
Natural attenuation processes of arsenic in the groundwater of the Brahmaputra floodplain of Assam, India†
Lalsangzela
Sailo
*a and
Chandan
Mahanta
b
aSchool of Engineering and Technology, Mizoram University, India. E-mail: zelasailo@gmail.com
bDept. of Civil Engineering, Indian Institute of Technology Guwahati, India. E-mail: mahantaiit@gmail.com
First published on 12th November 2015
Abstract
Natural attenuation of higher arsenic (As) concentration in the groundwater of the Brahmaputra valley of Assam can be a viable option for its remediation. The assessment of attenuation capacities in aquifers was done using three sediment core samples (namely C_50, C_70 and C_150) representing the commonly found sediments and characterized as viz. reduced, oxidized and moderately oxidized sediments, respectively. The feasability of aquifers to release low As water depends on the sorption capacity of the sediments to directly adsorbed as As(III) and/or oxidation to As(V) on oxidative sites. The various batch sorption experiments along with competition of other anions for adsorption sites at different concentrations were investigated. The sediments were observed to adsorb both As(III) and As(V) with varying magnitudes depending on the species of As, pH and concentrations of competing anions. The Elovich kinetic model best described kinetic test data. The oxidized sediment (C_70) with the highest Fe and Mn content has the highest adsorption capacity. The competition for adsorption sites with As(V) in the sediment samples followed the order PO43− > SiO2 > HCO3−. The oxidation of As(III) to As(V) by the sediments was analyzed as it is also the most effective pathway to reduce the toxicity as well as the mobility of As in the subsurface environment. Sediment C_70 showed the highest potential to oxidize As(III) to As(V) in the oxidative sites of the sediments. The removal of As(III) in the oxidative site via As(V) was the dominant As removal mechanism for the oxidized sediment (C_70) and moderately oxidized sediment (C_150) than the direct adsorption of As(III) on the adsorptive sites as on reduced sediment sample C_50. Therefore, targeting the oxidized sediment with a high concentration of Fe-oxides and Mn-oxides might make natural attenuation of As viable in the study areas.
Environmental impactThis paper addresses the natural attenuation capacities of sediments commonly found in the Brahmaputra Floodplains of Assam. A batch sorption experiment was conducted to evaluate the mechanism of sorption of arsenic from three different types of natural sediments and the effects of co-anions in the adsorption capacities. Finally, the oxidation of As(III) to As(V) test suggested that oxidation to As(V) on the oxidative site was the main removal mechanism in the oxidized sediments. This suggested that not only reduction in toxicity but also immobilization is achieved, since As(V) has higher affinity for adsorption. Therefore, the study helps in targeting safe aquifers (i.e. oxidized sediment) which could naturally attenuate arsenic to lower levels. |
1. Introduction
Elevated concentrations of arsenic (As) in the shallow groundwater of Tokobari village, in Jorhat district of the Brahmaputra valley in the Northeastern part of India pose a potential As contaminated health risk. Due to the absence of main anthropogenic sources of As it was considered to be geogenic. The As contamination in the Assam floodplains of the Brahmaputra River has been a serious concern due to the widespread dependence of the population on groundwater for drinking, domestic and irrigation purposes.1 The sources of As in the region may be primarily the sediments derived from the weathering and erosion of lithified crystalline rocks of the Himalaya and adjacent regions of Bangladesh2 which have similar hydrogeologic deposition.3 These sediments were transported by the big river Brahmaputra, deposited along with organic matter, thus becoming sources of As to the groundwater.4,5 High As groundwater areas are usually associated with reduced aquifers with high organic carbon and composed of gray to black sand.6–10 The arsenite As(III) was found to be the dominant species compared to arsenate As(V) in reducing environment groundwater of the study.11The arsenic concentration in groundwater is usually controlled by natural geochemical processes, where the adsorption–desorption reaction of As species on mineral plays a key role.3,12,13 The adsorption–desorption processes are also important for natural attenuation and water purification technologies, with the aim of removing As from water.14,15 The viability of aquifers to release low As water depends on the capacity of the sediments to remove As from the groundwater that could infiltrate the zone of high organic content and strongly reduced sediments. Adsorption is one of the most commonly reported mechanisms and possibly the initial reaction to occur when As interacts with sediments.14,16,17 Since, adsorption and oxidation of As regulate its fate and transport in the sediment, factors controlling the sorption process is of paramount importance. Numerous factors affect the efficiency of As adsorption, but the adsorption study of As on sediments has been mainly linked to environmental factors such as pH,18–22 the redox potential, reaction time,16,23 oxidation state of As16,17,20 and presence of competing anions.17,24–26 The competition for sorption sites and various surface complexation models simulating the actual field conditions has also been attempted.7,21,22,27–30 Color characterization of the aquifer for targeting safe arsenic drinking water in shallow wells of the Bengal basin has been already reported.6–8
The oxidation of As(III) to As(V) by the sediment was studied as it not only decreased the toxicity of As, but also helped to immobilize it. There is evidence of oxidation of As(III) to As(V) by Mn-oxides, which also could be an important factor controlling the As species and the extent of As adsorption by the sediments.17,31 However, oxidation of As(III) by Fe(III) hydroxide in lake sediments has been reported previously,32 possibly because of the high Fe(III)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :As(III) ratio. As(III) oxidation by MnO2 was found to occur abiotically;31,33 however, several microbial respiratory and non-respiratory enzymatic systems for oxidation of As(III) have been reported.34
:As(III) ratio. As(III) oxidation by MnO2 was found to occur abiotically;31,33 however, several microbial respiratory and non-respiratory enzymatic systems for oxidation of As(III) have been reported.34
