
Location-dependent sensing of nitric oxide and calcium ions in living rat kidney using an amperometric/potentiometric dual microsensor†
Yee Seul
Kim
a,
Yejin
Ha
a,
Jungeun
Sim
b,
Minah
Suh
*bcd and
Youngmi
Lee
*a
aDepartment of Chemistry and Nano Science, Ewha Womans University, Seoul, 120-750, Korea. E-mail: youngmilee@ewha.ac.kr; Fax: +82-2-3277-2384
bCenter for Neuroscience Research Imaging (CNIR), Institute for Basic Science (IBS), Suwon 440-746, Republic of Korea. E-mail: minahsuh@skku.edu
cDepartment of Biomedical Engineering, Sungkyunkwan University, Suwon, 440-746, Republic of Korea
dSamsung Advanced Institute of Health Science and Technology (SAIHST), Sungkyunkwan University, Suwon, 440-746, Republic of Korea
First published on 9th November 2015
Abstract
In this paper, we report the fabrication of a dual microsensor for sensing nitric oxide (NO) and calcium ions (Ca2+) and its application for simultaneous NO/Ca2+ measurements in living rat kidney tissue. NO and Ca2+ have very important physiological functions and are both intricately involved in many biological processes. The dual NO/Ca2+ sensor is prepared based on a dual recessed electrode possessing Pt (diameter, 25 μm) and Ag (diameter, 76 μm) microdisks. The Pt disk surface (WE1) is electrodeposited with porous Pt black and then coated with fluorinated xerogel; and used for amperometric sensing of NO. The Ag disk surface (WE2) is chloridated to AgCl, followed by silanization and then Ca2+ selective membrane loading; and used for potentiometric sensing of Ca2+. The dual sensor exhibits high sensitivity of WE1 to NO (40.8 ± 6.5 pA μM−1, n = 10) and reliable Nerntian response of WE2 to Ca2+ changes (25.7 ± 0.5 mV pCa−1, n = 10) with excellent selectivity to only NO and Ca2+ over common interferents and reliable stability (up to ∼4 h tissue experiment). The prepared sensor is employed for real-time monitoring of the dynamic changes of NO and Ca2+ levels of a rat kidney, which is induced by the administration of 10 mM L-NG-nitroarginine methyl ester (L-NAME, a NO synthase inhibitor). Due to the small sensor dimension, location-dependent analyses of NO and Ca2+ are carried out at two different regions of a kidney (renal medulla and cortex). Higher NO and Ca2+ levels are observed at the medulla than at the cortex. This study verifies the feasibility for real-time monitoring of intimately connected Ca2+ and endogenous NO production; and also for localized concentration assessments of both NO and Ca2+.
Introduction
Developing quantitative, sensitive, and selective techniques for real-time nitric oxide (NO) analysis is a field of great interest since NO has been recognized as a vital messenger molecule in biological/physiological systems.1,2 Diverse biological roles of NO include neurotransmitter, vasodilator, bactericide, etc.2–6 NO is endogenously synthesized from L-arginine by NO synthase (NOS) enzyme of which three isoforms have been identified: neuronal (nNOS); endothelial (eNOS); and inducible NOS (iNOS).7 Influx and efflux of cytosolic Ca2+ are closely linked to the vasodilating effect of NO.8 In particular, eNOS and nNOS are activated by an increase in the intracellular calcium ion (Ca2+) concentration following Ca2+ influx, and eventually synthesize NO.9 Once generated, NO rapidly diffuses across cellular membranes and binds with soluble guanylate cyclase (sGC) in target cells, which catalyzes the synthesis of guanosine 3′,5′-cyclic monophosphate (cGMP), a second messenger mediating smooth muscle relaxation.10,11 The extracellular level of Ca2+ is known to be 1–3 mM.12,13Radical NO has a very short half-life (< several seconds) especially under in vivo biological conditions because it decays rapidly through the reactions with scavengers such as oxygen, hemoglobin, etc.14–16 In addition, NO levels under biological conditions are generally quite low (<1 μM).17,18 Thus, the detection of NO in a biological system can be problematic. An investigation of NO in biological systems, including fluids, tissues, and cells, requires a faster, more selective and sensitive method than other existing systems. Recently, amperometric NO sensors based on direct electrochemical oxidation of NO have been popularly used due to their attractive capabilities, such as real-time, direct analysis with high sensitivity.19
On the other hand, a non-electroactive Ca2+ ion is detected with potentiometric methods using ion selective electrodes (ISEs). ISEs are one of the most well-established chemical sensors and have been popularly used for the determination of a wide range of analytes and routinely utilized in clinical analysis.20–23 In fact, ISE based analytical procedures have advantages such as wide dynamic range, fast response time, low detection limit, availability of cheap portable instrumentation and capability for miniaturization.24 Recently, new types of small all-solid-state ISEs based on microcavity25 or nanopore26 were reported. Compared with conventional ISEs with internal filling solutions, this cavity-based ISE configuration provided the robustness and made it easier to be miniaturized.
Aiming at simultaneous analysis of biologically important NO and Ca2+, we present the development and characterization of a novel NO/Ca2+ dual electrochemical microsensor. A dual sensor was fabricated by integrating an amperometric NO microsensor and a potentiometric all-solid-state Ca2+ selective electrode based on microcavity in a single sensing micro-device. In fact, NO is oxidized at the NO electrode to generate the anodic current signal proportional to the NO concentration and the sensor selectivity is obtained with the use of NO permselective membrane and optimized applied potential for NO oxidation. Ca2+ is detected with a Ca2+ selective ISE of the dual sensor by measuring the junction potential at the membrane/electronic conductor interface, and the selectivity toward Ca2+ is acquired by the use of Ca2+ selective ionophore-containing membranes. The developed sensor feasibility for real-time concurrent measurements of NO and Ca2+ was demonstrated in living rat kidney tissues. The effects of L-NG-nitroarginine methyl ester (L-NAME, a NOS inhibitor) administration on NO generation/Ca2+ uptake of rat kidney tissues were examined. Taking advantage of the small sensor dimensions, location-dependent analysis of NO and Ca2+ was carried out at two different regions of a kidney (renal medulla and cortex). To the best of our knowledge, there has been no research reported on sensors quantifying NO and Ca2+ at the same time, despite the aforementioned very tight links between NO and Ca2+.
