of small nanocrystals was induced by the addition of the surfactant (g–i) and citrate (j–l). The dark-field diffraction analysis of tungsten trioxide (f), (i), and (l) revealed that the microstructure of the particles was consistent with those produced using oxalic acid.1 Moreover, the XRD pattern (Fig. 4b) was also consistent with the observed tendency of amorphous particles to form upon the addition of the surfactant and acid because the level spacing between adjacent electron states is inversely proportional to the nanoparticle diameter. A decrease in the size of the oxide particles resulted in the broadening of the band gap between the highest occupied and lowest unoccupied electron levels. Thus, more energy is needed to induce an electron transition during the photochromic process for smaller particles (i.e., quantum-size effects), leading to a blue shift in the optical absorption maximum. As the concentrations of the surfactant and acid increased, the oxide becomes more amorphous, implying the presence of more defect sites that facilitate the charge-transfer process. The effects of both surfactant and acid in aqueous solution enhance the photoinduced effects due to particle size and boundary conditions. The gel can also be further condensed into a film or block without losing its enhanced photochromic properties.
3.4 Photochromic behavior of the hybrid film
Photochromic enhancement was illustrated by UV-Vis spectra as shown in Fig. 6. It indicates that as the concentration of F127 increases in Fig. 6a, the peak corresponding to tungstate absorbance tends to red shift as the crystalline structure became more regular. The ordered composition of the tungstate anion resulting from the combination of poly- or mono-tungstate with EO had a much broader absorbance bandwidth.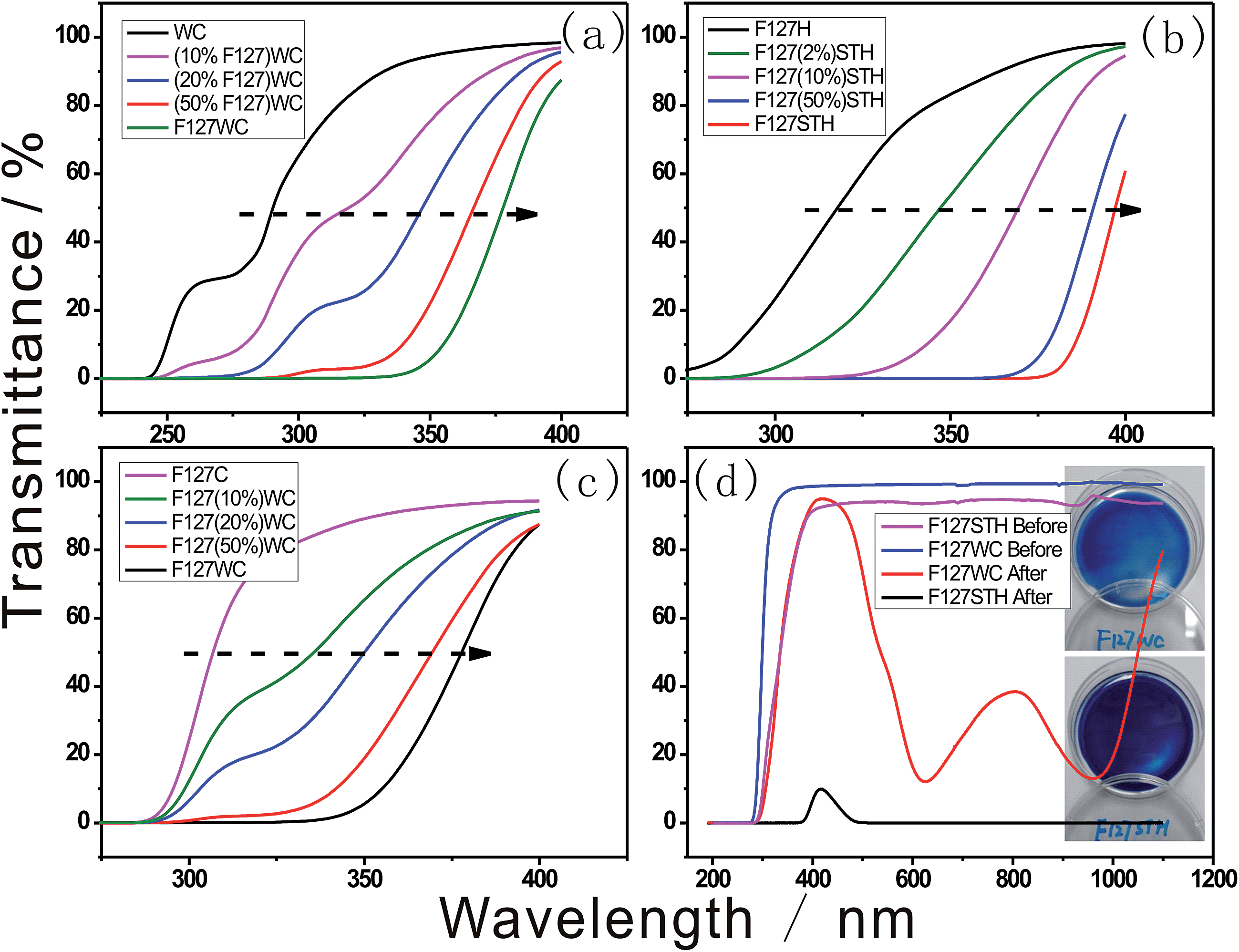 | ||
| Fig. 6 UV-Vis spectra of polytungstate under acidic conditions with varying concentrations of (a) F127, (b) sodium tungstate, (c) paratungstate and (d) UV-Vis spectra of before and after fully reduced F127WC and F127STH samples with their photographs nearby. | ||
