
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A proteomic approach for the rapid, multi-informative and reliable identification of blood†
E.
Patel‡
a,
P.
Cicatiello‡
b,
L.
Deininger
a,
M. R.
Clench
a,
G.
Marino
b,
P.
Giardina
b,
G.
Langenburg
c,
A.
West
d,
P.
Marshall
d,
V.
Sears
e and
S.
Francese
*a
aBiomolecular Research Centre, Sheffield Hallam University, Howard Street S1 1WB, Sheffield, UK. E-mail: s.francese@shu.ac.uk; Tel: +44 (0)1142256165
bDipartimento di Scienze Chimiche, Universita’ di Napoli Federico II, via Cinthia I-80126 Naples, Italy
cBureau of Criminal Apprehension, 1430 Maryland Avenue East, St Paul, Minnesota MN 55106, USA
dGlaxoSmithKline, Gunnels Wood Road, Stevenage, SG1 2NY, UK
eCentre for Applied Science and Technology, Home Office, St Albans, AL4 9HQ, UK
First published on 24th November 2015
Abstract
Blood evidence is frequently encountered at the scene of violent crimes and can provide valuable intelligence in the forensic investigation of serious offences. Because many of the current enhancement methods used by crime scene investigators are presumptive, the visualisation of blood is not always reliable nor does it bear additional information. In the work presented here, two methods employing a shotgun bottom up proteomic approach for the detection of blood are reported; the developed protocols employ both an in solution digestion method and a recently proposed procedure involving immobilization of trypsin on hydrophobin Vmh2 coated MALDI sample plate. The methods are complementary as whilst one yields more identifiable proteins (as biomolecular signatures), the other is extremely rapid (5 minutes). Additionally, data demonstrate the opportunity to discriminate blood provenance even when two different blood sources are present in a mixture. This approach is also suitable for old bloodstains which had been previously chemically enhanced, as experiments conducted on a 9-year-old bloodstain deposited on a ceramic tile demonstrate.
Introduction
The detection of blood in stains or fingermarks at crime scenes can be an invaluable piece of evidence in the investigation of violent crimes. Crime Scene Investigators (CSI) have several enhancement classes of techniques available to visualize the presence of blood including optical, spectroscopic and chemical development methods.1 In addition to limitations in common to all of the three classes of methods, chemical techniques are actually only presumptive methods thus occasionally leading to false positives. These methods have been extensively reviewed by Sears1 and all were reported to exhibit a lack of specificity; even haem-reactive compounds, the most specific class of blood reagents, may give false positives as horseradish, leather and other extracts from plant material2 show the same peroxidase activity exhibited by haem in human blood. For this reason, we have previously reported a rapid and specific Matrix Assisted Laser Desorption Ionisation mass spectrometric method to detect blood in stains and map this biofluid in bloodied fingermarks.3 With this method, the mass-to-charge ratio (m/z) of both haem and intact Haemoglobin were employed to reliably confirm the presence of blood. The method was applied to a real crime scene stain proving successful in less than five minutes of preparation and acquisition time. Since blood provenance is also a forensic question of interest and as the m/z of haem would not permit the determination of the blood source, the m/z of intact Haemoglobin chains were exploited to distinguish between equine, human and bovine blood, based on the small differences in the protein amino acid sequence.3 However, although the detection of blood at a molecular level provides much higher specificity and reliability, intact protein analysis by MALDI mass spectrometry suffers from mass resolution and mass accuracy issues which may become significant, especially if blood is mixed with other biofluids or protein sources.The use of a bottom up proteomic approach increases the reliability of protein identification because the mass accuracy that can be achieved on the protein-deriving peptides is much higher (a few parts per million). This approach would also enable the detection of additional blood specific proteins, besides Haemoglobin, allowing specificity and confidence in the determination of the blood presence to be further enhanced. The literature already contains many reports attempting to map the proteome of plasma and serum. Different authors concur on the extreme complexity of these matrices with plasma being particularly challenging due to the wide4 range of concentrations of the proteins present (spanning 9 orders of magnitude) and the huge heterogeneity due to a variety of protein glycoisoforms. In 2010, Liumbruno et al.5 extensively reviewed the literature covering the mapping of the blood proteome with all the techniques employed up to that point in time and the corresponding number of obtained protein identifications.5 The majority of the methods employed separation techniques (gel based or liquid chromatography) hyphenated with mass spectrometry, in both on-line and off-line approaches, employing Electrospray and MALDI respectively as mass spectrometry techniques. Amongst the techniques used, the combination of 2D gel electrophoresis and mass spectrometry was reported to be able to identify 289 plasma proteins in 2002;4 cation exchange coupled to capillary gradient reverse phase liquid chromatography combined to mass spectrometry of digested peptides contributed to the identification of 490 blood serum proteins.6 These numbers have further increased when depletion and sample enrichment methods were preliminarily employed. In a 2005 collaborative study coordinated by HUPO involving 35 laboratories, up to 3020 plasma/serum proteins were identified using a range of hyphenated techniques;7 since the start of the HUPO project the number of identified proteins has rapidly increased to populate a database (http://www.plasmaproteomedatabase.org/) of 10546 proteins.8 None of the approaches reported in the literature so far has involved the direct application of MALDI MS on enzymatically digested blood. This is understandable as in all of the previous reports the aim was to map the entirety of the blood proteome for medical and diagnostic purposes. However, in a forensic context, the detection of a handful of blood specific proteins via the more reliable bottom up proteomic approach using MALDI MS would be more than appropriate. Furthermore, in forensic science, provided that reliability of the evidence is not compromised, speed is paramount to investigations; the hyphenated methods reported can be very labour intensive and time consuming, especially since some of them have employed preliminary purification to remove the most abundant proteins (e.g. albumin and Haemoglobin). For these reasons, in our laboratories, we have optimized a method for the digestion of bloodstains followed by direct MALDI MS analysis; the method couples high mass accuracy, within the peptide mass fingerprinting stage, as well as further confirmatory analysis by Tandem Mass Spectrometry. A classical in-solution digestion protocol was optimized for blood stains by investigating the optimal concentration of trypsin to employ as well as the optimal digestion time. The performance of this method was then critically compared to that of a second method employing Vmh2 hydrophobin to preliminarily coat the MALDI target plate. This protein belongs to the class I hydrophobins and it has been demonstrated to homogeneously self-assemble on hydrophilic or hydrophobic surfaces9 and to subsequently strongly bind proteins, including enzymes in their active form such as trypsin.10 The use of Vmh2 has been recently proposed as a lab-on-plate approach as a simple and effective desalting method enabling decrease in the proteolysis time and increase of the peptides signal-to-noise (S/N) for tryptic digestion.11
It was found that both methods could be successfully used to: (i) reliably detect the presence of blood in stains, (ii) determine the blood provenance even when two different blood sources were mixed and (iii) to identify the presence of this biofluid in a 9-year-old sample that had been pre-treated with acid black 1,12,13 a protein dye used for the unspecific enhancement/visualisation of blood. As it is discussed in this manuscript, the present data will no doubt impact on the effectiveness of forensic practice by providing much more reliable and informative evidence, thus empowering both investigations (of cold cases too) and judicial debates.