Experimental
Chemicals and materials
Theta-type glass capillaries (diameter, 1.5 mm) were from WPI Inc. (Sarasota, FL). Pt microwire (diameter, 25 μm) and Teflon-coated Ag microwire (diameter, 76 μm) were purchased from Good Fellow (Huntingdon, England) and A-M System, Inc. (Carlsborg, WA), respectively. Platinizing solution (3% H2PtCl6) was from YSI Inc. (Yellow Spring, Ohio). Carbon paste was purchased from BASi (West Lafayette, IN). CaCl2, MgCl2, NaCl, KCl, NaCN, NaOH, NaNO2, HCl, FeCl3, ethanol, toluene, tris(hydroxymethyl)aminomethane (Tris), L-ascorbic acid (AA), uric acid (UA), dopamine (DP), 4-acetamidophenol (AP), D-glucose, tetrahydrofuran (THF), methyltrimethoxysilane (MTMOS), 3-aminopropyl triethoxysilane (APTES), chlorotrimethyl silane (TMCS), polyurethane (PU, selectophore grade), poly(vinyl chloride) carboxylated (c-PVC), 2-nitrophenyl octyl ether (NPOE), calcium ionophore (ETH1001), Trizma® base, and potassium tetrakis(4-chlorophenyl)borate (KTpClPB) were from Sigma-Aldrich (St. Louis, MO). (Heptadecafluoro-1,1,2,2,-tetrahydrodecyl)trimethoxysilane (17FTMS) was purchased from Gelest (Tullytown, PA). Phosphate buffered saline (PBS) was from Fisher Scientific (Rochester, NY). Argon (Ar) and nitric oxide (NO) gases were from Dong-A Gas Co. (Korea). All aqueous solutions were prepared with deionized water (>18 MΩ cm) using reagent grade compounds without further purification.Preparation and characterization of a NO/Ca2+ dual microsensor
A NO/Ca2+ dual microsensor was prepared based on a dual Pt microdisk/Ag microdisk working electrode (WE). The dual Pt/Au electrode was fabricated as follows: Pt microwire (25 μm diameter) and Ag microwire (76 μm diameter) were separately inserted into two openings of a pulled theta-glass capillary, one wire each, and the tapered region of the capillary was thermally fused under vacuum. The end of the thermally sealed region was carefully polished using a sand paper and 3-, 1-, 0.05 μm diamond papers successively to expose the Pt and Ag microdisks’ surface. The Pt and Ag disks were etched individually by applying 3.35 V ac voltage (60 Hz) for 1 s vs. Pt wire counter electrode: in 1.2 M CaCl2 in H2O–acetone (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) for Pt etching; and in 6 M NaCN and 0.1 M NaOH in water for Ag etching. The recessed Pt microdisk surface (WE1) was modified with porous Pt layer electrodeposited by cyclic voltammetry (CV, from +0.6 V to −0.35 V vs. Ag/AgCl, scan rate, 20 mV s−1, 1 cycle) in 3% chloroplatinic acid solution. Then, 1 vol% APTES (in ethanol) was loaded only over the platinized Pt electrode using a fluid dispenser (Ultimus, Nordson Co., USA, 3.0 psi, 0.0003 s) and then dried overnight. The Pt electrode end surface was further coated with a fluorinated xerogel solution (a mixture of 14.5 μL of 17FTMS, 18 μL of MTMOS, 10 μL of 0.5 M HCl, 160 μL of water, and 727.5 μL of ethanol) using the same dispenser to obtain a selectivity to NO.27
:1, v/v) for Pt etching; and in 6 M NaCN and 0.1 M NaOH in water for Ag etching. The recessed Pt microdisk surface (WE1) was modified with porous Pt layer electrodeposited by cyclic voltammetry (CV, from +0.6 V to −0.35 V vs. Ag/AgCl, scan rate, 20 mV s−1, 1 cycle) in 3% chloroplatinic acid solution. Then, 1 vol% APTES (in ethanol) was loaded only over the platinized Pt electrode using a fluid dispenser (Ultimus, Nordson Co., USA, 3.0 psi, 0.0003 s) and then dried overnight. The Pt electrode end surface was further coated with a fluorinated xerogel solution (a mixture of 14.5 μL of 17FTMS, 18 μL of MTMOS, 10 μL of 0.5 M HCl, 160 μL of water, and 727.5 μL of ethanol) using the same dispenser to obtain a selectivity to NO.27
On the other hand, the recessed Ag microdisk (WE2) was treated with a drop of 1 M FeCl3 solution for ∼1 h to form an Ag/AgCl layer and then rinsed thoroughly with deionized water. Then, 50 vol% of TMCS in toluene was dropped only onto the Ag/AgCl electrode. After being dried overnight, a Ca2+ ion selective polymer membrane cocktail was loaded onto the modified Ag/AgCl electrode using a fluid dispenser (7.0 psi, 0.0100 s) followed by drying for 8 h. The Ca2+ selective polymer membrane cocktail was prepared by dissolving 36.0 mg of c-PVC, 4.0 mg of PU, 2.0 mg of calcium selective ionophore (ETH1001), and 126.8 μL of o-NPOE, 1.0 mg of KTpClPB in 0.8 mL of THF. Two refillable miniature Ag/AgCl reference electrodes (eDAQ, electrode diameter = 2 mm) were used as the reference/counter electrodes: one for amperometric NO measurement (WE1) and the other for potentiometric calcium ion measurement (WE2).
The modified dual microelectrodes were presoaked in PBS solution (pH 7.4) while the WE1 was polarized for >3 h at +0.85 V vs. Ag/AgCl. All membranes were loaded on the electrode surfaces by Ultimus™ dispenser (NORDSON EFD, Ohio). The prepared dual sensor diagram is shown in Fig. 1. The sensor overall end plane diameter was ca. 400–500 μm (n = 10).
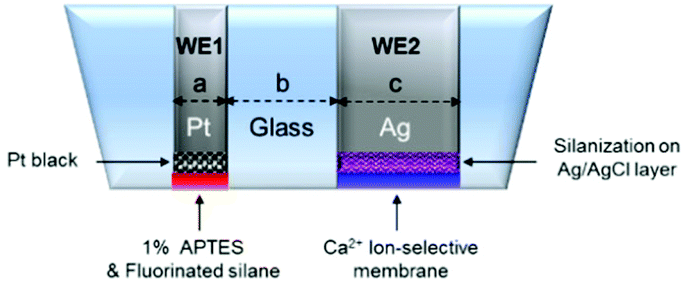 | ||
| Fig. 1 A schematic diagram of a NO/Ca2+ dual microsensor. The dual sensor possesses two working electrodes, WE1 and WE2. The electrode dimension is as follows: a = 25 μm, b = 95–150 μm, c = 76 μm, electrode overall end plane diameter = 400–500 μm (n = 10). | ||
WE1 and WE2 of the dual electrode were calibrated in amperometric and potentiometric modes, respectively. The dual electrode was immersed in a PBS solution and WE1 was prepolarized at +0.85 V vs. Ag/AgCl for 3–5 h to attain a stable background current before the initial use. Then, WE1 of a NO/Ca2+ dual microsensor was calibrated in a deoxygenated PBS solution by monitoring the WE1 current changes (at +0.85 V vs. Ag/AgCl) responding to a stepwise increase of NO concentration.28 The current of WE1 was measured using a CHI900B (CH Instruments Inc., TX). WE2 of the dual sensor was calibrated in a 0.1 M Tris-buffer solution by measuring the electrode potential changes (vs. Ag/AgCl) responding to a successive Ca2+ concentration increase (10−6 M to 10−2 M), which was achieved by adding standard CaCl2 solution into a test solution. The potential difference between the WE2 and Ag/AgCl reference electrode was measured using a 16-channel multi pH/ion meter (KOSENTECH Inc., Korea).