In this study, we focused on examining As sorption as well as oxidation to As(V) processes in the selected three sediment samples which are commonly found in the area namely C_50, C_70 and C_150 (at a depth of 50 ft, 70 ft and 150 ft), to gain an understanding for possible remediation and for delineating sources of low As groundwater. The characteristics of the sediment samples derived from SSE are shown in Table 1. The effects of pH, phosphate (PO43−), silica (Si) and bicarbonate (HCO3−) and combined anion effects on As adsorption were evaluated using batch experiments. Finally, the oxidation of As(III) to As(V) test was performed to analyze the sediment capacity to attenuate the As pollution and its mobility in the sub-surface environments.
| Sample ID | Color | Depth (m) | Reductively extracted Fe (mg kg−1) | Reductively extracted Mn (mg kg−1) |
|---|---|---|---|---|
| C_50 | Gray | 15 | 3000 | 8 |
| C_70 | Brown | 21 | 28000 |
200 |
| C_150 | Ash-greenish | 46 | 7000 | 160 |
2. Materials and methods
2.1 Study area
Samples of groundwater and sediments were collected from the Titabor (Tokobari) area of Jorhat District in Assam, India. The study area is situated in the eastern part of Assam state of India between latitude 26°27.3′ North and 26°30.8′ N, longitude 94°6.3′ E and 94°9.8′ E (Fig. 1). The site is located at an elevation slightly higher than the present day floodplains with younger alluvial deposits and Brahmaputra flows on the northern side of the district. The mean annual rainfall in the district is 2197 mm. The sub-surface material consists of gravels, sands, silts and clays.1 The depth of hand tubewells is within 40 m to 50 m. Old meanders, paleochannels (abandoned channels), natural levees, ox-bow lakes and water bodies are very common in the area.1 From the study area, 44 groundwater samples were collected during November 2008 to January 2010 from existing tubewells. The tubewells were classified as shallow tubewells as the depths were mostly between 40 and 50 m.1 Sediment sampling was simultaneously done.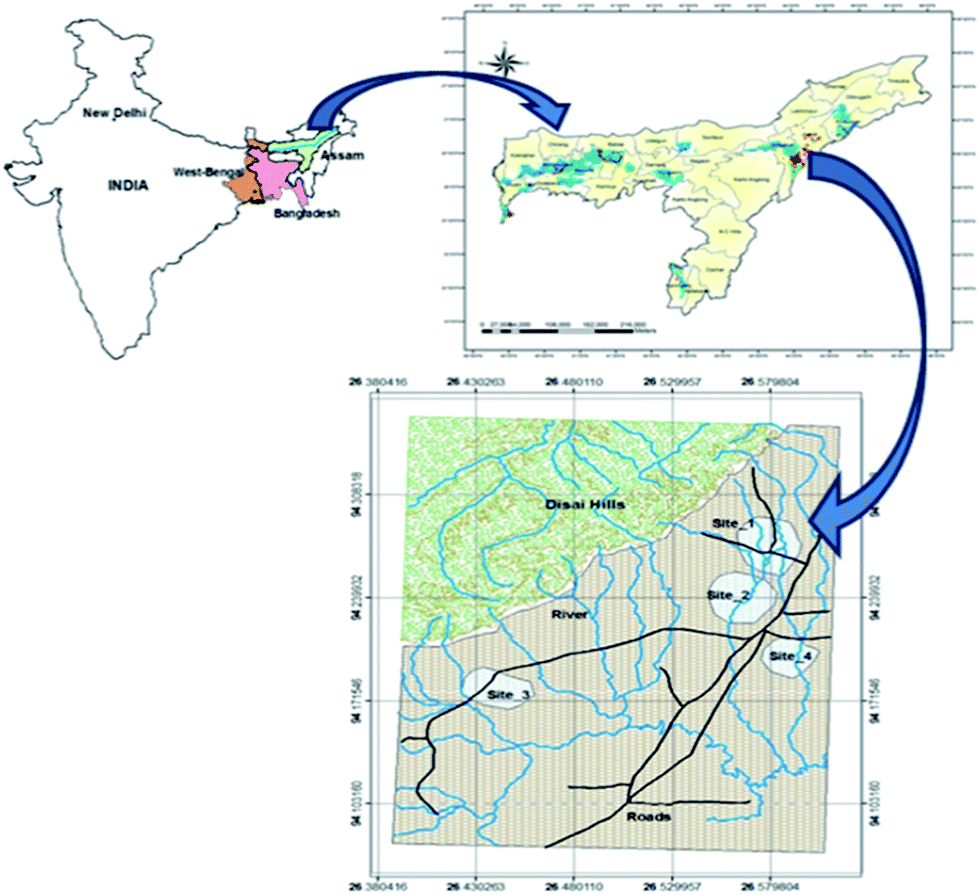 | ||
| Fig. 1 Location of the study area Tokobari, Jorhat district. | ||
2.2 Sample collection
Based on the results of groundwater and sediment samples, six boreholes were drilled adjacent to two higher As contaminated tubewells (T1 and T2 > 300 μg L−1 as shown in Fig. A1) in the latter part of January 2010 to understand the mobilization and immobilization processes governing the sub-surface. During drilling, the color and texture of the sediment were noted and samples were collected in clean sealed airtight polyethylene bags and stored in the dark until analysis was complete. The sediment samples were analysed for its physical, morphological and chemical properties.352.3 Batch experiment
The aim of the batch sorption study was to examine the mechanism and extent of arsenic uptake in the laboratory batch experiment using field sediments from cores taken at the 1C from the study site in order to quantify the ability of natural sediments to attenuate As present in groundwater. Three sediment samples C_50, C_70 and C_150 were chosen for the batch experiment, and characteristics of these sediments were evaluated (Table 1). The reduced sediment (C_50) was characterized by gray color with low Fe and Mn, while the oxidized sediment (C_70) was brown in colour with high concentrations of Fe and Mn, while the moderately oxidized sediment (C_150) had moderate values of Fe and Mn. Most tubewell screens were located at this depth (∼46 m) i.e. C_150. These sediment samples that were analysed were present in most of the sediment core samples from the area, which may have different geochemical properties for the release of As into the groundwater.All the experiments were performed with synthetic, arsenic spiked groundwater with equivalent characteristics of groundwater of the study area as far as possible. The solutions contained Ca2+ 16 mg L−1, Mg2+ 10 mg L−1, and K+ 3 mg L−1 which were broadly comparable to the natural groundwater of the study area that was used in all the batch experiments.11 For preparing the solutions, chloride forms were used (viz. CaCl2, MgCl2, KCl and NaCl) in these experiments to avoid the adsorption effect from HCO3− salts and a slightly higher Cl concentration was observed. The desired concentrations of cations were prepared in the laboratory using its molarity in order to find the competition of anions onto sorption. The pH of the solution was set to 6.5 which was the mean pH of the groundwater samples. The stock solutions were prepared from reagent grade sources i.e. As(V) from Na2HAsO4·7H2O, As(III) from NaAsO2, PO43− from K2HPO4, Si (silica) from Na2SiO3·9H2O and HCO3− from NaHCO3.
:20 soil/water ratio. The two initial As concentrations chosen to replicate the groundwater were 300 μg L−1 and 500 μg L−1 for the competition of ions. The samples were gently shaken in a horizontal shaker at an rpm of 180 for 1440 minutes (24 hours). The tubes along with its duplicates were withdrawn at predefined time interval (i.e. 5, 10, 20, 30, 60, 120, 240, 360, 600, and 1440 minutes) and then centrifuged at 9000 rpm for 15 minutes and supernatants were collected using a syringe. The amount of As adsorbed per unit mass of sediment (μg g−1) was calculated from eqn (1) and the sorption percentage was calculated using eqn (2). The various adsorption models viz. the pseudo-second order kinetic model, Elovich model and intraparticle diffusion model were fitted for each of the As species.
The amount of As adsorbed per unit mass of sediment (μg g−1) was calculated from the difference between the concentration of the supernatant and that of the initial solutions using the following equation:
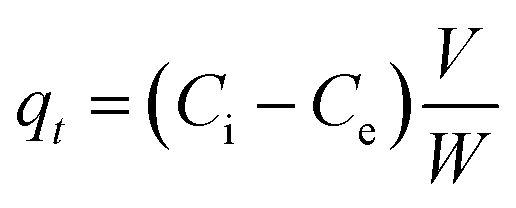 | (1) |
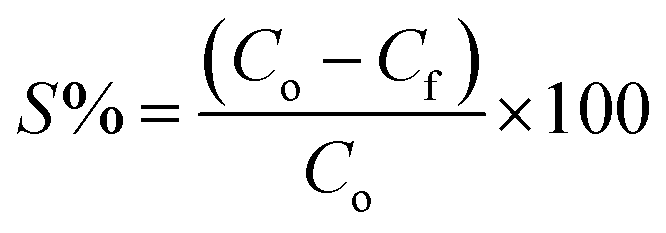 | (2) |
2.3.1.1 Elovich kinetic model. The Elovich equation has been widely used in adsorption kinetics, which described chemical adsorption in nature such as soils.36 The Elovich equation can be written in the form:37
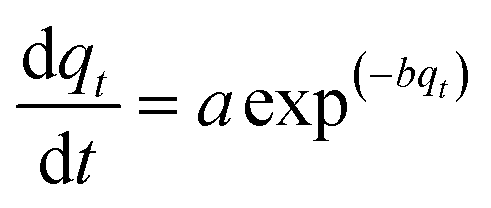 | (3) |
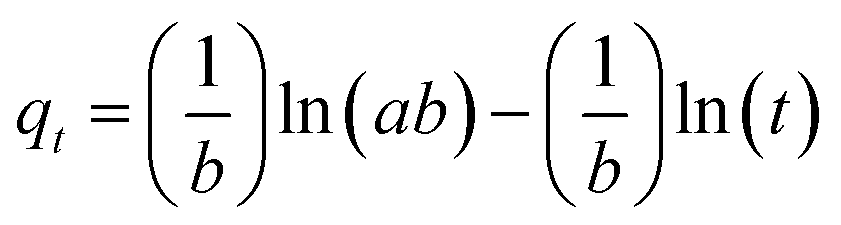 | (4) |
The constants a and b were calculated from the slope and intercept of plot qtversus ln(t).
2.3.1.2 Intraparticle diffusion model. It is usually difficult to apply simple kinetic equations to describe sediment chemical reactions, since the sediment surfaces are heterogeneous and one cannot differentiate the transport effect from chemical kinetics. The most-widely applied intraparticle diffusion equation was given by Weber and Morris (1963):38
| qt = Kit0.5 + c | (5) |
Freundlich and Langmuir adsorption isotherms were fitted on the observed experimental data. The Langmuir equation can be written in the following linear form:39
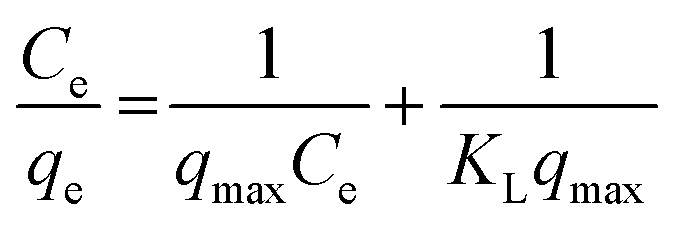 | (6) |
In the Freundlich adsorption model the adsorbed mass per mass of the adsorbent can be expressed by a power law function of the solute concentration:40
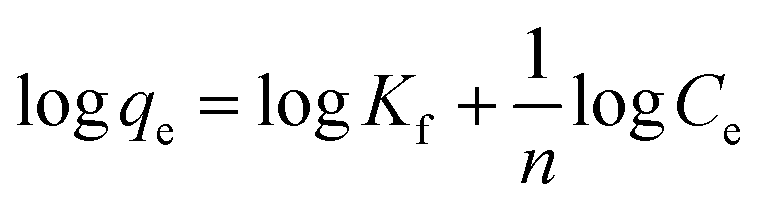 | (7) |
qeversus logCe has a slope with a value of 1/n and an intercept magnitude of logKf.