As shown in Fig. 6b and c, the addition of the tungstate anion induced a tunable shift in the transmittance wavelength. The size of the tungstate compounds formed changed as the concentration of tungstate increased, which in turn altered the size of the clusters formed. In particular, the sodium tungstate clusters formed in the presence of sulfate could be tuned to absorb in the visible light region. The blue color of the composite was created by electron hopping between W–O bonds in the cluster through IVCT. Therefore, the combination of the clusters is vital for the electromagnetic wave resonance. This combined structure is influenced by the folding of the long EO chains and the acidic anion.
In the surfactant and tungstate system, according to the Hofmeister series, citrate and sulfate tend to salt biomacromolecules out of solutions and increase the stability of biomacromolecules toward their folded natural state. These two well-hydrated anions interact with the hydrophilic portion of the polymer by changing the entropy of hydration water around the F127 micelle.25
The hydrated anions increase the stability according to the following order: citrate3− > sulfate2− > phosphate2− > F− > Cl−.
For the same tungstate species, citrate buffer has the greatest stabilizing effect, particularly for the relatively large paratungstate. A transparent, stable solution is a necessary precursor for fabricating flexible composite films. In aqueous solution, only the citrate and sulfate systems can achieve this photochromic effect. The interaction of the acid anion with the EO chain is vital to this process, as is the important influence the anions exert in terms of controlling the tungstate anion.
In Fig. 6d, the different transmittance spectra of the samples revealed that they were composed of different-sized tungstate anions and acid species, resulting in different depths of blue color in each sample. The –CH2–O– chain surfactant was used to form both samples, indicating that –CH2–O– hybridization with tungstate is ubiquitous under acidic conditions. As shown in Fig. 6d, the F127STH sample not only contains a higher concentration of tungstate owing to its saturation with sodium tungstate but also exhibits another type of self-assembly behavior. The black color of the F127STH sample resulted in an increase in its temperature upon IR irradiation. Besides that, sample F127STH's absorption over the entire range of the solar spectrum (including the IR region) could induce photo-thermal coloration.26 In the film containing the PT network, extraction of the hydrogen oxide caused IR-photochromic behavior. The hybrid film afforded the advantages of long durability of the colored state, which was maintained for a longer lifetime.