Simultaneous detection of NO and Ca2+ ions in rat kidney tissue
Male rats (Sprague-Dawley, 340–360 g) were anesthetized with Zoletil (100 μL per 100 g) and then were perfused transcardially with a saline solution. Both kidneys of a rat were excised and stored immediately in a cold Euro-Collins solution (32.7 g L−1D-glucose, 2.05 g L−1 KH2PO4, 7.40 g L−1 K2HPO4, 1.12 g L−1 KCl, 0.84 g L−1 NaHNO3) on ice. The kidneys were used for experiments within 24 h after being harvested. All surgical procedures were approved by the Institutional Animal Care and Use Committee of Sungkyunkwan University.NO and Ca2+ levels were measured concurrently at the surface of the kidney tissue slice immersed in a PBS solution (pH 7.4). The tissue slice (∼3 mm thick) was prepared by cutting out the most central section of a kidney longitudinally. WE1 current (at 0.85 V vs. Ag/AgCl) and WE2 potential (vs. Ag/AgCl) of the sensor were monitored simultaneously and continuously while the sensor was positioned at 10 μm vertical separation from the surface of the inner medulla region of the kidney with the administration of 10 mM L-NAME, which is an inhibitor of NOS enzyme.
To study the location-dependent NO and Ca2+ levels, these concentrations were measured at two different locations of the kidney tissue, the renal medulla and the cortex as shown in Fig. 2. First, a NO/Ca2+ dual microsensor was located at 10 μm vertical separation from the surface of the kidney medulla region (Position 1); and WE1 current and WE2 potentials were measured for ∼10 min. Once the measured sensor signals reached the relatively stable ones, the sensor was moved upward by 4000 μm using a z-piezoelectric step motor of a SECM instrument. At this position far from the kidney (Position 2), the sensor signals were measured until it reached the stable base levels. This whole procedure was repeated three times while measuring the sensor signals continuously. Then, the sensor at 4000 μm vertical distance from the kidney was moved horizontally toward the cortex region (Position 3) using a x-piezoelectric step motor of the SECM instrument and was moved toward the cortex surface down to 10 μm vertical separation. At this position near the cortex (Position 4), the sensor signals were measured until stable signals were observed. Then, the sensor was vertically retracted upward by 4000 μm (Position 3) and the sensor signals were measured until they reached quite stable levels. Again, this whole procedure was repeated three times over the cortex region. Finally the sensor was moved back to the cortex region and the sensor measurements were carried out three times at Positions 1 and 2. The sensor scan rate applied for all the sensor movements was 2000 μm s−1. Two Ag/AgCl reference electrodes (one for WE1; the other for WE2) were placed near the kidney slice in a glass container (diameter = 46 mm) containing 15 mL PBS solution. The current at WE1 and the potential at WE2 were recorded concurrently using CHI900B and a 16-channel multi pH/ion meter, respectively. The WE1 and WE2 were individually calibrated for NO and Ca2+ before and after the experiments. The measured current and potential were converted to NO and Ca2+ concentrations using the calibration curves, which were acquired immediately before and after the tissue experiments.
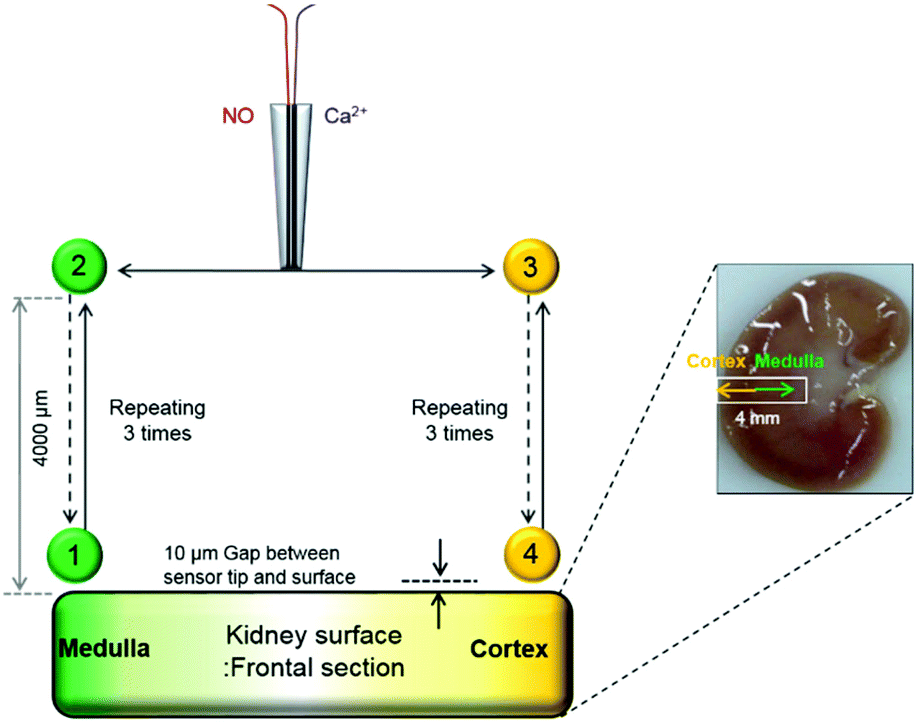 | ||
| Fig. 2 A scheme of experimental procedures to measure the NO and Ca2+ levels simultaneously for a kidney cross-sectional surface. All the sensor movements were conducted by x, y, z-piezoelectric step motors of a SECM instrument. | ||
Results and discussion
Characterization and performances of a NO/Ca2+ dual microsensor
Linear sweep voltammetry (LSV) carried out with WE1 of a dual sensor in a deaerated PBS solution containing 8.7 μM NO confirmed that +0.85 V (vs. Ag/AgCl) is a sufficient and proper applied potential to WE1 for NO oxidative detection (Fig. S1 in the ESI†).The changes of WE1 current at +0.85 V and WE2 potential of a dual sensor were monitored concurrently when either NO or Ca2+ concentration was increased stepwise in a test solution. As presented in Fig. 3A and B, the current measured at WE1 increased proportionally responding to the NO concentration increase (0 to 8.74 μM), while the potential of WE2 did not change noticeably regardless of the NO concentration elevation. On the other hand, the potential of WE2 increased linearly in proportion to the Ca2+ concentration (10−6 M to 10−2 M), while the current of WE1 changed negligibly (Fig. 3C and D). This verifies that the WE1 and WE2 signals respond to NO and Ca2+ separately and do not crosstalk with each other. Despite the small sensing dimension, the sensitivities of dual sensors were quite good: 40.8 ± 6.5 pA μM−1 for NO at WE1 (n = 10) and 25.7 ± 0.5 mV pCa−1 for Ca2+ at WE2 (n = 10). The sensor response times (t90%, time to reach 90% of the steady-state signals) were estimated to be 3.0 ± 0.8 s (n = 10) for NO at WE1 and 2.2 ± 0.9 s for Ca2+ (n = 10) at WE2 in typical calibration curves. Because the calibration procedure requires a certain time to homogenize a solution via stirring after the injection of a standard solution, the practical t90% could be shorter in a pre-equilibrated system than the ones estimated based on calibration curves. Detection limits of the sensors were determined as ∼20 nM NO at WE1 and ∼10−6 M Ca2+ at WE2 (S/N = 3).