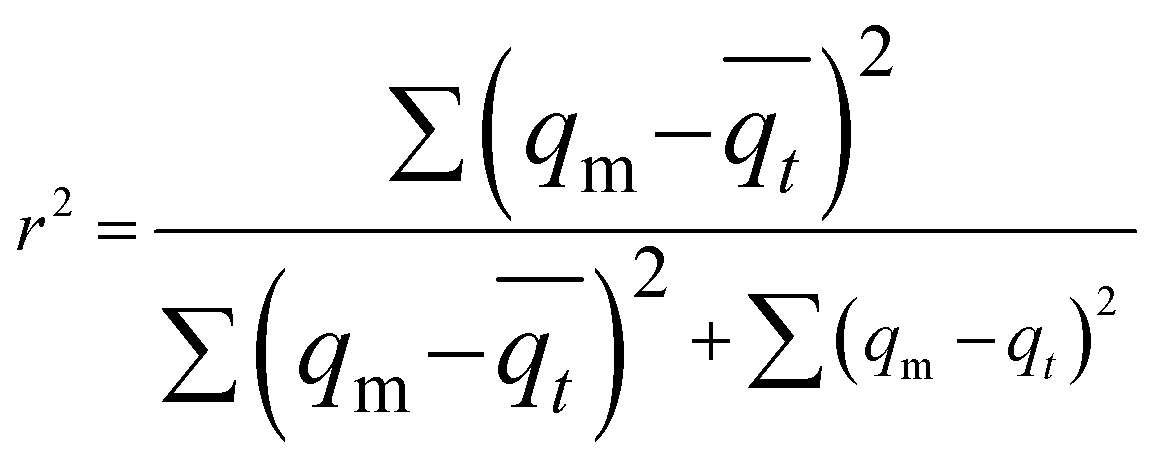 | (8) |
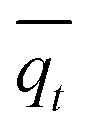 is the average of qt (μg mg−1).
is the average of qt (μg mg−1).
2.4 Oxidation kinetics of As(III) to As(V)
The experiments for oxidation kinetics of As(III) to As(V) were carried out under similar conditions for the sorption kinetics. The supernatant was collected from the tube by decantation after rotating in the dark in a horizontal shaker @ 180 rpm and the solid was used for the analysis of solid phase As. Then the solid-bound arsenic species were desorbed by adding 25 mL of 0.2 M oxalic acid after the removal of the supernatant.31 The tubes were then rotated in the dark for 15 min and centrifuged to remove the supernatant containing the desorbed arsenic species. The solid bound As was speciated to As(III) and As(V) to determine the adsorption and oxidation in the soil sediment samples.The speciation of As species was performed with disposable cartridges (MetalSoft Centre). The speciation of the dissolved arsenic was determined by passing 50 mL of the supernatant through the cartridges at a flow rate of 60 ± 30 mL per minutes using a 50 mL syringe. Under our experimental conditions, As(V) is present predominantly as H2AsO4− and is removed by using the cartridge, whereas As(III) is present predominantly as H3AsO3 and elutes through the cartridge column.
Speciation of the solid phase As species was determined by adjusting the pH of the supernatant from the oxalic acid extract to approximately 4.5 by adding 10 M NaOH. The As(III) concentration was determined from the eluting solution from the cartridge, and the As(V) concentration was determined using the difference between the total arsenic concentration in the supernatant and the As(III) concentration. The mass balance of arsenic species was achieved as a summation of the solid-phase As(III) and As(V) concentrations and total aqueous As(III) concentration.
3. Results and discussion
The sediments from the study areas contain a range of reactive functional groups that originate from the mixture of mineral phases and their surface coatings. The surface coatings were generally comprised of Fe, Al and Si, with lesser amounts of Mn as can be seen from SEM/EDX, likely in the form of Fe(III) hydroxides, Al hydroxides and silicates, and Mn oxides (Appendix Fig. A2 and A3).3.1 Arsenic adsorption isotherm
From the batch experimental study, it was found that the sediments can adsorb As(III) and As(V), for the three sediment samples viz. C_50, C_70 and C_150 (Fig. 2a and b). Freundlich and Langmuir adsorption isotherms were fitted into the experimental data (Table 2). It revealed that the highest As adsorption capacity was obtained for sediment sample C_70 which is consistent with the highest Fe and Mn content. The Fe-amorphous and Fe-crystalline oxides had high affinity for As sorption.18,20,41,42 Lower adsorption was observed for samples C_50 and C_150, and they had nearly comparable adsorption capacities. The adsorption of As(V) was found to be higher than that of As(III) and the adsorption behaviour of As(V) and As(III) onto Fe(III) oxide minerals is similar at pH 6.5 to 8.20 Since the pH of the groundwater was found to be near neutral conditions, it was expected that the oxidized sediments would also have significant capacity to adsorb As(III) as shown in Fig. 2a and b.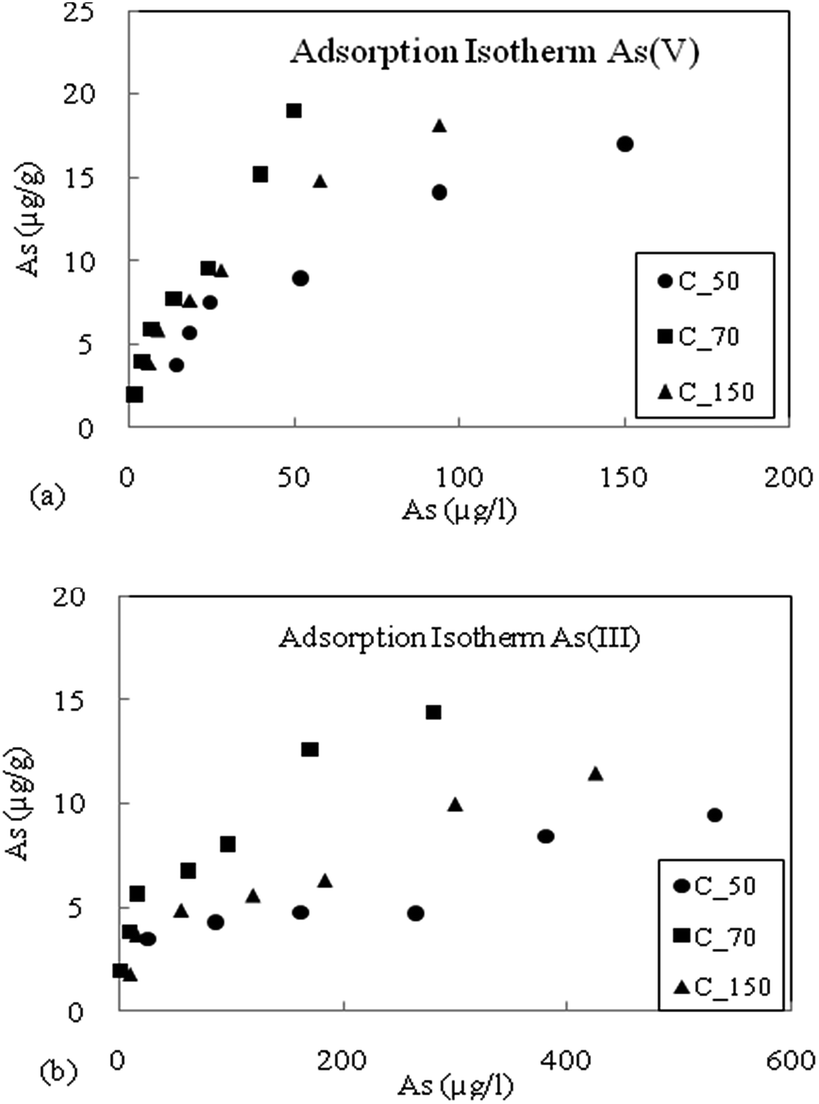 | ||
| Fig. 2 (a) Adsorption isotherm of As(V), for the three sediment samples C_50, C_70 and C_150. (b) Adsorption isotherm of As(III) for the three sediment samples C_50, C_70 and C_150. | ||
| As | Isotherm | Parameters | C_50 | C_70 | C_150 |
|---|---|---|---|---|---|
| As(III) | Freundlich | logKf |
0.2 | 0.25 | −0.02 |
| 1/n | 0.24 | 0.36 | 0.42 | ||
| r 2 | 0.66 | 0.95 | 0.89 | ||
| As(V) | Freundlich | logKf |
0.17 | 0.184 | 0.21 |
| 1/n | 0.47 | 0.626 | 0.52 | ||
| r 2 | 0.94 | 0.97 | 0.97 | ||
| As(III) | Langmuir | q max | 7.7 | 15.6 | 12.5 |
| K L | 0.01 | 0.02 | 0.01 | ||
| r 2 | 0.69 | 0.87 | 0.85 | ||
| As(V) | Langmuir | q max | 21.7 | 26.3 | 22.2 |
| K L | 0.02 | 0.03 | 0.03 | ||
| r 2 | 0.95 | 0.85 | 0.97 |
The quantification of As(III) was complicated by the fact that some of the As(III) was oxidized to As(V).17,31 In the batch adsorption study of As(III), a low concentration of aqueous As(V) was found. The experiment revealed that the oxidation might have taken place in the solid phase (sediment) oxidative sites and get adsorbed onto the surface without releasing the As(V) into the solution.