Experimental
Materials
ALUGRAMSIL G/UV254 aluminium sheets, acetonitrile (ACN), Ammonium Bicarbonate (AmBic), trifluoroacetic acid (TFA), trypsin from bovine pancreas and alpha-cyano 4 hydroxycinnamic acid (CHCA) were obtained from Sigma-Aldrich (Dorset, UK). Trypsin Gold was purchased from Promega, Southampton (UK) whereas Rapigest™ SF was purchased from Waters (Elstree, UK). Defibrinated horse blood was obtained from FisherScientific (USA). Unistik® 3 Neonatal & Laboratory single use lancet were obtained from Owen Mumford (Oxford, UK). Vmh2 ethanolic solution was prepared as previously described.10Instrumentation and data acquisition
Calibration over a 600–2800 Da mass range was performed prior to analysis using phosphorous red. MALDI IMS/MS data were acquired in positive ion mode from 600 to 3000 Da at a mass resolution of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 FWHM using a SYNAPT G2™ HDMS system (Waters Corporation, Manchester, UK) operating with a 1 KHz Nd:YAG laser. Full scan mass spectra were manually acquired over 45 seconds; all experiments were carried out in duplicate. The laser energy was set to 250 arbitrary units on the instrument; with laser energy increased to 270 arbitrary units for MALDI IMS-MS/MS experiments. MS/MS analyses were conducted in situ on the most intense peaks. Fragmentation was carried out in the transfer region of the instrument, post ion mobility separation, therefore product ions retain the same drift time as the precursor ion. Collision energies ranging between 60–80 eV were used to obtain the best signal to noise ratio for product ions.
000 FWHM using a SYNAPT G2™ HDMS system (Waters Corporation, Manchester, UK) operating with a 1 KHz Nd:YAG laser. Full scan mass spectra were manually acquired over 45 seconds; all experiments were carried out in duplicate. The laser energy was set to 250 arbitrary units on the instrument; with laser energy increased to 270 arbitrary units for MALDI IMS-MS/MS experiments. MS/MS analyses were conducted in situ on the most intense peaks. Fragmentation was carried out in the transfer region of the instrument, post ion mobility separation, therefore product ions retain the same drift time as the precursor ion. Collision energies ranging between 60–80 eV were used to obtain the best signal to noise ratio for product ions.
Methods
For enzymatic digestions performed using the lab-on-plate approach, 10 μl of defibrinated horse blood was spread across pre-cut 2 cm2 ALUGRAMSIL G/UV254 aluminium sheets pre-treated as previously described.14 These were sealed in petri dishes with parafilm and placed in an environmental chamber for 5 hours at 25 °C and 60% relative humidity. Under full ethical approval (HWB-BRERG23-13-14), human blood was obtained from the tip of the index finger using a Unistik® 3 Neonatal & Laboratory single use lancet UK) and blood was then prepared as described for horse blood. The MALDI plates were preliminarily functionalized with Vmh2 hydrophobin and subsequently immobilized with trypsin from bovine pancreas as previously described.10 The aluminium sheets with dried blood were carefully rolled into a glass vial, covered with 1 mL 50% ACN solution and ultra-sonicated for 10 min. One μl of sample was spotted on Vmh2-adsorbed enzyme wells (MALDI plate) contained immobilized trypsin. The on plate digest reaction was carried out for 5 min at room temperature. The reaction was stopped by the addition of 0.5 μl 10 mg mL−1 CHCA matrix solution. After mass spectrometric analysis the Vmh2 coating was removed by washing the MALDI plate with 10% TFA (and gently polishing the surface) followed by washing with 100% acetonitrile, water, and 100% acetone.
:1. Spectral processing consisted of smoothing, baseline correction and lock mass based mass correction. Prior to performing an MS/MS Mascot (Matrix Science, London, UK) search, spectra were processed using MassLynx™ with the MaxEnt 3 algorithm to deisotope and enhance the S/N.17 Queries were searched against the “Swiss-Prot” database with parent and fragment ion tolerances set to 50 ppm and 0.1 Da respectively. Two missed cleavages were also selected.