In contrast to the citrate-containing sample, in the sulfate-containing sample, the color change occurred at the air–water interface during both the coloration and bleaching processes. There are two major reasons for this phenomenon. First, the paratungstate cluster anion is partitioned more fully by citrate, which suspends the particles more uniformly within the system. Second, as a type of surfactant, the EPE molecule tends to condense at the air–water interface. As surfactant condensation increases, the degree of folding of –CH2–O– intensifies, resulting in coloration of the air–water interface. Tungstate anion clusters need more compact space to form the hybridization structure shown in Scheme 1. Therefore, the film exhibits an enhanced photochromic interface because the condensed gel developed by the dip-coating method advantageously improves the photochromic behavior by increasing the folding level.
In addition to EPE, formaldehyde was used for W5d–O2p hybridization. Formaldehyde-containing films exhibited photochromic enhancement similar to that observed for F127-containing films. However, after drying under ambient conditions for 1 day, the photochromism of the formaldehyde-based films was lost due to the lack of C–O hybridization once formaldehyde degassed from the material.
Images of highly transparent flexible thin films are shown in Fig. 7a and b. The as-prepared films had smooth surfaces and were highly transparent and scatter-free due to the good miscibility of the tungstate anion and F127, the homogeneous distribution of tungstate in the films, and the elimination of microbubbles. In addition, oxygen defects were preserved during the fabrication process, enhancing the photochromic character of the system. Moreover, the efficiency and density of the color change were both enhanced significantly by film formation due to further condensation of the EO chains in the gel.27
 | ||
| Fig. 7 Hybrid films (a) before and (b) after UV irradiation; (c) dynamic optical transmittance of the F127STH gel. | ||
The appearance of dark-blue coloring occurred immediately upon irradiation of this sample under a 50 mW cm−2 UV lamp for 1 second, this efficiency was much higher than that of neat WO3 or other hybrid films. The dynamic contrast of the hybrid gel was approximately 80%. With respect to the switching kinetics, bleaching was observed when exposed to a sufficient amount of O2 under ambient conditions.28 The reversible change processes for large-scale thin film precursor in the transmittance of the gel exposed to a series of optical excitation cycles are shown in Fig. 7c. There was no obvious difference in the maximum and minimum transmittances corresponding to the color-bleaching process (recorded at 600 nm and 640 nm, respectively) after many cycles. The hybrid gel exhibited remarkably improved stability, retaining 98% of its original contrast after 10 cycles. The enhanced stability of the hybrid can be attributed to the fact that defect sites caused by hybridization may not involve the oxidation or reduction of tungstate. During the coloration process, an electron–hole pair is generated in hybrid H0.2WO3 by solar irradiation: the hole splits a water molecule, and the electron reduces W6+ to W5+, and a proton is intercalated following the intercalation of the electron. During the bleaching process, the electron reacts with oxygen molecules to form reactive oxygen species. Thus it is observed that the EPE molecule is only involved in electron state displacement, not in any reduction or redox process. Therefore, the reversible and sustainable photo-electrochemical behaviors observed are fast, steady and reproducible.
4. Conclusions
In summary, hybrid films consisting of EPE and tungstate were readily prepared using a one-pot self-assembly sol–gel method. The method provides a simple and efficient means of producing hybrid films over a large area. Moreover, the oxygen from EPE hybridizes the W5d–O2p electron state and generates defect sites in the W–O framework, enhancing the photochromic efficiency of the film. The fabrication and morphology of WO3 prepared from isopolytungstate also indicate that the formation of amorphous colloids is enhanced by the addition of acids and surfactants, which is beneficial for enhanced coloration. The characterization of films demonstrated that the observed photochromism is the result of electron transfer from W6+ to W5+ in the colloidal H0.2WO3 framework. Importantly, this method for enhancing the photochromic properties does not involve any redox or reduction processes that may destroy the polymer component of the films. Thus, the interaction between EPE and tungstate in the hybrid film not only enhances the photochromic response but also improves the durability of the hybrid film.Acknowledgements
The authors wish to acknowledge the support of NSFC/RGC joint grant No. N_HKUST601/11 and RGC grants No. HKUST 605411.References
- (a) T. He and J. N. Yao, Prog. Mater. Sci., 2006, 51, 810 CrossRef CAS PubMed; (b) H. Durr and H. Bouas-Laurent, Photochromism: Molecules and Systems, Elsevier, New York, 1990, vol. 1 Search PubMed; (c) G. M. Tsivgoulis and J. M. Lehn, Angew. Chem., Int. Ed., 1995, 34, 1119–1122 CrossRef CAS; (d) J. C. Crano and R. J. Guglielmetti, Organic Photochromic and Thermochromic Compound, Plenum, New York, 1999, vol. 1 Search PubMed; (e) M. Irie, Chem. Rev., 2000, 100, 1685 CrossRef CAS PubMed.