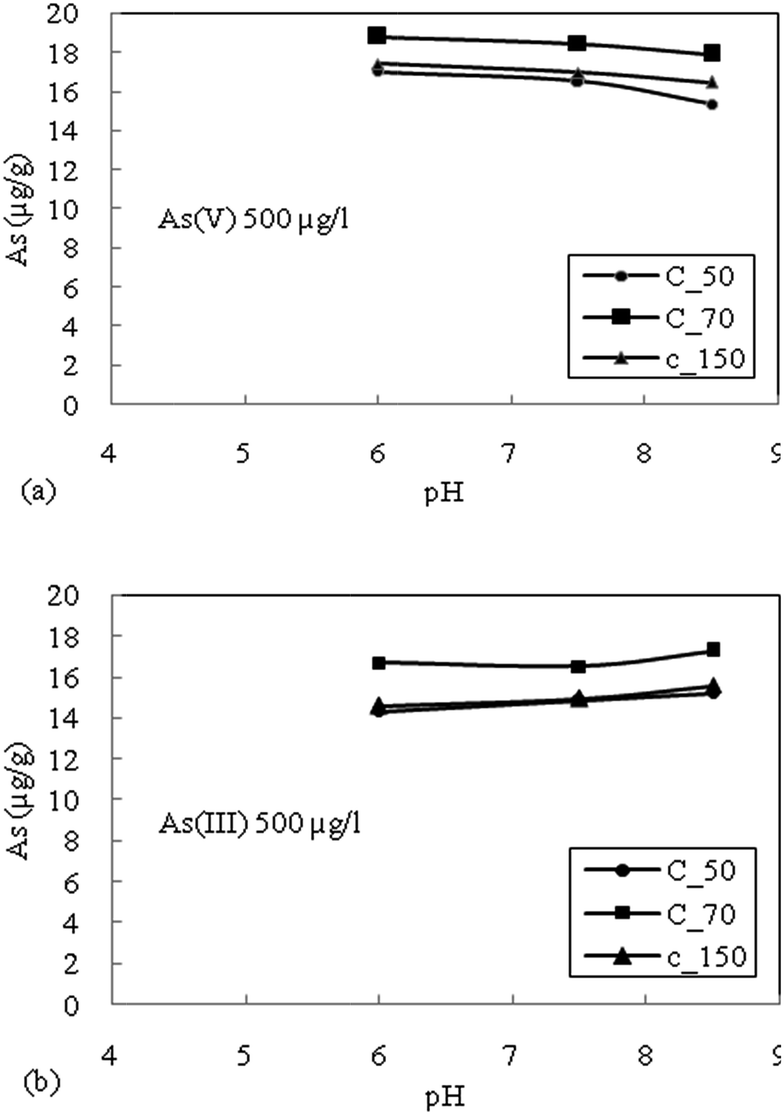 | ||
| Fig. 3 (a) Effect of pH on adsorption of As(V) with an initial concentration of 500 μg L−1 for sediment samples. (b) Effect of pH on adsorption of As(III) with an initial concentration of 500 μg L−1 for sediment samples. | ||
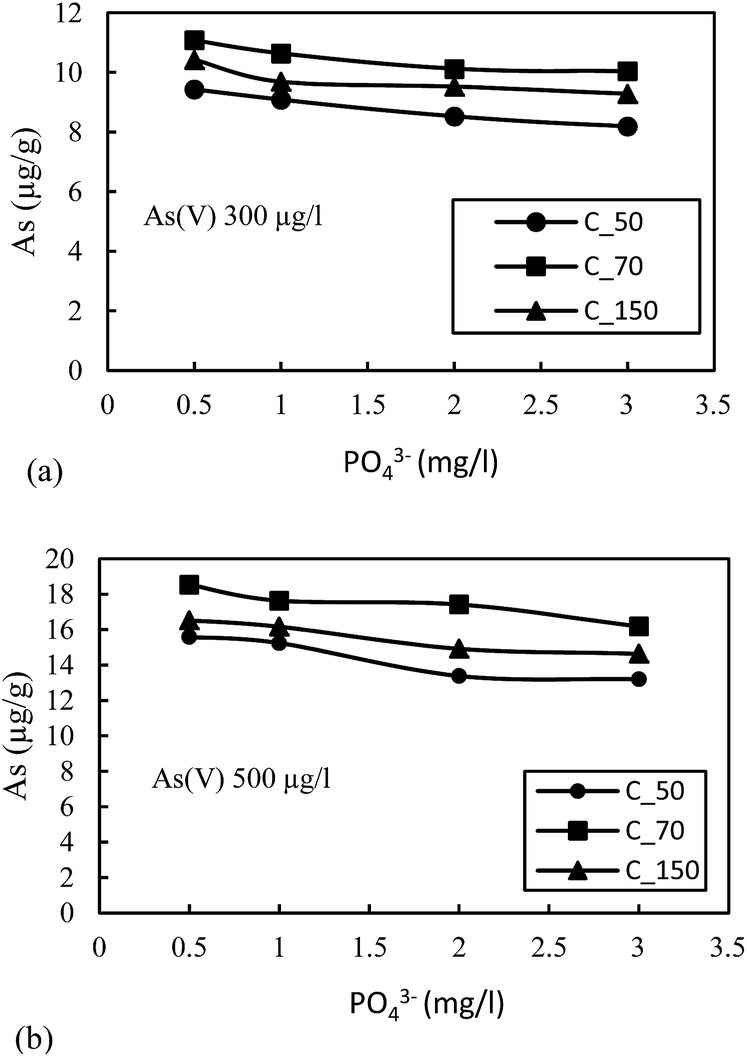 | ||
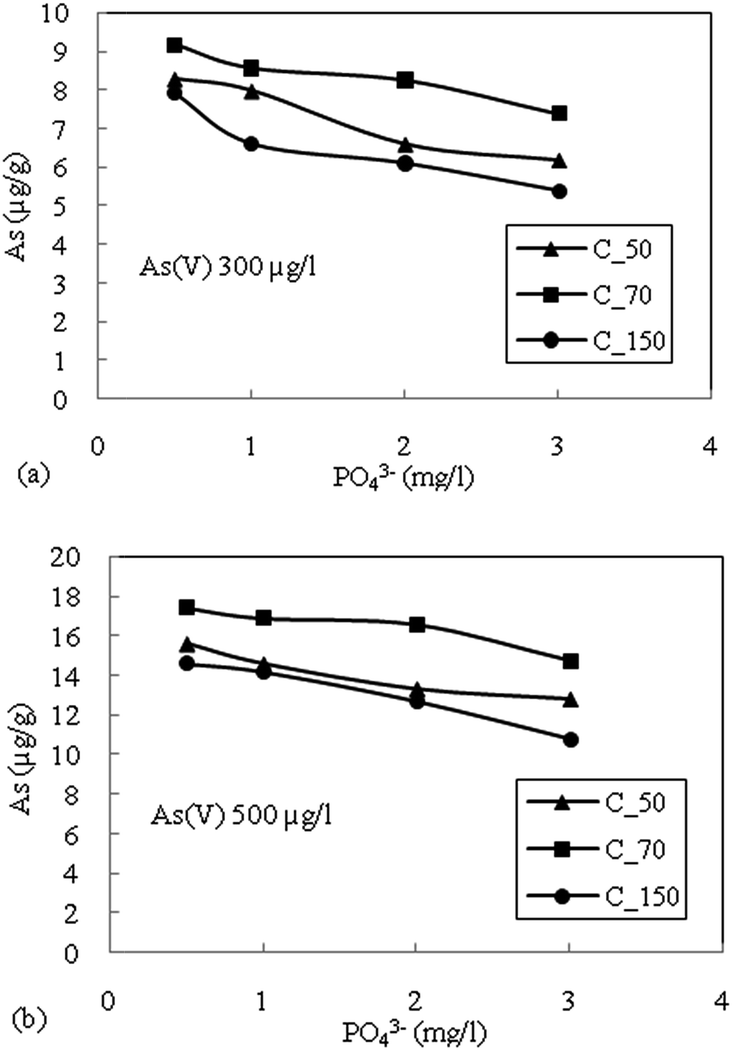
| Fig. 4 (a) Effect of phosphate (PO43−) on adsorption of As(V) with an initial concentration of 300 μg L−1. (b) Effect of phosphate (PO43−) on adsorption of As(V) with an initial concentration of 500 μg L−1 on sediment samples. | ||
The highest reduction in the adsorption capacity was noticed in the reduced sediment sample C_50 (gray color sediment). The reduction in adsorption of As(V) with 500 μg L−1 initial concentration was 16%, 11% and 13% for C_50, C_70 and C_150, respectively, and a similar trend was noticed for As(V) with an initial concentration of 300 μg L−1. The reduction of surface potential due to the formation of an inner sphere complex with Fe-oxides also reduces the adsorption of As(V) due to the electrostatic repulsion of similar charges on the sediment surface.16 Therefore, the accumulation or precipitation of PO43− on sediments might also promote the formation of negatively charged surface sites, which reduces the As(V) adsorption capacity.