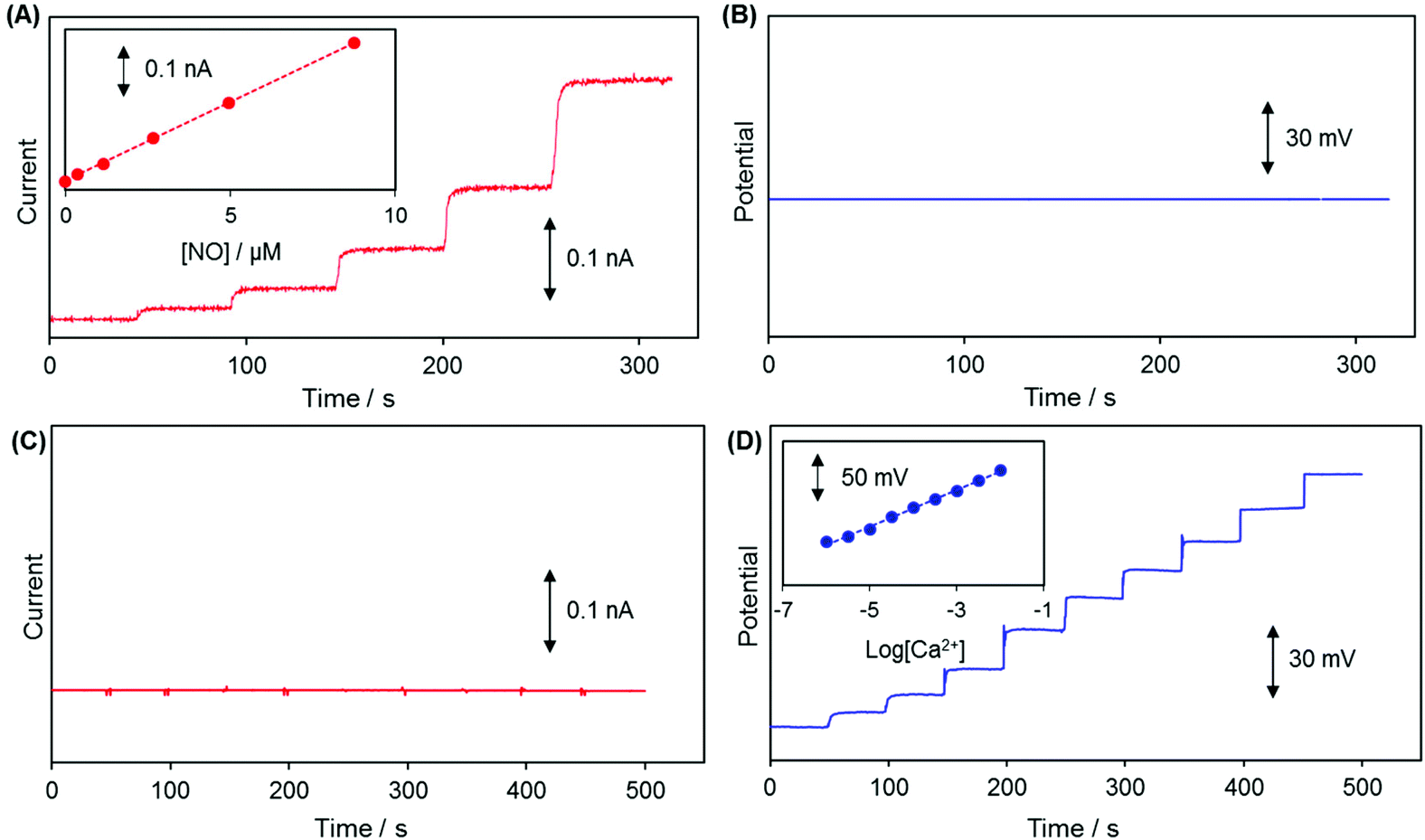 | ||
| Fig. 3 Typical dynamic response curves of (A) WE1 and (B) WE2 to NO concentration change (0 μM to 8.74 μM); and those of (C) WE1 and (D) WE2 to Ca2+ concentration change (10−6 M to 10−2 M). The applied potential to WE1 was +0.85 V vs. Ag/AgCl. (A and B) Changes in the WE1 current and WE2 potential were simultaneously recorded with the successive additions of a NO standard solution to a deaerated PBS (pH 7.4) solution. (C and D) Changes in the WE1 current and WE2 potential were simultaneously measured in 0.1 M Tris-buffer with the successive injections of a standard CaCl2 solution. Insets show the corresponding calibration curves. (A) WE1 sensitivity to NO is 40.8 ± 6.5 pA μM−1, (D) WE2 sensitivity to Ca2+ is 25.7 ± 0.5 mV pCa−1. | ||
The potential drift is a common problem observed particularly in small sized solid-state ISEs due to poorly defined junction potential at the membrane/electronic conductor interface.29 The pretreatment of the WE2 Ag/AgCl surface via silanization with TMCS prior to the loading of the ion selective membrane cocktail improved the potential stability of WE2. Compared to the electrode without pre-silanization, the one prepared with TMCS pretreatment exhibited a stable potential response without a potential drift (Fig. S2 in the ESI†). This improvement is presumably due to a better adhesion of the ion-selective membrane to the silanized hydrophobic electrode surface.