Results and discussion
Although detection of blood at crime scenes or on evidential items is often a crucial piece of intelligence in the investigation of criminal offences, current forensic visualization methods do not offer the desired level of specificity.3 This may result in incomplete or even in missing crucial information. In this paper the development of a rapid bottom up proteomic method offering blood-specific signatures is reported. The developed methodology employs a recently proposed procedure involving immobilization of trypsin on hydrophobin Vmh2 coated MALDI plates,10 (“lab-on-plate” approach). Although other methods for immobilizing trypsin for enzymatic digestion have been reported we have found the use of Vmh2 to be very straightforward and have optimized the reported protocols for the detection and identification of blood. MALDI MS profiles of blood were acquired from both in solution digest and the lab-on plate digest for comparative purposes. In order to optimise both methodologies, defibrinated horse blood was preliminarily employed. Both optimized methods yielded blood specific peptide signatures including those from myoglobin and the two chains of Haemoglobin with a mass accuracy lower than 8 ppm (Table 1). In general, relevant peptide intensities are greater within the 1 hour in solution digest; however the majority of peptides are still present employing the 5 minutes lab-on-plate digestion with generally a much better mass accuracy (Fig. 1A, B and Table 1). Since high throughput is always one of the “desirables” for any new forensic protocol, the method employing Vmh2 is highly relevant since it has been observed that the proteolysis is most efficient if the sample is allowed to digest for no longer than 5 minutes. The optimized methodologies were subsequently applied to whole human blood. The digestion of whole human blood using the classic in solution method resulted in a number of tentative protein identifications. In addition to peptides resulting from Haemglobin α (αHb) and β (βHb), a number of other proteins were detected including complement C3, apolipoprotein A-1, alpha-1-antitrypsin, haemopexin, serotransferrin and alpha-2-macroglobulin (Table 2). As seen in Table 2, the number of peptides originating from αHb and βHb is marginally greater in the in solution digest compared to the immobilized digest. However it is apparent that there are peptides from proteins such as myoglobin, haemopexin and serotransferrin detected only via the on lab-on-plate digest. Interestingly, using both methods, it was possible to tentatively assign multiple peptides to Erythrocyte membrane protein band (EPB) 3 and 4.2. The significance of this is that EPB 3 is specific to human blood. In the case of whole human blood, the overall relevant peptides intensities were lower within the in solution digest (Fig. 1C) in comparison to the on plate digest (Fig. 1D); this is probably due to the analyses being performed on whole human blood as opposed to a defibrinated sample (less complex) as in the case of the equine blood.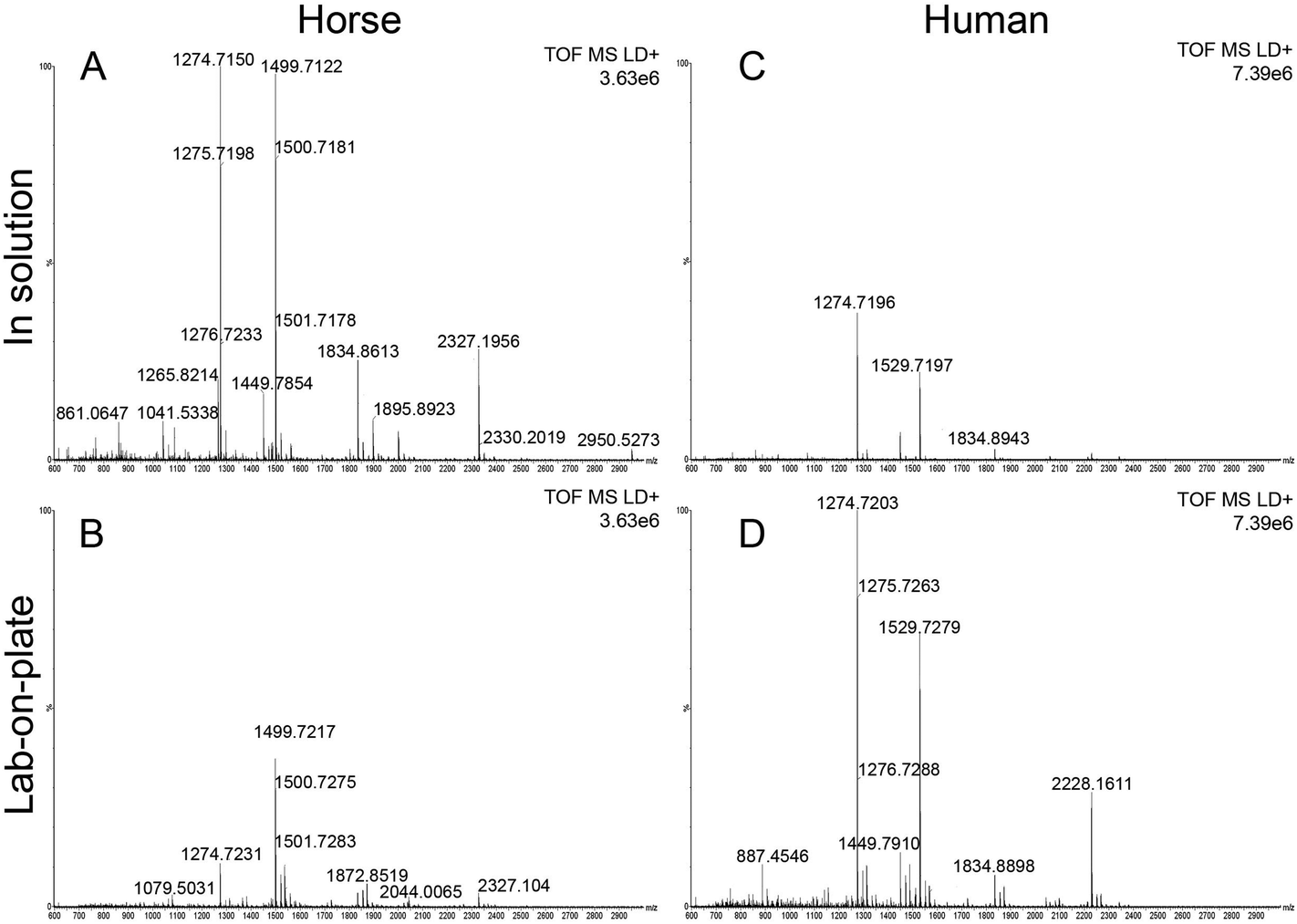 | ||
| Fig. 1 MALDI MS spectrum of digested blood. Panels 1A and 1B show the MALDI spectra of equine blood digested in solution and via the lab-on-plate approach respectively. Panels 1C and 1D show the MALDI spectra of whole human blood digested in solution and via the lab-on-plate approach respectively. | ||
| Horse proteins | Peptide m/z | Sequence | In solution relative error (ppm) | Lab-on-plate relative error (ppm) |
|---|---|---|---|---|
| Myoglobin | 2232.0865 | 120HPGDFGADAQGAMTKALELF R140 | — | −2.3296 |
| Haemoglobin beta | 2326.2037 | 9AAVLALWDKVNEEEVGGEALGR30 | −5.7174 | −0.2579 |
| 1999.9218 | 41FFDSFGDLSNPGAVMGNPK59 | −6.0002 | 6.3002 | |