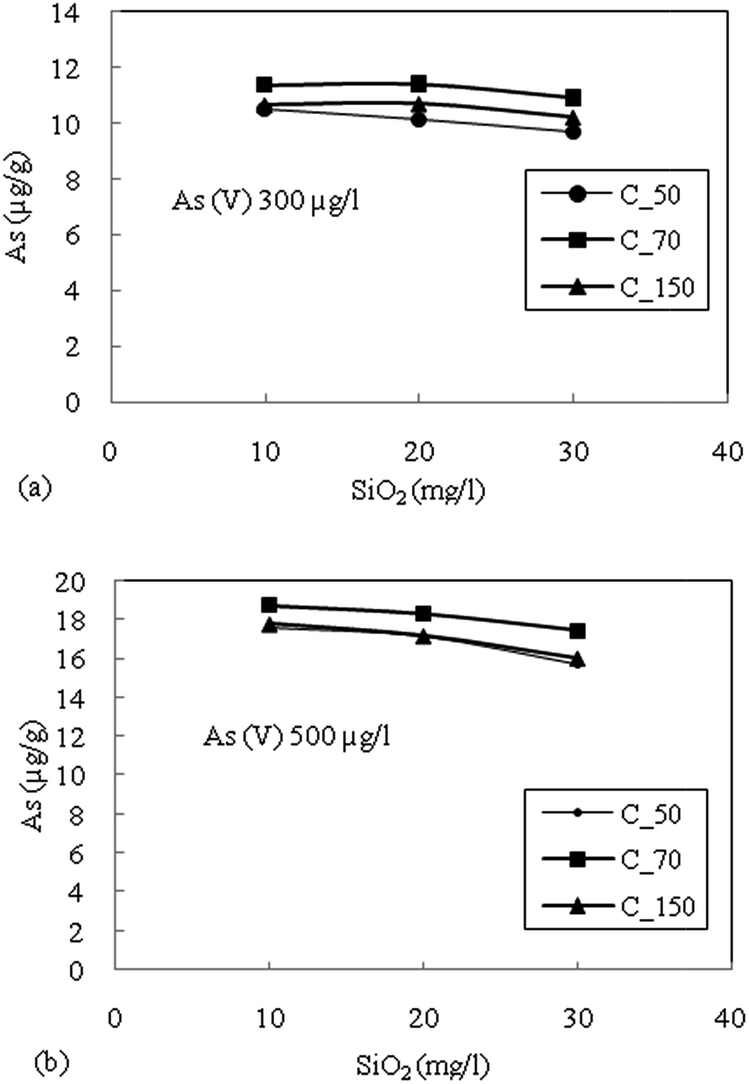 | ||
| Fig. 5 (a) Effect of silica (SiO2) on adsorption of As(V) with an initial concentration of 300 μg L−1. (b) Effect of silica (SiO2) on adsorption of As(V) with an initial concentration of 500 μg L−1 for sediment samples. | ||
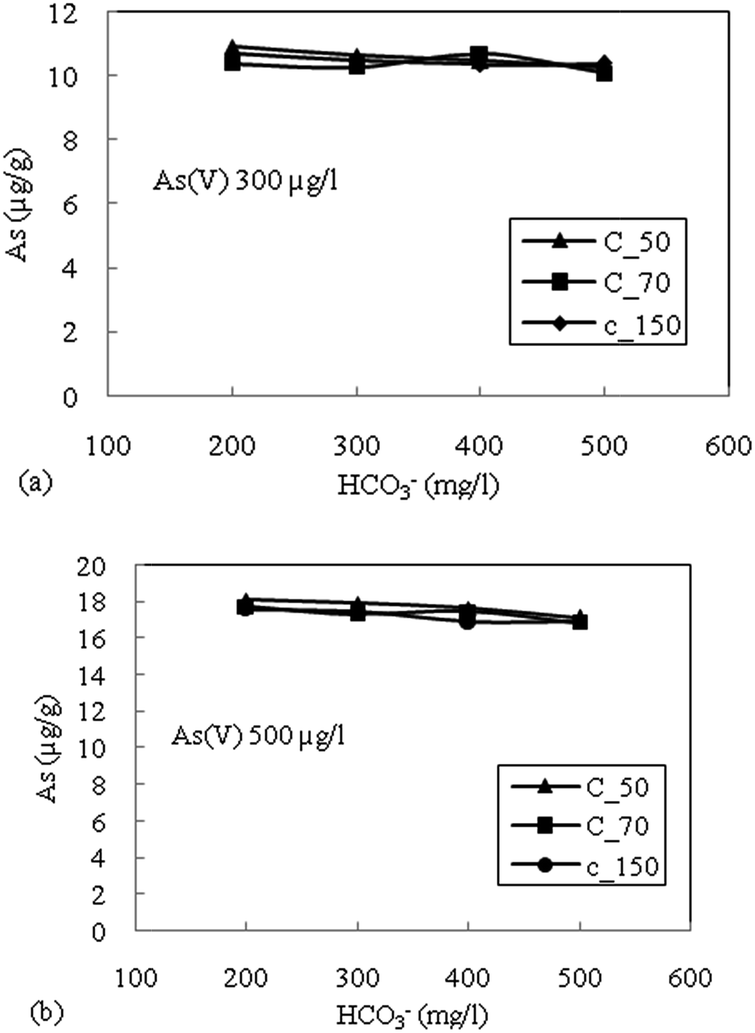 | ||
| Fig. 6 (a) Effect of bicarbonate (HCO3−) on adsorption of As(V) with an initial concentration of 300 μg L−1 for sediment samples. (b) Effect of bicarbonate (HCO3−) on adsorption of As(V) with an initial concentration of 500 μg L−1 for sediment samples. | ||
 | ||
| Fig. 7 (a) Combined effect of anions on adsorption of As(V) with an initial concentration of 300 μg L−1 for sediment samples with increasing phosphate (PO43−). (b) Combined effect of anions on adsorption of As(V) with an initial concentration of 500 μg L−1 for sediment samples with increasing phosphate (PO43−). | ||
3.2 Sorption kinetic of arsenic
The adsorption process of As was found to be rapid. It was found that about 80% of total As adsorption was observed in the initial 30 minutes.The Elovich adsorption kinetic model was found to be the best suitable kinetic model to describe the adsorption behaviour of As on the sediment. The model parameters were calculated from the linear plot of qtversus ln(t). The Elovich constants a and b are defined as initial rate constants, ‘a’ represents the rate of chemisorptions at zero coverage (μg g−1 min−1) and ‘b’ is the extent of surface coverage and the activation energy for chemisorptions (g μg−1) has been tabulated in Table 3.
| As species | Initial conc. | Parameters | C_50 | C_70 | C_150 |
|---|---|---|---|---|---|
| As(III) | 300 μg L−1 | a (μg (g min)−1) | 2.72 × 101 | 1.21 × 101 | 5.52 × 102 |
| b (g μg−1) | 1.62 | 1.12 | 1.9 | ||
| r 2 | 0.97 | 0.97 | 0.88 | ||
| As(V) | 300 μg L−1 | a (μg (g min)−1) | 7.73 × 105 | 1.59 × 105 | 7.74 × 109 |
| b (g μg−1) | 2.15 | 1.76 | 2.98 | ||
| r 2 | 0.89 | 0.94 | 0.91 |
A similar rapid initial adsorption process was reported by various researchers16,23 on natural geological materials. Goh and Lim (2004)16 analyzed the tropical soil from Singapore and reported the Elovich model parameter for As(V) as, a = 4.3 × 103 and b = 0.0366 with r2 = 0.995 and As(III) a = 4.78 × 102 and b = 0.0516 with r2 = 0.985. The values obtained from our experiments (Table 3) revealed that there is no consistency in the values of Elovich coefficients for the sediment samples. Similar inconsistencies of the Elovich coefficient have been reported by various authors and sometimes, apparently abnormal values are also found.37
The heterogeneity of sediments suggested pore diffusion or intra-particle diffusion to play a significant role along with surface adsorption. The plots of qtversus t0.5 (Fig. 8) do not pass through the origin, but the straight line observed confirms the presence of intraparticle diffusion. A deviation from the origin shows that intraparticle transport is not the only rate limiting step. The large intercept in the plot indicates the greater contribution of the surface sorption in the rate controlling step. Table 4 shows the various parameters of intraparticle diffusion models for various sediments. This reveals that the mechanism of metal ion adsorption by sediments is complex and both the surface adsorption as well as the intraparticle diffusion contributes to the rate determining step.