Selectivity of WE1 over common oxidizable biological interferents was confirmed. In fact, the current of WE1 did not change noticeably with the addition of 50 μM nitrite, 50 μM AA, 50 μM UA, 50 μM AP, 10 μM DA, 2 μM H2S, and 10 μM H2O2 in stirred PBS solution (Fig. S3 in the ESI†). The used concentrations of these interfering species were higher than the physiological levels.30 The hydrophobicity of the fluorinated xerogel films provided the selectivity to NO gas, while rejecting relatively hydrophilic interferents. The low resistance of the xerogel film seemingly enables WE1 to function amperometrically.27 Meanwhile, the selectivity coefficient (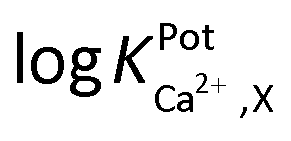 ) of WE2 to Ca2+ ion over other interfering cations (X = Na+, K+, and Mg2+) was determined using the separate solution method (SSM). Among the tested interfering cations, WE2 potential increased slightly responding to the Na+ ion at its high concentration >10−2.0 M with negligible responses to K+ and Mg2+ up to 10−1.5 M (Fig. S4 in the ESI†). The
) of WE2 to Ca2+ ion over other interfering cations (X = Na+, K+, and Mg2+) was determined using the separate solution method (SSM). Among the tested interfering cations, WE2 potential increased slightly responding to the Na+ ion at its high concentration >10−2.0 M with negligible responses to K+ and Mg2+ up to 10−1.5 M (Fig. S4 in the ESI†). The 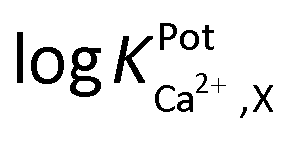 of an ISE is generally required to be under at least −2.0 to be applied for biological samples.31 WE2 of a NO/Ca2+ dual sensor exhibited
of an ISE is generally required to be under at least −2.0 to be applied for biological samples.31 WE2 of a NO/Ca2+ dual sensor exhibited 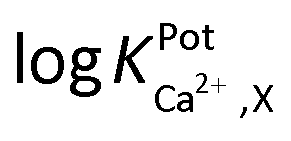 of −3.5, suggesting its possible use for biological applications.
of −3.5, suggesting its possible use for biological applications.
Simultaneous monitoring of NO and Ca2+ changes at renal medulla induced by L-NAME administration
Real-time simultaneous measurements of NO and Ca2+ were carried out for ∼2000 s at the surface of a kidney slice with a dual NO/Ca2+ microsensor, whose end plane was located at 10 μm vertical separation from the surface of the renal inner medulla region. Once the monitored sensor signals became stable, L-NAME stock solution was added to attain 10 mM concentration in the tissue bathing PBS solution. The measured WE1 current and WE2 potential were converted to the corresponding NO and Ca2+ levels with the use of the sensor pre-calibration curves. A typically observed result is shown in Fig. 4. After L-NAME addition, the NO concentration started to decrease while the Ca2+ concentrations begin to increase. Then NO decreased down to the minimum (Δ[NO] = −62.94 ± 6.37 nM, n = 5) and Ca2+ reached the peak value (Δ[Ca2+] = +8.45 ± 1.45 μM, n = 5) simultaneously, followed by the gradual recovery to their basal levels. No responses of WE1 current and WE2 potential to L-NAME itself were confirmed (Fig. S5 in the ESI†). This observation possibly indicates the tight biological interaction between NO and Ca2+. L-NAME inhibits NO production leading to vasoconstriction and then the system senses the need for NO to induce vasodilation. Since the activation of eNOS requires Ca2+ influx into cells,9,32 the dynamic changes of the NO production and extracellular Ca2+ level are expected to be opposite which is well matched with our observation. This verifies the dual sensor feasibility for real-time monitoring of intimately connected Ca2+ cellular uptake and endogenous NO production.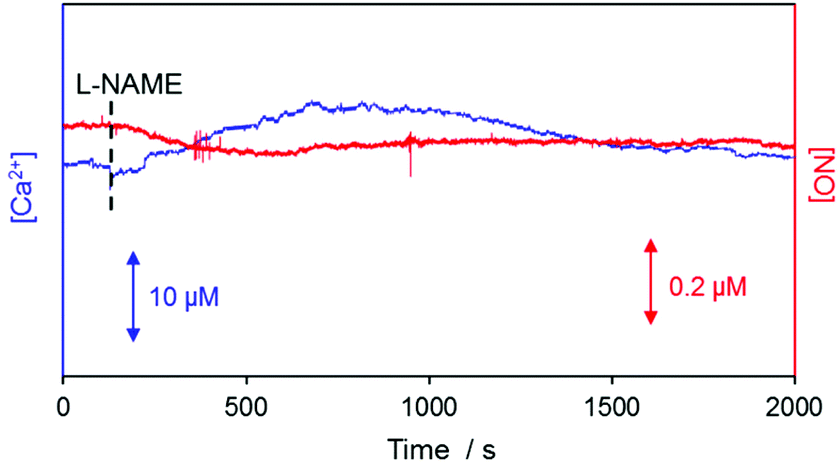 | ||
| Fig. 4 Representative simultaneous NO and Ca2+ measurements near the renal medulla of a rat kidney bathed in PBS solution (pH = 7.4) with respect to time. 10 mM L-NAME was added to the bathing solution at the time indicated with a black dashed vertical line. A NO/Ca2+ dual microsensor was positioned at 10 μm vertical distance above the kidney's surface. | ||
Location-dependent NO and Ca2+ levels at the surface of rat kidney
NO and Ca2+ levels were measured at two different locations of kidney tissue, the renal medulla and the cortex. Detailed experimental procedures are described in the Experimental section and illustrated in Fig. 2. As presented in Fig. 5, both NO and Ca2+ levels were higher near the kidney surface (both medulla and cortex, Positions 1 and 4, respectively) compared to that in the bulk solution phase (Positions 2 and 3). A close investigation of the data reveals that NO and Ca2+ levels were measured to be higher at the medulla (Position 1) than at the cortex (Position 4). First, three repeated measurements of NO and Ca2+ near the medulla (vertical distance = 10 μm) exhibited similar concentration values. Then, the sensor was moved to the cortex region, where NO and Ca2+ were measured. The three repeated measurements near the cortex (vertical distance = 10 μm) also exhibited similar patterns. In fact, both NO and Ca2+ concentrations at the cortex (Position 4) were monitored to be lower than those at the medulla (Position 1). When the sensor returned to the medulla region, higher NO and Ca2+ were recovered.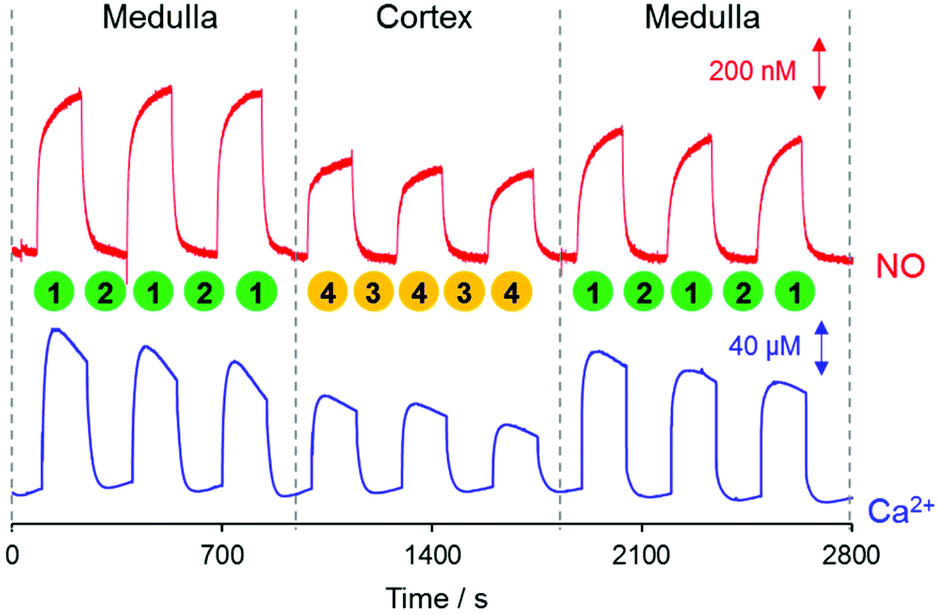 | ||