| 1930.0293 | 66KVLHSFGEGVHHLDNLK82 | −5.4403 | −7.9791 | |
| 1801.9343 | 67VLHSFGEGVHHLDNLK82 | −7.5474 | — | |
| 1449.7961 | 133VVAGVANALAHKYH146 | −7.3803 | −0.6207 | |
| 1426.6849 | 121DFTPELQASYQK132 | −4.2756 | — | |
| 1358.6546 | 18VNEEEVGGEALGR30 | −6.0353 | −1.6928 | |
| 1274.7255 | 31LLVVYPWTQR40 | −7.8448 | −1.0198 | |
| 1265.8303 | 105LLGNVLVVVLAR116 | −7.3469 | — | |
| Haemoglobin alpha | 2043.0042 | 13AAWSKVGGHAGEFGAEALER32 | −3.3773 | −0.0978 |
| 1499.7237 | 18VGGHAGEFGAEALER32 | −7.4680 | −1.1335 | |
| 1833.8918 | 42TYFPHFDLSHGSAQVK57 | −7.1432 | −0.0545 |
| Human proteins | Peptide m/z | Sequence | In solution relative error (ppm) | Lab-on-plate relative error (ppm) |
|---|---|---|---|---|
| Haemoglobin beta | 767.4886 | 61VKAHGKK67 | −4.5603 | −10.8144 |
| 952.5098 | 2VHLTPEEK9 | −4.5143 | −5.5642 | |
| 1274.7255 | 32LLVVYPWTQR41 | −1.8827 | −4.0793 | |
| 1314.6648 | 19VNVDEVGGEALGR31 | −4.3357 | 0.1521 | |
| 1378.7001 | 122EFTPPVQAAYQK133 | 2.8287 | −10.0094 | |
| 1449.7961 | 134VVAGVANALAHKYH147 | −3.5177 | −3.1728 | |
| 1669.8907 | 68VLGAFSDGLAHLDNLK83 | −5.0901 | −10.7192 | |
| 1866.0119 | 2VHLTPEEKSAVTALWGK18 | −1.1253 | — | |
| 2058.9477 | 42FFESFGDLSTPDAVMGNPK60 | −2.7198 | −2.3312 | |
| 2228.1669 | 10SAVTALWGKVNVDEVGGEAL GR31 | −2.2439 | −2.4683 | |
| 2529.2190 | 84GTFATLSELHCDKLHVDPEN FR105 | −0.0790 | −8.1052 | |
| Haemoglobin alpha | 1071.5543 | 33MFLSFPTTK41 | −1.7731 | −1.6798 |
| 1087.6258 | 92LRVDPVNFK100 | −1.6549 | −0.5516 | |
| 1171.6681 | 2VLSPADKTNVK12 | −6.9132 | — | |
| 1529.7342 | 18VGAHAGEYGAEALER32 | −4.5105 | −3.7915 | |
| 1833.8918 | 42TYFPHFDLSHGSAQVK57 | −2.3447 | −3.7624 | |
| 2043.0042 | 13AAWGKVGAHAGEYGAEALER32 | −5.9226 | −3.1815 | |
| 2341.1836 | 42TYFPHFDLSHGSAQVKGHGKK62 | −2.6055 | −2.5200 | |
| 2582.2707 | 18VGAHAGEYGAEALERMFLSFPTTK41 | −1.1230 | −6.5059 | |
| 2996.4894 | 63VADALTNAVAHVDDMPNALSALSDLHAHK91 | −3.5374 | −3.1370 | |
| Myoglobin | 1685.8679 | 135ALELFRKDMASNYK148 | — | −5.1012 |
| Complement C3 | 887.4581 | 842NEQVEIR848 | −3.0423 | −3.2677 |
| 1334.7096 | 672SVQLTEKRMDK682 | 8.1665 | −6.6681 | |
| 1087.6357 | 1592EALKLEEKK1600 | −10.7572 | −9.6539 | |
| Apolipoprotein A-I | 1215.6215 | 220ATEHLSTLSEK230 | −4.1131 | — |
| 1230.7092 | 240QGLLPVLESFK250 | −0.9750 | −2.1938 | |
| 1723.9449 | 141QKVEPLRAELQEGAR155 | −3.7704 | −4.0024 | |
| 1815.8507 | 48DSGRDYVSQFEGSALGK64 | 7.2693 | 7.8200 | |
| 1833.8918 | 42TYFPHFDLSHGSAQVK57 | −2.3447 | −3.7624 | |
| 1908.9847 | 158LHELQEKLSPLGEEMR173 | −4.0859 | — | |
| Alpha-1-antitrypsin | 1318.6758 | 248LGMFNIQHCKK258 | −0.3033 | 5.4600 |
| Haemopexin | 965.4430 | 403VDGALCMEK411 | −5.9040 | 9.4257 |
| 1060.5785 | 84ELISERWK91 | — | −1.8857 | |
| 1070.5741 | 214GEVPPRYPR222 | — | 2.6154 | |
| Serotransferrin | 1068.5506 | 61KASYLDCIR69 | — | 9.7328 |
| 1855.8683 | 531EGYYGYTGAFRCLVEK546 | −0.1616 | −0.6465 | |
| EPB 4.2 | 949.4771 | 454EKMEREK460 | 5.0554 | 8.3203 |
| 1048.5455 | 451VEKEKMER458 | −0.1907 | 5.2453 | |
| 1079.5745 | 205WSQPVHVAR213 | −9.4481 | — | |
| 1113.4881 | 428CEDITQNYK436 | 1.7063 | — | |
| 1258.7001 | 446EVLERVEKEK455 | −2.3834 | 1.9861 | |
| EPB 3 | 949.4771 | 284AAATLMSER292 | 5.0554 | 8.3203 |
| 1328.6852 | 731SVTHANALTVMGK743 | — | −2.7847 | |
| Alpha 2-Macroglobulin | 1334.7215 | 350LSFVKVDSHFR360 | −0.7492 | — |
A close evaluation of the data on its performance, in comparison with an optimized in solution digestion of the minimum duration of 1 hour (Fig. 1A and B), shows that the lab-on-plate protocol enabled the detection of the same number of blood proteins but less blood protein-derived peptides (10/13 of the peptides from myoglobin, αHb and βHb observed in in solution digest). However the slightly fewer number of peptides detected is outweighed by the considerably reduced digestion time for the lab-on-plate approach.