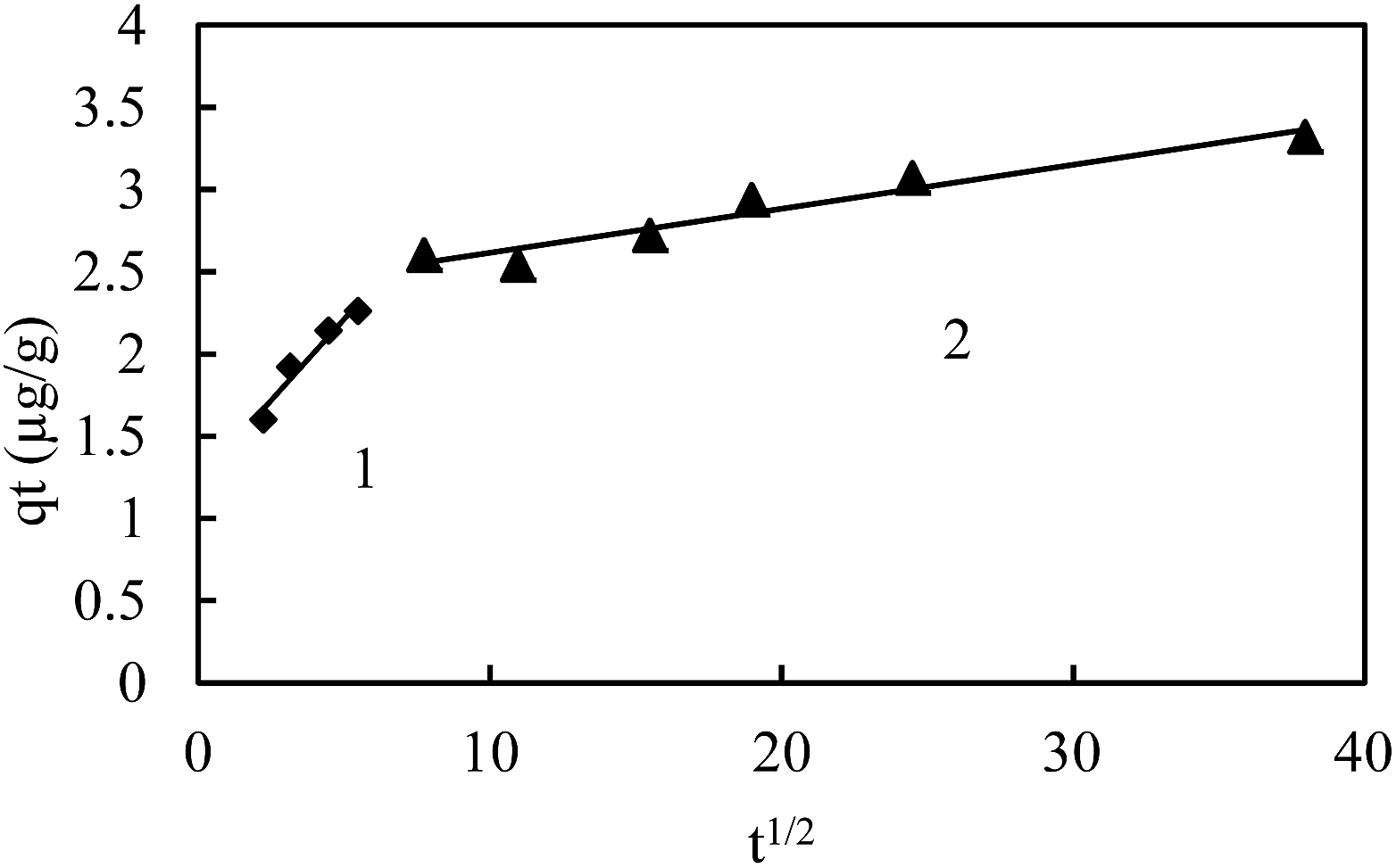 | ||
| Fig. 8 Intra-particle diffusion model for the C_50 sample with two linear segment for the initial As(III) concentration of 300 μg L−1. | ||
| As (μg L−1) | ID | K id (μg (g min0.5)−1) | C (μg g−1) | r 2 |
|---|---|---|---|---|
| As(III) 300 | C_50 | 0.096 | 3.81 | 0.86 |
| As(III) 300 | C_70 | 0.412 | 4.38 | 0.94 |
| As(III) 300 | C_150 | 0.086 | 4.86 | 0.89 |
| As(III) 500 | C_50 | 0.054 | 4.39 | 0.94 |
| As(III) 500 | C_70 | 0.06 | 8.95 | 0.79 |
| As(III) 500 | C_150 | 0.033 | 4.95 | 0.66 |
| As(V) 300 | C_50 | 0.06 | 7.85 | 0.71 |
| As(V) 300 | C_70 | 0.088 | 8.488 | 0.86 |
| As(V) 300 | C_150 | 0.045 | 8.91 | 0.63 |
| As(V) 500 | C_50 | 0.079 | 14.37 | 0.83 |
| As(V) 500 | C_70 | 0.134 | 14 | 0.87 |
| As(V) 500 | C_150 | 0.107 | 14.01 | 0.62 |
The multi linearity of the plot (Fig. 8) shows that different mechanisms of adsorption take place for sample C_50. The initial part was attributed to the surface adsorption occurring instantaneously, while the gentle slope part was attributed to intraparticle diffusion, which was slow and likely to be rate-controlled.36,37,48
3.3 Oxidation of As(III) to As(V)
The As(III) displayed varying transformation rates depending on the aquifer material. Since, sediment samples were found to adsorb both As(III) and As(V), therefore, it was necessary to quantify redox speciation of arsenic in both dissolved and solid phases. The oxidation kinetics of As(III) for the initial concentration 300 μg L−1 for three different types of sediments namely C_50, C_70 and C_150 are shown in Fig. 9a–c. The sediment sample C_50 shows that the removal of aqueous As(III) was low (Fig. 9a). The greater aqueous As(III) removal (25%) from the solution takes place via adsorption (higher solid As(III)) and lesser amount (20%) in oxidation of As(III) to As(V), which is in the form of solid As(V) in the sediment.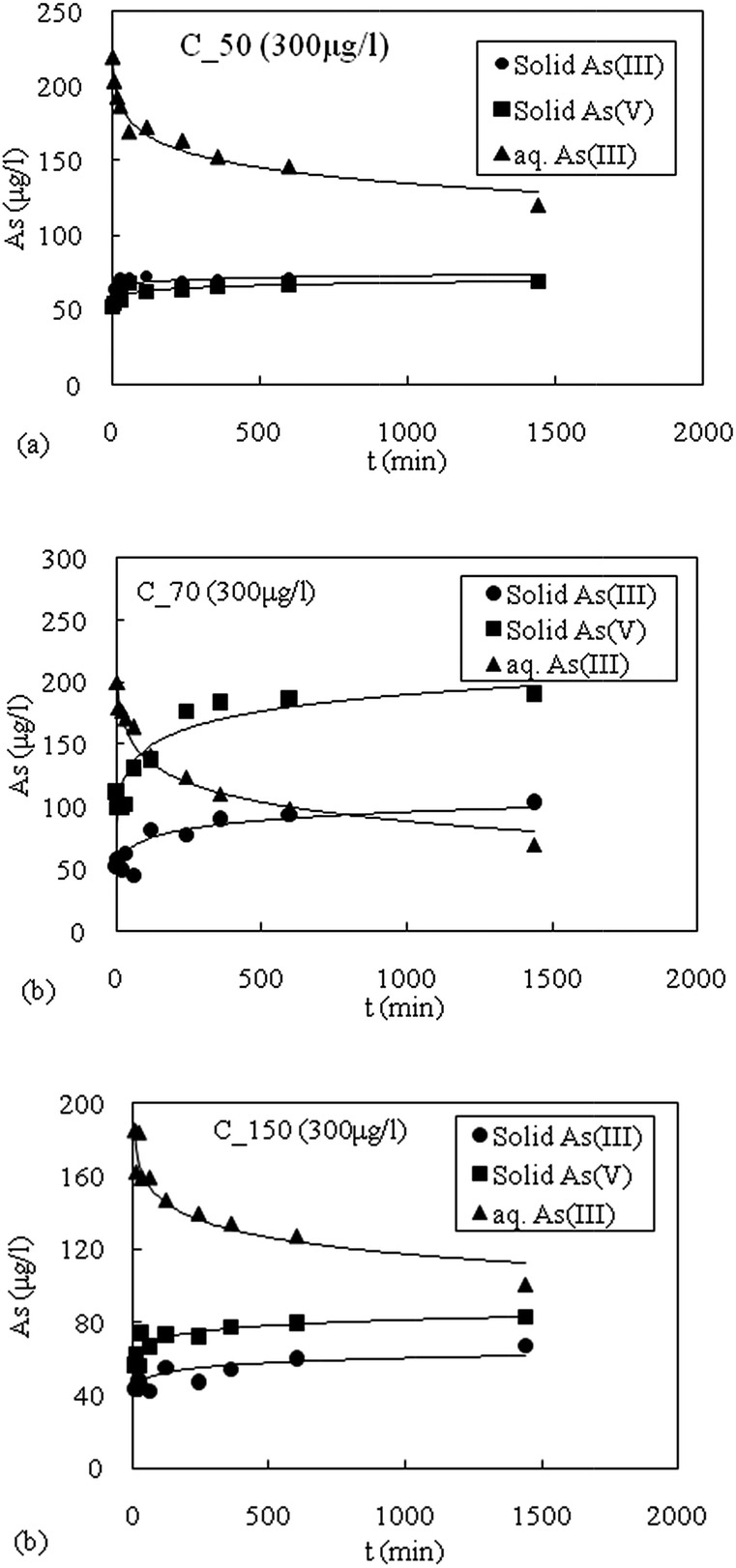 | ||
| Fig. 9 (a) Oxidation kinetics of As(III) with a concentration of 300 μg L−1 for sediment sample C_50. (b) Oxidation kinetics of As(III) with a concentration of 300 μg L−1 for sediment sample C_70. (c) Oxidation kinetics of As(III) with a concentration of 300 μg L−1 for sediment sample C_150. | ||
The oxidized sediment sample C_70 has the highest capacity to oxidize As(III) to As(V) as shown in Fig. 9b. The decrease in the aqueous As(III) concentration with time was caused by a combination of oxidation and adsorption. The sediment of this zone has a high concentration of Fe and Mn (Table 1). The Fe-oxides (amorphous and crystalline) have high affinity for As sorption.18,20,41,42 The aqueous As(III) removal from the solution was found to be highest among the sediment samples tested. It was observed that oxidation as compared to adsorption, was clearly the dominant mechanism for aqueous As(III) removal. From sample C_70 with an initial concentration of 300 μg L−1 for As(III), about 60% was removed via oxidation of As(III) to As(V) while 32% through adsorption onto the sediment samples, respectively.
The sediment sample C_150 removal mechanism of As(III) from solution is shown in Fig. 9c. Sediment sample C_150 was found to have higher aqueous As(III) removal via oxidation and a lower amount of adsorption. For 300 μg L−1 of As(III) concentration, the oxidation of As(III) to As(V) accounted for 27% whereas the adsorption of As(III) onto the aquifer material accounted for about 23%.