| Fig. 5 The representative simultaneous measurement of NO and Ca2+ for the renal medulla and the cortex. The sensor vertical separations from the kidney surface were 4000 μm at Positions 2 and 3; and were 10 μm at Positions 1 (medulla) and 4 (cortex). After the measurements over the medulla with the three-time repeated sensor up/down movement between Positions 1 and 2, the sensor was moved to the cortex and the measurement was carried out with similar three-time repeated sensor up/down movement between Positions 3 and 4. Then, the sensor was returned to the former spot of the medulla and repeated the measurement three times. | ||
For statistical analysis, NO and Ca2+ levels were measured six times both at the medulla and at the cortex of each kidney, and this total 12 measurements were carried out on five different kidneys. Location dependent measurements of NO and Ca2+ concentrations of the five kidneys (overall 30 measured values either at the medulla or at the cortex) were averaged and are shown in Fig. 6A and C. The averaged NO levels were 0.38 ± 0.026 μM at the cortex, and 0.57 ± 0.051 μM at the medulla; and the averaged Ca2+ levels were 0.15 ± 0.012 mM at the cortex, and 0.19 ± 0.016 mM at the medulla. These levels are obviously above the detection limits of the sensors (∼20 nM NO at WE1, ∼10−6 M Ca2+ at WE2). P-Values calculated with paired t-test were <0.01 for both NO and Ca2+, indicating that the NO and Ca2+ concentration differences between the medulla and cortex are statistically significant. Fig. 6B and D present the averaged NO and Ca2+ levels of the six times repeated measurements at the medulla and at the cortex of each kidney. Within each individual kidney of five kidneys, the levels of both NO and Ca2+ were higher at the medulla than at the cortex without exception. By virtue of the small dimension, the dual sensor could analyze the location-dependent NO and Ca2+ levels at the medulla and the cortex; and the sensor sensitivity was sufficient to differentiate the NO/Ca2+ concentration differences between the medulla and the cortex.
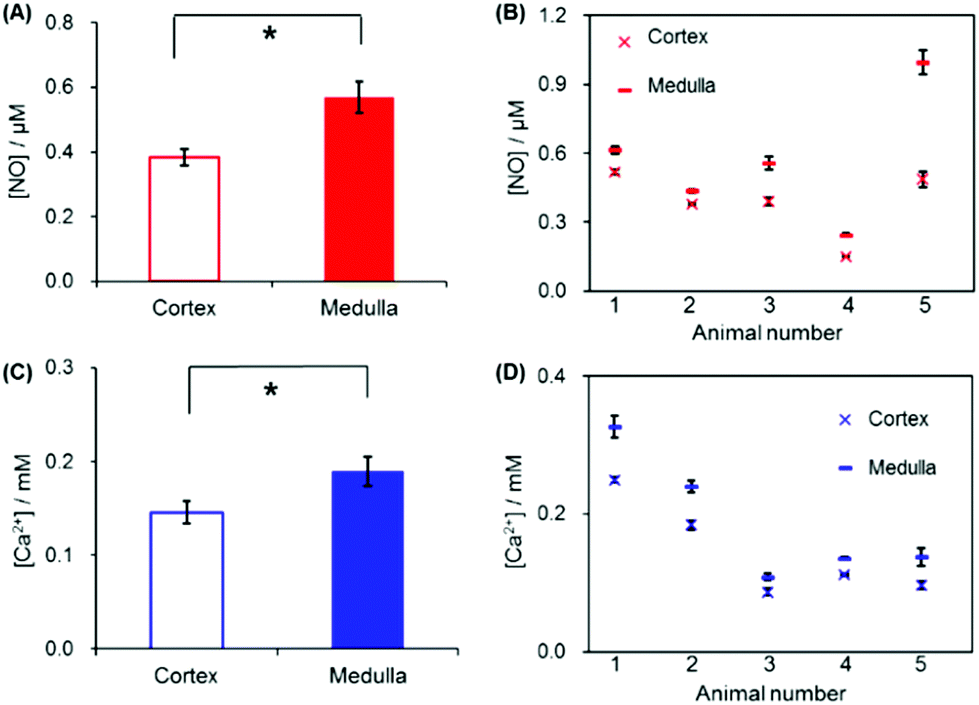 | ||
| Fig. 6 (A, C) The average of basal concentrations of NO and Ca2+ measured at the medulla and also at the cortex of five different kidneys (n = 30: six-time measurements for each kidney and total five kidneys). *p values <0.01 based on paired t-test. (B, D) The averaged NO and Ca2+ levels of the six-time repeated measurements at the medulla and the cortex of each individual kidney. | ||
The observed higher NO and Ca2+ levels at the medulla than at the cortex are well matched with previous reports on rat kidneys even though indirect methods were used in these previous studies as follows. For example, higher NOS distribution in the medulla region than the cortex of a rat kidney was analyzed with RT-PCR33 and western blot.34 Methemoglobin (NO-bound hemoglobin) analysis35 and microdialysis-hemoglobin trapping assay36 also demonstrated a higher NO level at the medulla (57.1 ± 5.5 nM,35 105.2 ± 18.7 nM36) than at the cortex (31.2 ± 5.7 nM,35 62.6 ± 16.1 nM36). Measurement of L-citrulline (a by-product of NO synthesis via NOS activity) also reported higher L-citrulline formation in the medulla than in the cortex suggesting higher NO generation in the medulla.37 Measurements of Ca2+ concentrations in rat kidneys have been rarely reported. One study using radiometric photometry reported a higher Ca2+ level in the medulla (1.93 ± 0.12 mM) than in the cortex (1.62 ± 0.19 mM), though the concentration difference is rather small.38 One of the reasons for the lower Ca2+ measured could be attributed to the binding of Ca2+ to phosphate species in PBS bathing solution. As mentioned in the previous section discussing NO generation related to the Ca2+ ion influx, we may anticipate that a higher NO level accompanies a lower extracellular Ca2+ level. However, the location-dependent study of a kidney exhibited that both NO and Ca2+ levels were higher at the medulla than at the cortex (Fig. 6). This suggests that the observed concentration difference between the medulla and the cortex is induced by the locally different kidney function rather than NO generation linked to Ca2+ influx. In fact, Ca2+ absorption is known to take place mainly in proximal tubules, thick ascending limbs of Henle's loop, and distal tubules which are located within the cortex and outer medulla areas.39 Because more uptake occurs at the cortex than medulla, the extracellular Ca2+ level is seemingly expected to be higher in the inner medulla region as we measured. The actual values of NO and Ca2+ concentrations determined at the medulla and the cortex in the current study are not directly comparable to the previously reported values due to the completely different analyzing techniques: direct surface concentration measurements for the living organ in the current study vs. postmortem or indirect bulk analysis in previous reports.