As can be seen in Table 2 there are instances in which only one peptide could be putatively assigned to a protein (i.e. in the case of myoglobin, alpha-1-antitrypsin and alpha-2-macroglobulin). This is not standard practice in proteomics whereby, for increased identification reliability, at least two peptides should be assigned to a single protein. In the view of these authors, this is not an issue preventing to claim the presence of blood; based on the experiments carried out, we suggest the presence of two or more peptides from αHb and βHb and another blood protein (i.e. myoglobin or serotransferrin) to be the proposed minimum for the confident identification of blood.
Encouraged by these data, the focus was moved onto investigating the opportunity to provide information of the provenance of blood. These authors have already reported preliminary data on blood provenance by MALDI-MS;3 an intact protein detection approach that was employed that, whilst successful in the instances investigated, may suffer from mass resolution and mass accuracy issues, thus reducing the level of reliability of the scientific evidence provided. At least one criminal case has been widely reported in the UK (Regina vs. Mrs Susan May),18 in which determining with certainty the provenance of the blood detected would have resulted in a better informed or speedier outcome. The importance of determining blood provenance is further testified by a case from the USA reported 1996. Here the blood of the dog shot together with his owners aided the conviction of two men of murder; in this case it took a DNA test (in the first trial ever in the country to use animal DNA as evidence) to prove the presence of canine blood on the jacket of one of the murderers.19 Already the comparison of the peptides obtained for equine and human blood (Fig. 1A, D and Tables 1, 2) demonstrate this as a feasible approach to determine blood provenance with a much higher specificity than previously shown.3 To further demonstrate robustness of the method, the lab-on-plate approach was applied to a sample made from mixing both equine and human blood.
Fig. 2 shows the peptide mass spectral profiles obtained from in solution (Fig. 2A) and lab-on-plate (Fig. 2B) digests of a mixture of human and equine blood. Although overall signal intensity is higher within the in solution digest spectrum, both digestion protocols enabled the detection of blood peptide markers specific to each species and putatively assigned peptides are shown in Table S1 (ESI†). A number of tryptic peptides originating from αHb and βHb were present. However, due to the extensive sequence homology between the two species, it was not possible to solely use the m/z of these protein derived peptides or even the confirmed presence of βHb tryptic peptide at m/z 1274.7260 via MALDI-IMS-MS/MS analysis of the peptide ion (Fig. 3A) as markers for species differentiation. However, subjected to MS/MS analysis, the tryptic peptide at m/z 1499.7237 was identified as equine αHb with Mascot score of 99 (Fig. 3B). Furthermore, the tryptic peptide m/z 1815.9024 originating from myoglobin was also detected in the same spectra. This peptide is specific to the equine protein sequence thus more robustly confirming the presence of blood from equine provenance. Additionally, as expected from the in silico digestions, the detection of the human EPB 4.2 peptides, at m/z 949.4771 and 1113.4881 (present in the 1 hour in solution digest and via the rapid lab-on-plate hydrolysis), as well as that of serotransferrin at m/z 1529.7529, indicated the further presence of human blood thus enabling to claim the sample to be of mixed provenance, as well as indicating the individual species contributing to the blood sample under investigation. The authors would like to note that although there is a significant sequence homology between EPB 4.2 and α-2-macroglobulin within humans and chimpanzees, the indication of EPB 4.2 to be specific to human within this discussion is only with respect to equine blood. Both the in solution and the lab-on-plate approaches were successful in determining the double source of blood, and the considerably shorter digestion time within the lab-on-plate makes this the preferred method once again.