In all the samples, the mass balance was within ±15% of the initial As(III) concentration. This suggested that 15 minutes of 0.2 M oxalic acid extraction is an effective method for desorption of As species from aquifer materials.17,31 Oxidation of As(III) to As(V) by Mn-oxides in natural sediments has been reported.17,31,49
Thus, taping water from the zone of oxidized sediments of relatively good thickness will be able to reduce the As concentration in the groundwater. Since, As chemistry is governed by complex sets of biogeochemical processes, therefore, quantification of As desorbed from the aquifer leading to groundwater As concentration requires more advanced experimental studies and extensive field investigations to develop a model and assess the attenuation capacity of the sediments for As.
A comparison was made with the studies in Bengal Delta and Red River Delta, Vietnam where intensive studies on the geochemical modelling using a surface complexation model have been published on the Bengal Basin22 and in the Red River floodplains of Vietnam.21 Biswas et al. (2014)22 reported the role of competing ions in the As mobilization process. They observed that reductive dissolution of only Fe-oxyhydroxide cannot explain the higher As concentration in the groundwater of the Bengal Basin. The As release by reductive dissolution of Fe-oxyhydroxide might have been readsorbed in sediment materials. Biswas et al. (2014)22 accounted the reductive dissolution followed by competitive ion adsorption, mainly (PO43−) responsible for the higher concentration of As in the aquifer of the Bengal Basin. Jessen et al. (2012)21 on goethite found that As(V) adsorbs much stronger than As(III) and PO43−- and Fe(II) form the main competing species in the Red River flood plains, Vietnam. Similarly, this paper discusses the role of competing ions in the release of As in the Brahmaputra floodplains of Assam. The results also indicated that PO43− is the main competing ion for surface adsorption onto the sediments. And among the three generic sediments found in the area, more oxidized sediments have the highest capacity for As adsorption. The attenuation of arsenic occurs when As(III) gets oxidized to As(V) followed by adsorption on the sediments. Therefore, target safe aquifers in the area should incorporate the color identification of sediments as the depth of aquifer media vary to a large extent in the vertical direction.
4. Conclusions
The batch sorption studies for the attenuation of As in groundwater were performed on sediment samples viz. C_50, C_70 and C_150 representing common sedimentary deposits in the area of high As contaminated areas. All the sediment samples were able to adsorbed As(V) and As(III) at pH where groundwater was observed. The minor reduction in adsorption of As(V) with increasing pH (6–8.5) suggested electrostatic repulsion due to soil surface charge density. A small increase in the adsorption of As(III) with increasing pH could be due to the relatively less negative charge, which prohibited electrostatic repulsion between As(III) and the hydroxylic functional group. As(V) exhibits higher adsorption than As(III) species for all types of sediments. The Freundlich adsorption isotherm fits relatively better for both As(III) and As(V).The oxidized sediment sample C_70 demonstrated the highest As adsorption capacity, as this sample has a higher Fe and Mn content. PO43− was found to have the highest competition for As(V) adsorption sites on the reduced sediment (C_50) as well as the moderately oxidized sediment (C_150), and showed lesser effects on the oxidized sediment (C_70). The competition of the anions for adsorption sites was considered to generally follow the order PO43− > SiO2 > HCO3−. The combined effects of anions showed the highest reduction in the adsorption capacity of all the sediment samples. The adsorption kinetics could be best described with the Elovich kinetic model. A significant correlation was observed for the intra-particle diffusion model due to the heterogeneity of the sediments. The rate determining step was found to be controlled mainly by surface sorption and partly through intra-particle diffusion.
The transformation of As(III) to As(V) not only decreases the toxicity but also enhances As removal from drinking water and sequestration to soils and sediments. The oxidation of As(III) to As(V) experiment showed that aqueous As(III) oxidation to solid As(V) in the oxidative sites of the sediments was the dominant As removal mechanism for the oxidized sediment (C_70) and moderately oxidized sediment (C_150). The direct adsorption of aqueous As(III) on the adsorptive sites as solid As(III) was the main process of As removal for reduced sediment sample C_50.
The findings of the study can be used for exploring the integrated mitigation strategy for As contamination e.g. through targeting oxidizing sediments by placing well screens at specific depths or targeting specific aquifers with As free sediment architecture to obtain As safe drinking water for rural communities. Thus suggested that natural aquifer materials might make natural attenuation of As viable sources of drinking water.
References
- CGWB-Central Ground Water Board, 2004 “Groundwater Resources of Assam” CGWB, North East Region, Ministry of Water Resource, Guwahati.
- G. N. Breit, J. W. Whitney, A. L. Foster, K. G. Stollenwerk, J. C. Yount and A. H. Welch, Geochemical changes affecting arsenic in the sediment of the Ganges-Brahmaputra-Meghna river system, Geological Society of America Abstracts with Programs, 2003, 35(6), 271 Search PubMed.
- R. Nickson, C. Sengupta, P. Mitra, S. N. Dave, A. K. Banerjee and A. Bhattacharya, Current knowledge on the distribution of arsenic in groundwater in five states of India, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2007, 42, 1707–1718 CrossRef CAS PubMed.
- C. F. Harvey, C. H. Swartz, A. B. M. Badruzzaman, B. N. Keon, W. Yu, A. M. Ashraf, J. Jay, R. Beckie, V. Niedan, D. Brabander, P. M. Oates, K. N. Ashfaque, S. Islam, H. F. Hemond and M. F. Ahmed, Arsenic mobility and groundwater extraction in Bangladesh, Science, 2002, 298, 1602–1606 CrossRef CAS PubMed.
- C. F. Harvey, C. H. Swartz, A. B. M. Badruzzaman, B. N. Keon, W. Yu and M. A. Ali, Groundwater arsenic contamination on the Ganges delta: biogeochemistry, hydrology, human perturbations, and human suffering on a large scale, C. R. Geosci., 2005, 337, 285–296 CrossRef CAS.
- A. Biswas, B. Nath, P. Bhattacharya, D. Halder, A. K. Kundu, U. Mandal, A. Mukherjee, D. Chatterjee, C. M. Mörth and G. Jacks, Hydrogeochemical contrast between brown and grey sand aquifers in shallow depth of Bengal Basin: consequences for sustainable drinking water supply, Sci. Total Environ., 2012, 431, 402–412 CrossRef CAS PubMed.
- C. Robinson, M. von Bromssen, P. Bhattacharya, S. Haller, A. Biven, M. Hossain, G. Jacks, K. M. Ahmed, M. A. Hasan and R. Thunvik, Dynamic of arsenic adsorption in the targeted arsenic-safe aquifers in Matlab, south-eastern Bangladesh: insight from experimental studies, Appl. Geochem., 2011, 26, 624–635 CrossRef CAS.
- M. van-Bromssen, S. H. Larsson, P. Bhattacharya, M. A. Hasan, K. M. Ahmed, M. Jakariya, M. A. Sikder, O. Sracek, A. Bivén, B. Doušová, C. Patriarca, R. Thunvik and G. Jacks, Geochemical characterisation of shallow aquifer sediments of Matlab Upazila, Southeastern Bangladesh – implications for targeting low-As aquifers, J. Contam. Hydrol., 2008, 99(602), 137–149 CrossRef PubMed.
- J. M. McArthur, P. Ravenscroft, S. Safiullah and M. F. Thirlwall, Arsenic in groundwater: testing pollution mechanisms for sedimentary aquifers in Bangladesh, Water Resour. Res., 2001, 37, 109–117 CrossRef CAS.
- J. M. McArthur, B. Nath, D. M. Banerjee, R. Purohit and N. Grassineau, Palaeosol Control on Groundwater Flow and Pollutant Distribution: The Example of Arsenic, Environ. Sci. Technol., 2011, 45, 1376–1383 CrossRef CAS PubMed.
- L. Sailo and C. Mahanta, Hydro-geochemical factors affecting the mobilization of As into the groundwater of the Brahmaputra alluvial plains of Assam, Northeast India, Environ. Sci.: Processes Impacts, 2013, 15, 1775–1782 CAS.
- P. Bhattacharya, D. Chatterjee and G. Jacks, Occurrence of arsenic contaminated groundwater in alluvial aquifers from Delta plains, Eastern India: options for safe drinking water supply, Water Resource Development, 1997, 13, 79–92 CrossRef.
- D. Postma, S. Jessen, M. Hue, M. T. Duc, C. B. Koch, N. T., P. H. Viet, P. Q. Nhan and F. Larsen, Mobilization of arsenic and iron from Red River floodplains sediments, Vietnam, Geochim. Cosmochim. Acta, 2010, 74, 3367–3381 CrossRef CAS.
- S. Wang and C. N. Mulligan, Natural attenuation processes for remediation of arsenic contaminated soils and groundwater, J. Hazard. Mater., 2006, B138, 459–470 CrossRef PubMed.
- Y. Wang and Y. Deng, Environmental Geochemistry of High-Arsenic Aquifer Systems, Heavy Metal in the Environment, ed. L. K. Wang, J. P. Chen, Y. T. Hung and N. K. Shammas, Taylor and Francis group, CRC press, 2009 Search PubMed.
- K. H. Goh and T. T. Lim, Geochemistry of inorganic arsenic and selenium in a tropical soil: effect of reaction time, pH, and competitive anions on arsenic and selenium adsorption, Chemosphere, 2004, 55, 849–859 CrossRef CAS PubMed.