Fig. 7A and B present the correlation of the NO/Ca2+ levels between the medulla and the cortex within an individual kidney. For total five kidneys, it was observed that one kidney with the higher NO level at the cortex also exhibited the higher value at the medulla than the other kidneys and vice versa. A similar pattern, the high level at the cortex linked to the high one at the medulla, was observed for Ca2+ more clearly. This indicates that the subject variation and individual physiological condition mainly determine the NO and Ca2+ basal levels at both the renal medulla and the cortex. The correlation between the NO and Ca2+ levels either at the cortex or the medulla was also investigated. As shown in Fig. 7C and D, an intimate linkage between the NO levels and Ca2+ levels was not obviously observed at both the cortex and the medulla. This supports the fact that the observed NO levels and Ca2+ levels of the kidneys are not dominantly governed by vasodilation/vasoconstriction, mediated via NO endogenous production linked to the cellular uptake of Ca2+. Instead, the locally different kidney functions possibly determine the basal levels of NO and Ca2+ in kidneys.
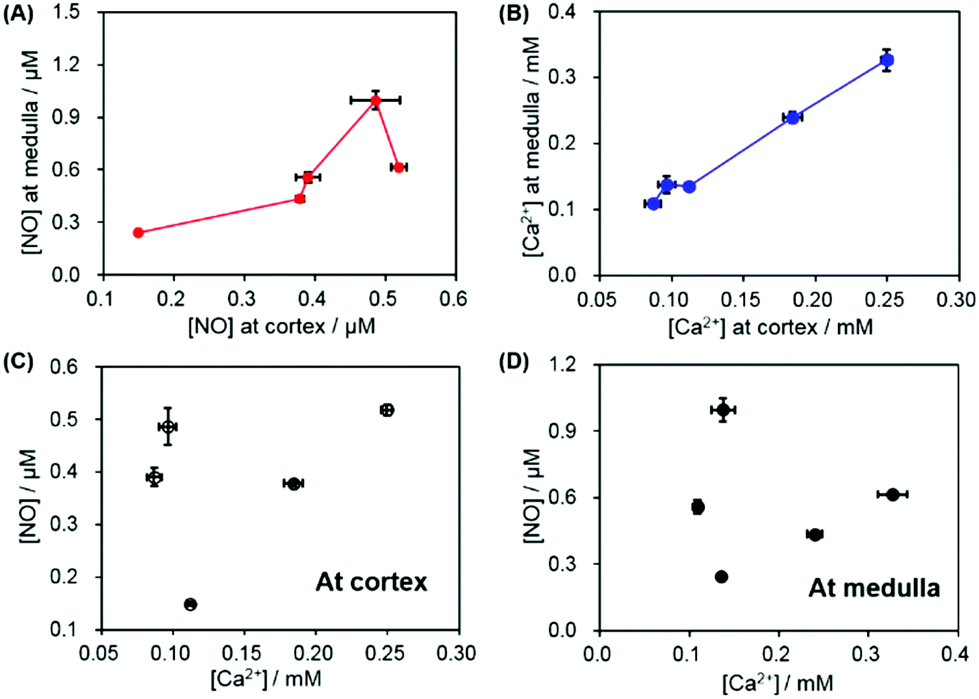 | ||
| Fig. 7 (A) The correlation between the cortex and the medulla of NO levels and (B) those of Ca2+ levels. The correlation between NO and Ca2+ levels (C) at the cortex and (D) at the medulla. | ||
The sensor stability was accessed by comparing the calibration curves obtained before and after the tissue experiment. As seen in Fig. S6 (in the ESI†), WE1 and WE2 maintained ∼92% and ∼98% of the initial sensitivities after ∼4 h tissue experiment, indicating high stability. This decent stability of a sensor supports the fact that the simultaneously measured NO and Ca2+ levels in the kidneys are reliable.
Conclusions
A dual sensor was fabricated by integrating an amperometric NO microsensor (WE1) and a potentiometric all-solid-state Ca2+ ISE (WE2) in a single sensing micro-device: WE1 was a porous Pt/fluorinated xerogel coated Pt microdisk electrode for the selective and sensitive NO sensing; and WE2 was an Ag/AgCl electrode pre-silanized and then loaded with a Ca2+ ion selective membrane. The pretreatment of the Ag/AgCl surface via silanization using TMCS improved the potential stability of WE2 greatly, due to a better adhesion of the ion selective membrane on the electrode surface. The sensor showed real-time concurrent measurements of NO and Ca2+ without crosstalk between the two sensing signals of WE1 and WE2. The dual sensor was successfully used to monitor the dynamic changes of the NO and Ca2+ levels at the renal medulla of a rat kidney. L-NAME administration induced the decrease of NO along with the increase of Ca2+, showing an intimately connected Ca2+ cellular uptake and endogenous NO production.The small dimension of a sensor made the location-dependent analysis feasible. The measurement of NO and Ca2+ at two different regions, the renal medulla and the cortex, showed higher NO and Ca2+ levels at the medulla than at the cortex. This suggests the potential applicability of the dual sensor in many research areas in which NO and Ca2+ are required to be measured simultaneously.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (2014R1A2A2A05003769) and by IBS-R015-D1.Notes and references
- R. F. Furchgott and J. V. Zawadzki, Nature, 1980, 288, 373–376 CrossRef CAS PubMed.