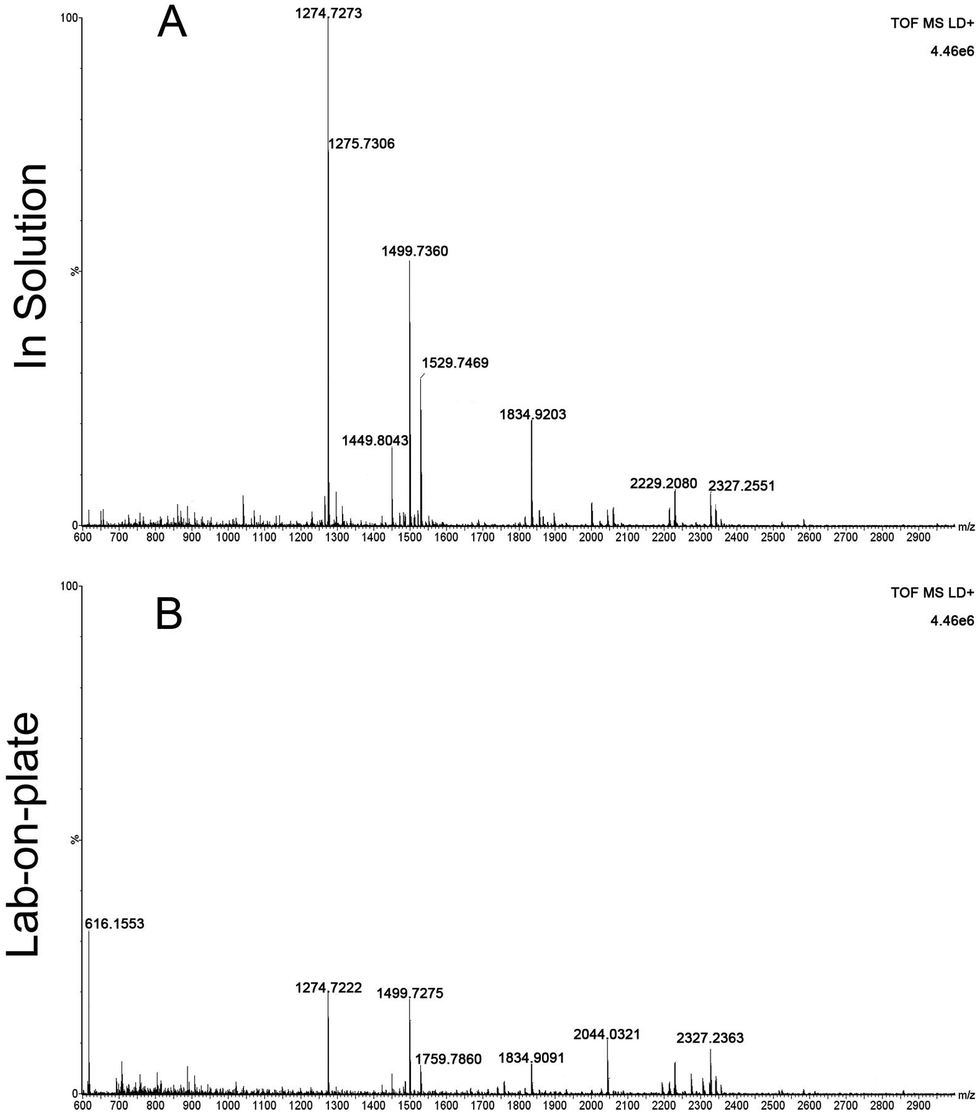 | ||
| Fig. 2 MALDI MS spectrum of mixed digested blood. Panels A and B show the mass spectral profile of whole human blood mixed with defibrinated horse blood using the in solution and the lab-on-plate approach respectively. | ||
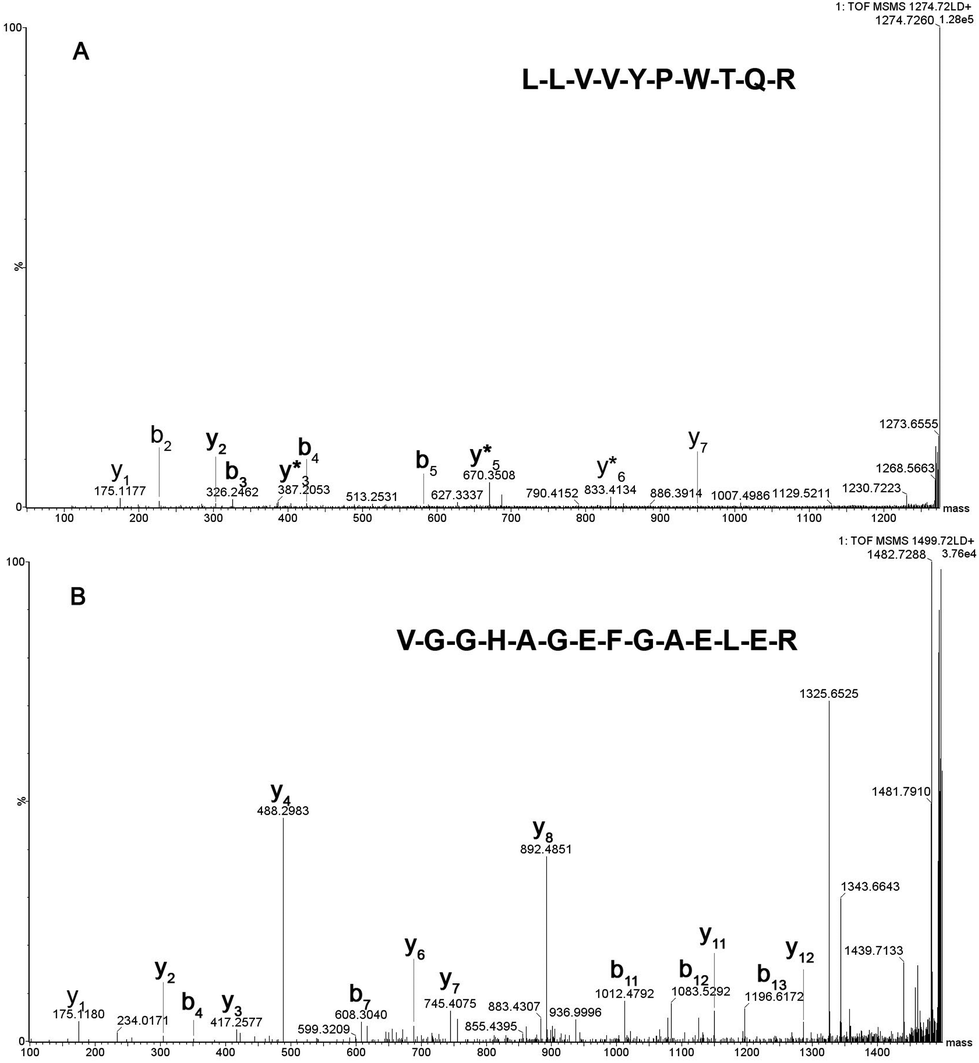 | ||
| Fig. 3 MALDI-IMS-MS/MS of tryptic peptides m/z 1274 (3A) and m/z 1499.7237 (3B), identified via Mascot as βHb and αHb respectively. Both b and y ions are annotated with y* representing the y-NH3 fragment ion. | ||
Finally, a method that is applicable not only to fresh bloodstains but also to much older ones would be highly desirable in the review of cold cases. Therefore the Vmh2 lab-on-plate method was tested, in comparison with the classic optimized in solution protocol, on a 9-year-old bloody handprint which was deposited on a ceramic tile and stored at room temperature (Fig. 4A(i and ii)). Spectra acquired from the analysis of the extract digested in solution (Fig. 4B) and via on plate hydrolysis (Fig. 4C) are shown, with corresponding expanded mass regions in the m/z range 1000–2000. A number of relevant tryptic peptides are present including αHb peptides m/z 1087.6258, 1529.7342 and βHb peptides m/z 1274.7255 and 1449.7961 to name a few (Table S2†). Data obtained indicated that blood presence confirmation was possible with the in solution approach, though both EBP 4.2 (indicating that the blood may be of human origin) and Complement C3 were identified by one peptide only each. The lab-on-plate approach did not allow the detection of the Complement C3 protein (which is not highly specific to blood in any case) and also enabled the detection only one EBP 4.2 peptide. The authors suggest that in these cases, the lab-on-plate approach should still be used first for its rapidity. However for confirmatory purposes, as a tryptic digestion generates numerous peptides resulting in complex mixtures, often with overlapping signals, cross validation and identification using LC/MS/MS may be beneficial.