- K. G. Stollenwerk, G. N. Breit, A. H. Welch, J. C. Yount, J. W. Whitney, A. L. Foster, M. N. Uddin, R. K. Majumder and N. Ahmed, Arsenic attenuation by oxidized aquifer sediments in Bangladesh, Sci. Total Environ., 2007, 379, 133–150 CrossRef CAS PubMed.
- B. A. Manning and S. Goldberg, Adsorption and stability of arsenic at the clay mineral–water interface, Environ. Sci. Technol., 1997, 31, 2005–2011 CrossRef CAS.
- S. Goldberg, Competitive adsorption of arsenate and arsenite on oxides and clay minerals, Soil Sci. Soc. Am. J., 2002, 66, 413–421 CrossRef CAS.
- S. Dixit and J. G. Hering, Comparison of arsenic(V) and arsenic(III) sorption onto iron oxide minerals: implications for arsenic mobility, Environ. Sci. Technol., 2003, 37, 4182–4189 CrossRef CAS PubMed.
- S. Jessen, D. Postma, F. Larsen, P. Q. Nhan, L. Q. Hoa, P. T. K. Trang, T. V. Long, P. H. Viet and R. Jakobsen, Surface complexation modeling of groundwater arsenic mobility: results of a forced gradient experiment in a Red River flood plain aquifer, Vietnam, Geochim. Cosmochim. Acta, 2012, 98, 186–201 CrossRef CAS.
- A. Biswas, J. P. Gustafsson, H. Neidhardt, D. Halder, A. K. Kundu, D. Chattarjee, Z. Berner and P. Bhattacharya, Role of competing ions in the mobilization of arsenic in groundwater of Bengal Basin: insight from surface complexation modelling, Water Res., 2014, 55, 30–39 CrossRef CAS PubMed.
- C. A. A. Barrachina, F. B. Carbonell and J. M. Beneyto, Kinetics of arsenite sorption and desorption in Spanish soils, Commun. Soil Sci. Plant Anal., 1996, 27, 3101–3117 CrossRef.
- S. K. Acharyya, S. Lahiri, B. C. Raymahashay and A. Bhowmik, Arsenic toxicity of groundwater in parts of the Bengal basin in India and Bangladesh: the role of Quaternary stratigraphy and Holocene sea-level fluctuation, Environ. Geol., 2000, 39, 1127–1137 CrossRef CAS.
- H. M. Anawar, J. Akai, K. Komaki, H. Terao, T. Yoshioka, T. Ishizuka, S. Safiullah and K. Kato, Geochemical occurrences of arsenic in groundwater of Bangladesh: sources and mobilization processes, J. Geochem. Explor., 2003, 77, 109–131 CrossRef CAS.
- M. Berg, P. T. K. Trang, C. Stengel, J. Buschmann, P. H. Viet, N. V. Dan, W. Giger and D. Stuben, Hydrological and Sedimentary controls leading to arsenic contamination of groundwater in the Hanoi area, Vietnam: the impact of iron-arsenic ratios, peat, river bank deposits and excessive groundwater abstraction, Chem. Geol., 2008, 249, 91–112 CrossRef CAS.
- O. Sracek, P. Bhattacharya, G. Jacks, J. P. Gustafsson and M. von Brömssen, Behavior of arsenic and geochemical modeling of arsenic enrichment in aqueous environments, Appl. Geochem., 2004, 19, 169–180 CrossRef CAS.
- C. H. Swartz, N. K. Blute, B. Badruzzman, A. Ali, D. Brabander, J. Jay, J. Besancon, S. Islam, H. F. Hemond and C. Harvey, Mobility of arsenic in a Bangladesh aquifer: inferences from geochemical profiles, leaching data, and mineralogical characterization, Geochim. Cosmochim. Acta, 2004, 68, 539–4557 CrossRef.
- D. Postma, F. Larsen, T. M. H. Nguyen, T. D. Mai, H. V. Pham, Q. N. Pham and S. Jessen, Arsenic in groundwater of the Red River floodplain, Vietnam: controlling geochemical processes and reactive transport modeling, Geochim. Cosmochim. Acta, 2007, 71(562), 5054–5071 CrossRef CAS.
- M. Stachowicz, T. Hiemstra and W. H. van Riemsdijk, Multi-competitive interactions of As(III) and As(V) oxyanions with Ca2+, Mg2+, PO43−, and CO32− ions on goethite, J. Colloid Interface Sci., 2008, 320, 400–414 CrossRef CAS PubMed.
- A. Amirbahman, D. B. Kent, G. P. Curtis and J. A. Davis, Kinetics of sorption and abiotic oxidation of arsenic(III) by aquifer materials, Geochim. Cosmochim. Acta, 2006, 70, 533–547 CrossRef CAS.
- R. de Vitre, N. Belzile and A. Tessier, Speciation and adsorption of arsenic on diagenetic iron oxyhydroxides, Limnol. Oceanogr., 1991, 36(7), 1480–1485 CrossRef CAS.
- D. W. Oscarson, P. M. Huang, D. Defosse and A. Herbillion, Oxidative power of Mn(IV) and Fe(III) oxides with respect to As(III) in terrestrial and aquatic environments, Nature, 1981, 291, 50–51 CrossRef CAS.
- R. S. Oremland and J. F. Stolz, The ecology of arsenic, Science, 2003, 300, 939–944 CrossRef CAS PubMed.
- L. Sailo and C. Mahanta, Arsenic mobilization in the Brahmaputra plains of Assam: groundwater and sedimentary controls, Environ. Monit. Assess., 2014, 186, 6805–6820 CrossRef CAS PubMed.
- F. C. Wu, R. L. Tseng and R. S. Juang, Initial behavior of intraparticle diffusion model used in the description of adsorption kinetics, Chem. Eng. J., 2009, 153, 1–8 CrossRef CAS.
- S. S. Gupta and K. G. Bhattacharyya, Kinetics of adsorption of metal ions on inorganic materials: a review, Adv. Colloid Interface Sci., 2011, 162, 39–58 CrossRef PubMed.
- W. J. Weber and C. J. Morris, Kinetics of adsorption on carbon from solution, J. Sanit. Eng. Div., Am. Soc. Civ. Eng., 1963, 89, SA2 31 Search PubMed.
- J. Febrianto, A. N. Kosasih, J. Sunarso, Y. Ju, N. Indraswati and S. Ismadji, Equilibrium and kinetic studies in adsorption of heavy metals using biosorbent: a summary of recent study, J. Hazard. Mater., 2009, 162, 616–645 CrossRef CAS PubMed.
- H. Freundlich, Uber die adsorption in lusungen, J. Phys. Chem., 1906, 57, 385 CAS , (in German).
- P. L. Smedley and D. G. Kinniburgh, A review of the source, behavior and distribution of arsenic in natural waters, Appl. Geochem., 2002, 17, 517–568 CrossRef CAS.
- Y. Arai, D. L. Sparks and J. A. Davis, Arsenate adsorption mechanisms at the allophane–water interface, Environ. Sci. Technol., 2005, 39, 2537–2544 CrossRef CAS PubMed.
- X. G. Meng, S. Bang and G. P. Korfiatis, Effects of silicate, sulfate and carbonate on arsenic removal by ferric chloride, Water Res., 2000, 34, 1255–1261 CrossRef CAS.
- W. Stumm, Chemistry of the Solid-water Interface. Wiley and Sons, New York, 1992 Search PubMed.
- Y. Zheng, A. van Geen, M. Stute, R. Dhar, Z. Mo, Z. Cheng, A. Horneman, I. Gavrieli, H. J. Simpson, R. Versteeg, M. Steckler, A. Grazioli-Venier, S. Goodbred, M. Shahnewaz, M. Shamsudduha, M. A. Hoque and K. M. Ahmed, Geochemical and hydrogeological contrasts between shallow and deeper aquifers in two villages of Araihazar, Bangladesh: implications for deeper aquifers as drinking water sources, Geochim. Cosmochim. Acta, 2005, 69, 5203–5218 CrossRef CAS.
- E. Eiche, T. Neumann, M. Berg, B. Weinman, A. van Geen, S. Norra, Z. Berner, P. T. K. Trang, P. Hung Viet and D. Stüben, Geochemical processes underlying a sharp contrast in groundwater arsenic concentrations in a village on the Red River delta, Vietnam, Appl. Geochem., 2008, 23, 3143–3154 CrossRef CAS.
- C. A. J. Appelo, M. J. J. van-der-Weiden, C. Tournassat and L. Charlet, Surface complexation of ferrous iron and carbonate on ferrihydrite and the mobilization of arsenic, Environ. Sci. Technol., 2002, 36, 3096–3103 CrossRef CAS PubMed.
- Y. S. Ho, W. T. Chiu, C. S. Hsu and C. T. Huang, Sorption of lead ions from aqueous solution using tree fern as a sorbent, Hydrometallurgy, 2004, 73(1–2), 55–61 CrossRef CAS.
- X. Han, Y. L. Li and J. D. Gu, Oxidation of As(III) by MnO2 in the absence and presence of Fe(II) under acidic conditions, Geochim. Cosmochim. Acta, 2011, 75, 368–379 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5em00401b |
| This journal is © The Royal Society of Chemistry 2016 |