- L. Jia, C. Bonaventura, J. Bonaventura and J. S. Stamler, Nature, 1996, 380, 221–226 CrossRef CAS PubMed.
- G. C. Brown, Science, 2003, 299, 838 CrossRef PubMed.
- K. Shibuki, Neurosci. Res., 1990, 9, 69–76 CrossRef CAS PubMed.
- M. N. Friedemann, S. W. Robinson and G. A. Gerhardt, Anal. Chem., 1996, 68, 2621–2628 CrossRef CAS PubMed.
- T. Malinski and Z. Taha, Nature, 1992, 358, 676–678 CrossRef CAS PubMed.
- J. Stamler, D. Singel and J. Loscalzo, Science, 1992, 258, 1898–2001 CAS.
- J. A. Carvajal, A. M. Germain, J. P. Huidobro-Toro and C. P. Weiner, J. Cell. Physiol., 2000, 184, 409–420 CrossRef CAS PubMed.
- B. C. Kone and C. Baylis, Am. J. Physiol. Renal Physiol., 1997, 272, F561–F578 CAS.
- L. J. Ignarro, Nitric oxide: biology and pathobiology, Academic press, 2000 Search PubMed.
- J. W. Denninger and M. A. Marletta, Biochim. Biophys. Acta, Bioenerg., 1999, 1411, 334–350 CrossRef CAS.
- D. Rouse and W. N. Suki, Kidney Int., 1990, 38, 700–708 CrossRef CAS PubMed.
- R. Masuyama, M. Uehara, K. Suzuki and S. Goto, J. Bone Miner. Metab., 1996, 14, 26–30 CrossRef CAS.
- S. H. Snyder and D. S. Bredt, Sci. Am., 1992, 266, 68–71 CrossRef CAS PubMed.
- M. Kelm, M. Feelisch, R. Spahr, H.-M. Piper, E. Noack and J. Schrader, Biochem. Biophys. Res. Commun., 1988, 154, 236–244 CrossRef CAS PubMed.
- D. D. Thomas, X. Liu, S. P. Kantrow and J. R. Lancaster, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 355–360 CrossRef CAS.
- T. Malinski, F. Bailey, Z. Zhang and M. Chopp, J. Cereb. Blood Flow Metab., 1993, 13, 355–358 CrossRef CAS PubMed.
- H. Wakatsuki, H. Gomi, M. Kudoh, S. Kimura, K. Takahashi, M. Takeda and K. Shibuki, J. Physiol., 1998, 513, 71–81 CrossRef CAS.
- F. Bedioui and N. Villeneuve, Electroanalysis, 2003, 15, 5–18 CrossRef CAS.
- Y. Wang, H. Xu, J. Zhang and G. Li, Sensors, 2008, 8, 2043–2081 CrossRef CAS.
- P. Bühlmann, E. Pretsch and E. Bakker, Chem. Rev., 1998, 98, 1593–1688 CrossRef.
- E. Bakker and Y. Qin, Anal. Chem., 2006, 78, 3965–3984 CrossRef CAS PubMed.
- A. J. Bard and C. Zoski, Electroanalytical chemistry: a series of advances, CRC Press, 2011 Search PubMed.
- S. Walsh, D. Diamond, J. McLaughlin, E. McAdams, D. Woolfson, D. Jones and M. Bonner, Electroanalysis, 1997, 9, 1318–1324 CrossRef CAS.
- F. Sundfors, R. Bereczki, J. Bobacka, K. Tóth, A. Ivaska and R. E. Gyurcsányi, Electroanalysis, 2006, 18, 1372–1378 CrossRef CAS.
- J. H. Shim, J. Kim, G. S. Cha, H. Nam, R. J. White, H. S. White and R. B. Brown, Anal. Chem., 2007, 79, 3568–3574 CrossRef CAS PubMed.
- J. H. Shin, B. J. Privett, J. M. Kita, R. M. Wightman and M. H. Schoenfisch, Anal. Chem., 2008, 80, 6850–6859 CrossRef CAS PubMed.
- Y. Lee, J. Yang, S. M. Rudich, R. J. Schreiner and M. E. Meyerhoff, Anal. Chem., 2004, 76, 545–551 CrossRef CAS PubMed.
- J. Bobacka, A. Ivaska and A. Lewenstam, Chem. Rev., 2008, 108, 329–351 CrossRef CAS PubMed.
- B. J. Privett, J. H. Shin and M. H. Schoenfisch, Chem. Soc. Rev., 2010, 39, 1925–1935 RSC.
- J. R. Allen, T. Cynkowski, J. Desai and L. G. Bachas, Electroanalysis, 1992, 4, 533–537 CrossRef CAS.
- T. R. Uhrenholt, J. Schjerning, P. M. Vanhoutte, B. L. Jensen and O. Skøtt, Am. J. Physiol. Renal Physiol., 2007, 292, F1124–F1131 CrossRef CAS PubMed.
- F. Wu, F. Park, A. W. Cowley and D. L. Mattson, Am. J. Physiol. Renal Physiol., 1999, 276, F874–F881 CAS.
- D. L. Mattson and D. J. Higgins, Hypertension, 1996, 27, 688–692 CrossRef CAS PubMed.
- A.-P. Zou and A. W. Cowley, Hypertension, 1997, 29, 194–198 CrossRef CAS PubMed.
- A.-P. Zou, F. Wu and A. W. Cowley, Hypertension, 1998, 31, 271–276 CrossRef CAS PubMed.
- D. Mattson and F. Wu, Acta Physiol. Scand., 2000, 168, 149–154 CrossRef CAS PubMed.
- M. M. Mupanomunda, B. Tian, N. Ishioka and R. D. Bukoski, Am. J. Physiol. Renal Physiol., 2000, 278, F644–F649 CAS.
- P. A. Friedman, Nephron, 2000, 8, 343–350 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: LSV for NO oxidation of WE1 (Fig. S1); dynamic potential response of WE2 to Ca2+ (Fig. S2); amperometric responses of WE1 to typical interfering agents (Fig. S3); potentiometric responses of WE2 to various cations (Fig. S4); responses of a dual sensor to L-NAME (Fig. S5); and calibration curves before and after tissue experiments (Fig. S6). See DOI: 10.1039/c5an01804h |
| This journal is © The Royal Society of Chemistry 2016 |