- (a) M. T. Pope and A. Muller, Angew Chem., Int. Ed., 1991, 30, 34–36 CrossRef; (b) A. Proust, B. Matt, R. Villanneau, G. Guillemot, P. Gouzerha and G. Izzet, Chem. Soc. Rev., 2012, 41, 7605–7622 RSC; (c) D. L. Long, R. Tsunashima and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 1736–1758 CrossRef CAS PubMed; (d) T. Yamase and M. T. Pope, Polyoxometalate Chemistry for Nano-composite Design, Springer, New York, USA, 2002 Search PubMed.
- (a) M. Gratzel, Nature, 2001, 414, 338 CrossRef CAS PubMed; (b) C. L. Lin, C. C. Lee and K. C. Ho, J. Electroanal. Chem., 2002, 524–525, 81–89 CrossRef CAS; (c) S. Vogel and R. Holze, Electrochim. Acta, 2005, 50, 1587–1596 CrossRef CAS PubMed; (d) A. Hagfeldt and M. Gratzel, Acc. Chem. Res., 2000, 33, 269–278 CrossRef CAS PubMed; (e) P. M. Beaujuge and J. R. Reynolds, Chem. Rev., 2010, 110, 268–320 CrossRef CAS PubMed; (f) C. L. Hill, D. A. Bouchard, M. Kadkhodayan, M. M. Williamson, A. Schmidt and E. F. Hilinski, J. Am. Chem. Soc., 1988, 110, 5471–5419 CrossRef CAS.
- (a) A. Hiskia, A. Mylonas and E. Papaconstantinou, Chem. Soc. Rev., 2001, 30, 62–69 RSC; (b) M. Lu, B. H. Xie, J. Kang, F. C. Chen, Y. Yang and Z. H. Peng, Chem. Mater., 2005, 17, 402–408 CrossRef CAS; (c) Y. B. Yang, L. Xu, F. Li, X. G. Du and Z. X. Sun, J. Mater. Chem., 2010, 20, 10835–10840 RSC; (d) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- (a) S. M. Wang, L. Liu, W. L. Chen, Z. M. Zhang, Z. M. Su and E. B. Wang, J. Mater. Chem. A, 2013, 1, 216–220 RSC; (b) X. Q. Gong, J. X. Li, S. Y. Chen and W. J. Wen, Appl. Phys. Lett., 2009, 95, 251907–251911 CrossRef PubMed.
- Z. B. Han, E. B. Wang, G. Y. Luan, Y. G. Li, H. Zhang, Y. B. Duan, C. W. Hu and N. H. Hu, J. Mater. Chem., 2002, 12, 1169–1173 RSC.
- X. Zhang, X. Dong and W. Yang, Thin Solid Films, 2006, 496, 533–538 CrossRef PubMed.
- X. J. Luo and C. Yang, Phys. Chem. Chem. Phys., 2011, 13, 7892–7902 RSC.
- Z. L. Wang, R. L. Zhang, Y. Ma, L. Zheng, A. D. Peng, H. B. Fu and J. N. Yao, J. Mater. Chem., 2010, 20, 1107–1111 RSC.
- G. Zhang, W. Yang and J. N. Yao, Adv. Funct. Mater., 2005, 15, 1255–1259 CrossRef CAS.