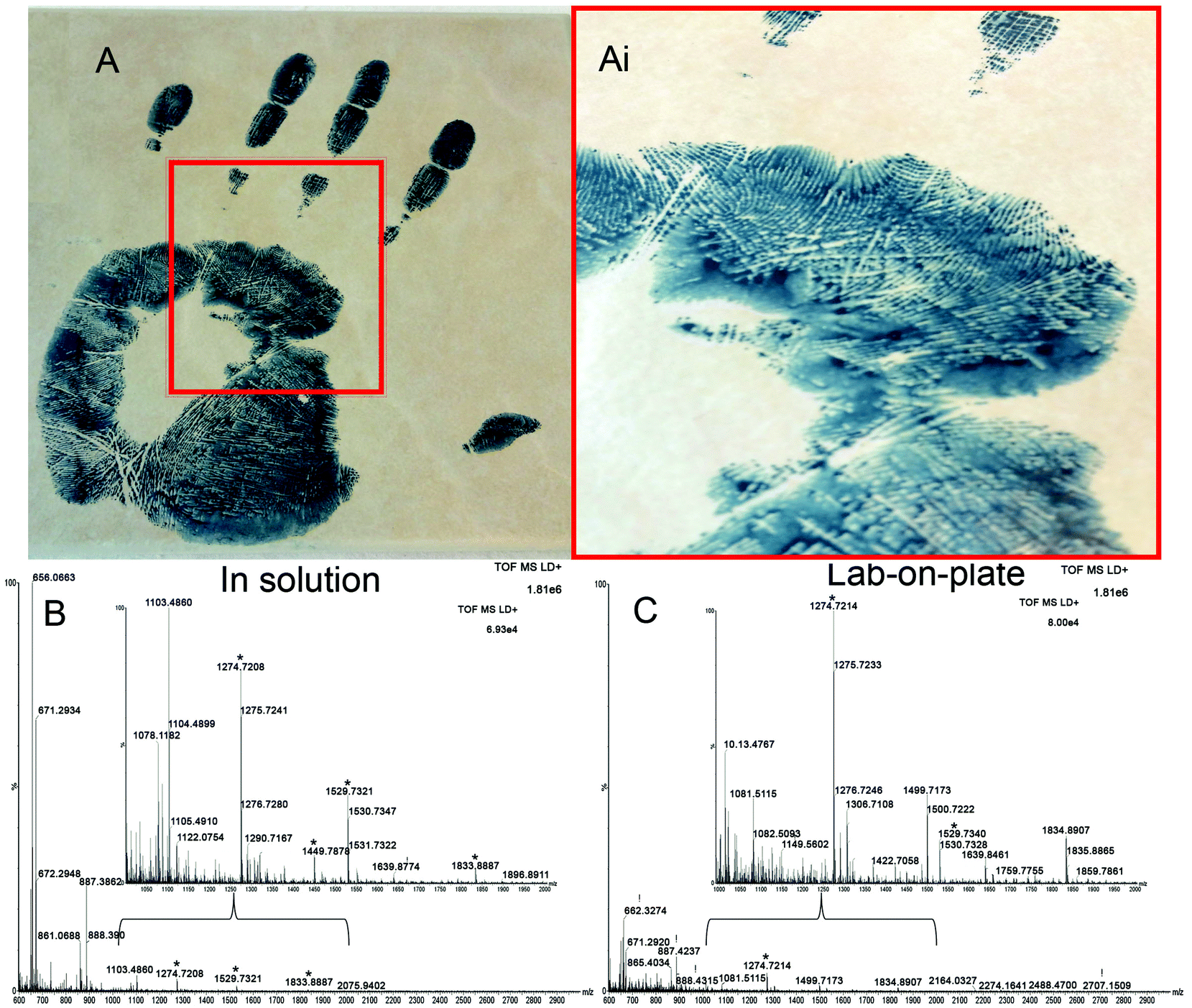 | ||
| Fig. 4 Confirmation of the presence of blood from a 9-year-old forensically treated sample. Panels A and Ai show the bloodied handprint and magnified region from which the blood was swabbed respectively (the blue-black colour is due to the treatment with the protein stain Acid black 1). Panels B and C show the mass spectral profiles of the extracts digested in solution and via the lab-on-plate approach respectively. | ||
In addition to the ability to detect blood reliably and from such an old sample, it is very important to note that the bloodied handprint was preliminarily, 9 years ago, enhanced with acid black 1, a commonly used protein stain for blood enhancement. Successful blood confirmation in this instance demonstrates feasibility of the protocol to be integrated in the forensic workflow for blood enhancement/visualisation. The data obtained suggest that the acid black 1 does not interfere with the analyses, rather, that it may slow down degradation of the blood proteins over time.
Conclusions
The shotgun method illustrated in this report will have a significant impact on forensic practice as well as on the overall criminal justice system by generating more robust and informative evidence. This is due to the high specificity of the method against current presumptive tests prone to generate false positives. Furthermore the recovery of simultaneous information on blood provenance will both empower and speed up investigations as well as strengthening judicial debates. The study also crucially highlights compatibility with the necessary and prior application of blood enhancement techniques in combination with the analysis of very old blood samples, thus opening up new forensic opportunities for the review of cold cases. The lab-on-plate approach was shown to additionally offer rapid results (5 minutes only proteolysis time) which, in an operational forensic context, is a highly desirable feature. These studies are currently being expanded in our laboratories and include the reliable mapping of blood signatures on fingermark ridges using MALDI MS Imaging in order to link the suspect (through the biometric information) to the crime. Finally, validation has also been planned whereby the requirement for the minimum number of blood peptide signatures for both blood detection and blood provenance determination will be provided through a blind study in collaboration with the Minnesota Bureau of Criminal Apprehension.Acknowledgements
The authors gratefully acknowledge BBSRC and GlaxoSmithKline for the BBSRC Industrial Case Award funding the Ph.D. studentship of E. P. The European COST Action programme BM1104 ‘Mass Spectrometry Imaging: New Tools for Healthcare Research’ is also gratefully acknowledged for funding a 3 month Short Term Scientific Mission of PhD student P. C. at the Biomolecular Research Centre, Sheffield Hallam University.References
- V. Sears, in Advances in Fingerprint Technology, ed. H. Lee and R. Gaensslen, CRC Press, 2013 Search PubMed.
- M. Stoilovic, Forensic Sci. Int., 1991, 51, 289–296 CrossRef CAS PubMed.
- R. Bradshaw, S. Bleay, M. R. Clench and S. Francese, Sci. Justice, 2014, 54, 110–117 CrossRef CAS PubMed.
- N. L. Anderson and N. G. Anderson, Mol. Cell. Proteomics, 2002, 1, 845 CAS.
- G. Liumbruno, A. D'Alessandro, G. Grazzini and L. Zolla, J. Proteomics, 2010, 73, 483–507 CrossRef CAS PubMed.
- J. N. Adkins, S. M. Varnum, K. J. Auberry, R. J. Moore, N. H. Angell, R. D. Smith, D. L. Springer and J. G. Pounds, Mol. Cell. Proteomics, 2002, 1, 947 CAS.
- G. S. Omenn, Proteomics: Clin. Appl., 2007, 1, 769–779 CrossRef CAS PubMed.
- V. Nanjappa, J. K. Thomas, A. Marimuthu, B. Muthusamy, A. Radhakrishnan, R. Sharma, A. Ahmad Khan, L. Balakrishnan, N. A. Sahasrabuddhe, S. Kumar, B. N. Jhaveri, K. V. Sheth, R. Kumar Khatana, P. G. Shaw, S. M. Srikanth, P. P. Mathur, S. Shankar, D. Nagaraja, R. Christopher, S. Mathivanan, R. Raju, R. Sirdeshmukh, A. Chatterjee, R. J. Simpson, H. C. Harsha, A. Pandey and T. S. K. Prasad, Nucleic Acids Res., 2014, 42, D959 CrossRef CAS PubMed.
- L. De Stefano, I. Rea, E. De Tommasi, I. Rendina, L. Rotiroti, M. Giocondo, S. Longobardi, A. Armenante and P. Giardina, Eur. Phys. J. E, 2009, 30, 181–185 CrossRef CAS.
- S. Longobardi, A. Gravagnuolo, R. Funari, B. Della Ventura, F. Pane, E. Galano, A. Amoresano, G. Marino and P. Giardina, Anal. Bioanal. Chem., 2015, 407, 487–496 CrossRef CAS PubMed.
- S. Longobardi, A. M. Gravagnuolo, I. Rea, L. De Stefano, G. Marino and P. Giardina, Anal. Biochem., 2014, 449, 9–16 CrossRef CAS PubMed.
- V. Sears and T. Prizeman, Journal of Forensic Identification, 2000, 50, 470–480 Search PubMed.
- V. Bowman, V. Sears, H. Bandey, S. Bleay, L. Fitzgerald, A. Gibson, S. Hardwick, A. Hart, D. Hewlett, T. Kent and S. Walker, nonymous HOPSDB, 2nd edn, 1998 Search PubMed.
- R. Wolstenholme, R. Bradshaw, M. R. Clench and S. Francese, Rapid Commun. Mass Spectrom., 2009, 23, 3031–3039 CrossRef CAS PubMed.
- M. Strohalm, M. Hassman, B. Kosata and M. Kodicek, Rapid Commun. Mass Spectrom., 2008, 22, 905–908 CrossRef PubMed.
- M. Strohalm, D. Kavan, P. Novak, M. Volny and V. Havlicek, Anal. Chem., 2010, 82, 4648–4651 CrossRef CAS PubMed.
- M. C. Djidja, S. Francese, P. M. Loadman, C. W. Sutton, P. Scriven, E. Claude, M. F. Snel, J. Franck, M. Salzet and M. R. Clench, Proteomics, 2009, 9, 2750–2763, DOI:10.1002/pmic.200800624 DOI:10.1002/pmic.200800624.
- CCRC report-Statement of reasons, http://www.susanmay.co.uk/ccrc.htm, (accessed July 2015).
- Forensic Files-Historic cases, chief evidence, https://www.youtube.com/watch?v=q8jL8S2yIe8, (accessed July 2015).
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5an02016f |
| ‡ These authors have equally contributed to the manuscript. |
| This journal is © The Royal Society of Chemistry 2016 |