- S. Y. Park, J. M. Lee, C. Noh and S. U. Son, J. Mater. Chem., 2009, 19, 7959–7964 RSC.
- D. T. Gillaspie, R. C. Tenent and A. C. Dillon, J. Mater. Chem., 2010, 20, 9585–9592 RSC.
- L. Pérez-Lombarda, J. Ortizb and C. Poutb, Energ. Build., 2008, 40, 394–398 CrossRef PubMed.
- A. Azens and C. G. Granqvist, J. Solid State Electrochem., 2003, 7, 64–68 CrossRef CAS PubMed.
- C. G. Granqvist, Sol. Energy Mater. Sol. Cells, 2007, 91, 1529–1598 CrossRef CAS PubMed.
- (a) T. Ikawa, K. Abe, K. Honda and E. Tsuchida, J. Polym. Sci., Polym. Chem. Ed., 1975, 13(7), 1505–1514 CrossRef CAS; (b) P. Alexandridis and T. A. Hatton, Colloids Surf., A, 1995, 96, 1–46 CrossRef CAS.
- S. S. Kanu and R. Binions, Proc. R. Soc. A, 2010, 466, 19–44 CrossRef CAS.
- J. M. Parker, P. V. Wright and C. C. Lee, Polymer, 1981, 22, 1305–1307 CrossRef CAS.
- Z. G. Zhao and M. Miyauchi, Chem. Commun., 2009, 2204–2206 RSC.
- (a) L. Gao, E. B. Wang, Z. H. Kang, Y. L. Song, B. D. Mao and L. Xu, J. Phys. Chem. B, 2005, 109, 16587–16592 CrossRef CAS PubMed; (b) M. Picquart, S. Castro-Garcia, J. Livage, C. Julien and E. Haro-Poniatowski, J. Sol-Gel Sci. Technol., 2000, 18, 199–206 CrossRef CAS; (c) C. Rocchiccioli-Deltcheff, R. Thouvenot and M. Dabbabi, Spectrochim. Acta, Part A, 1977, 33, 143–153 CrossRef.
- (a) R. Gehlig and E. Salje, J. Solid State Chem., 1983, 49, 318–324 CrossRef CAS; (b) B. A. D. Angelis and M. Schiavello, J. Solid State Chem., 1977, 21, 67–72 CrossRef.
- (a) M. Miyauchi, A. Kondo, D. Atarashi and E. Sakai, J. Mater. Chem. C, 2014, 2, 3732–3737 RSC; (b) S. Bai, K. Zhang, L. Wang, J. Sun, R. Luo, D. Li and A. F. Chen, J. Mater. Chem. A, 2014, 2, 7927–7934 RSC; (c) H. Ling, J. L. Lu, S. Phua, H. Liu, L. Liu, Y. Z. Huang, D. Mandler, P. S. Lee and X. H. Lu, J. Mater. Chem. A, 2014, 2, 2708–2717 RSC.
- (a) A. Sanyal, T. Bala, S. Ahmed, A. Singh, V. Piterina, T. M. McGloughlin, F. R. Laffir and K. M. Ryan, J. Mater. Chem., 2009, 19, 8974–8981 RSC; (b) T. Wang, J. Tang, X. Fan, J. Zhou, H. Xue, H. Guo and J. P. He, Nanoscale, 2014, 6, 5359–5371 RSC.
- B. A. Deyerle and Y. Zhang, Langmuir, 2011, 27, 9203–9210 CrossRef CAS PubMed.
- Y. P. He and Y. P. Zhao, J. Phys. Chem. C, 2008, 112, 61–68 CAS.
- W. J. Kunz, J. Henle and B. W. Ninham, Curr. Opin. Colloid Interface Sci., 2004, 9, 19–37 CrossRef CAS PubMed.
- (a) C. Tanielian, R. Seghrouchni and C. Schweitzer, J. Phys. Chem. A, 2003, 107, 1102–1111 CrossRef CAS; (b) C. Tanielian, Coord. Chem. Rev., 1998, 178–180, 1165–1181 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |