
Oceanic source strength of carbon monoxide on the basis of basin-wide observations in the Atlantic†
Keyhong
Park ‡
and
Tae Siek
Rhee
*
Korea Polar Research Institute, Incheon, Korea. E-mail: rhee@kopri.re.kr; Fax: +82-32-760-5399; Tel: +82-32-760-5304
First published on 6th November 2015
Abstract
We measured the carbon monoxide (CO) concentrations in the marine boundary layer and the surface waters of the Atlantic Ocean from 50°N to 50°S during the UK Atlantic Meridional Transect expedition (AMT-7) in October 1998, covering the open ocean and coastal regions. Throughout the cruise track, atmospheric CO concentrations continually decreased southwards in the northern hemisphere with sporadic low and high concentrations encountered. South of the intertropical convergence zone (ITCZ) atmospheric CO was enhanced by ∼10 ppb compared to north of the ITCZ due likely to biomass burning emissions prevailing in the tropical continents. The remainder of the southern hemisphere remains nearly invariable except for the vicinity of Rio de la Plata. The surface seawater was supersaturated everywhere along the track and its saturation anomaly oscillated up to 90, exhibiting a typical diurnal cycle. The maximal dissolved CO concentration in the diurnal cycle appeared 2–5 hours behind the local maximum of solar insolation in the open ocean and the time lag further increased in the coastal region. The global ocean flux of CO to the atmosphere was estimated to be 14 Tg(CO) a−1 within the range of 4–24 Tg(CO) a−1. This is within uncertainty almost identical to what was estimated on the basis of the basin-wide observations in the Pacific and the Atlantic, but more than ∼4 times lower than the values appeared in the Intergovernmental Panel on Climate Change (IPCC) reports.
Environmental impactPhotochemical processes in the marine boundary layer impact the fate of atmospheric trace gases including pollutants and greenhouse gases. Carbon monoxide plays a key role in atmospheric chemistry because of the predominant sink of hydroxyl radicals which oxidize pollutants and greenhouse gases emitted to the atmosphere by human activities. Due to the limited accessibility to the remote oceans, few basin-wide observations have been conducted so far, leading to large uncertainties and even bias in the oceanic CO emissions in the atmospheric CO budget. Rectifying oceanic CO emissions will help constrain the atmospheric budget and understand the behavior of atmospheric pollutants and greenhouse gases, particularly in the remote ocean where anthropogenic impacts are limited. |
1. Introduction
Carbon monoxide (CO) has little direct radiative impact, but its strong reaction with the OH radical and the production or destruction of ozone along the photochemical chain reactions in the troposphere indirectly influence the budget of other important greenhouse gases.1–3 Because of its critical role in climate change and atmospheric chemistry, numerous studies have investigated the budget of CO in the atmosphere by means of measurements in the field4,5 and space,6,7 or modeling studies.8,9The open ocean has been identified as a minor contributor for CO. The oceanic CO flux was estimated to be 6–30 Tg(CO) a−1 based on the basin-wide observations in the Pacific10 and the Atlantic.11 The oceanic flux of CO corresponds to ∼2% of all natural sources.12 Since the Pacific occupies 50% of the world ocean,13 scaling the flux from the Pacific to the world ocean may not lead to a large bias. Even the emission estimated from the Atlantic expedition turned out to be lower than that from the Pacific.11 However, the CO flux from the ocean estimated by Bates et al.10 and Stubbins et al.11 is far lower than the estimations by a series of Intergovernmental Panel on Climate Change (IPCC) reports, which basically refer to the observations in the 1970s. Since CO is pertinent to climate change as well as atmospheric chemistry, it is urgently required to resolve these large discrepancies and further to evaluate reliably the budget in the atmosphere. The main goal of this paper is to accurately estimate the emission of CO from the ocean based on the direct basin-wide observations.
A campaign was conducted onboard RRS James Clark Ross as the 7th expedition of the Atlantic Meridional Transect research program (AMT-7).14 The details of the goal and activities of AMT-7 have been described in detail elsewhere.15,16 In brief, the cruise commenced in Grimsby, U.K, on September 12, and ended in the Falkland Islands, U.K. on October 25, 1998 (Fig. 1). During the cruise, we sailed several different oceanographic regimes in the Atlantic Ocean such as the coastal region, coastal and equatorial upwelling zones, and the open ocean. Various characteristic air masses in synoptic scales were encountered based on in situ measurements of meteorological parameters and 4 day backward trajectory analyses.15 During this period, over 2300 measurements of CO in the surface water and overlying air were conducted.
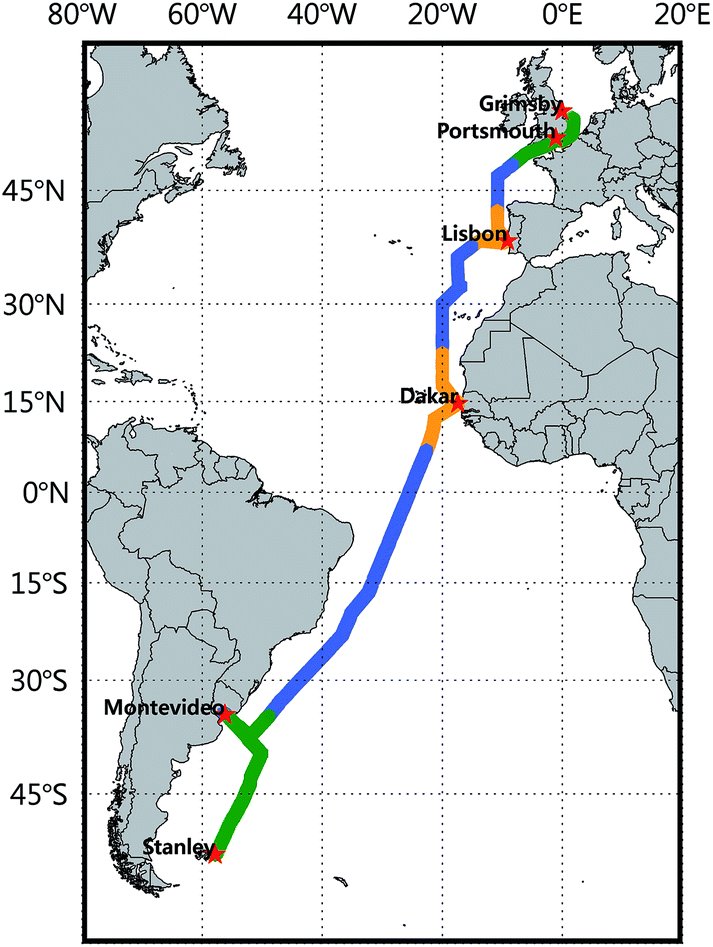 | ||
| Fig. 1 Cruise track of the 7th Atlantic Meridional Transect (AMT-7). Color code designates oceanographic provinces of the coastal region (green), the coastal upwelling (orange), and the open ocean (blue) which are defined by the chlorophyll a concentration along the track. | ||
2. Experimental
2.1. Underway measurement
Atmospheric and dissolved gas sampling methods have been described in detail elsewhere.15 In brief, the air sampling inlet was mounted on the foremast of the ship 19 m above the sea surface, located ∼53 m far from the exhaust of the ship so that the flow of contaminated air into the detector was minimized. Air samples were drawn through polyethylene inner-coated aluminum tubing (Dekabon) at a flow rate of ∼10 L min−1 using an Air-Cadet pump. No production of CO from the tubing was confirmed in the laboratory before going to sea and Bates et al.10 also reported the same results.Uncontaminated surface seawater drawn at a depth of ∼6 m was continuously supplied at 10–13 L min−1 to a Weiss-type equilibrator to sample dissolved CO molecules. The equilibrator was mounted inside the laboratory to avoid sunlight. 100 mL of the headspace air in the equilibrator was sampled every 40 or 50 minutes to flush the sample loop and to determine the dissolved CO concentration. Since the e-folding time of the equilibration of CO in the headspace is ∼30 minutes with a headspace volume of 3.7 L, 40 to 50 minutes of equilibration is enough to collect dissolved CO equilibrated in the seawater.17 To keep the analyzing system from being wet, water traps soaked in an ice bath were connected to the sampling lines before the pump.
2.2. Determination of gas concentrations
Sample gas from either the ambient air, the headspace gas in the equilibrator, or calibration gases was dried by passing it through Sicapent™ (P2O5) before entering the sample loop (Fig. 2). Laboratory tests showed that neither absorption nor production of CO from the Sicapent dryer was detected. Two different sizes of commercial sample loops (0.5 mL and 2 mL, VICI™) were installed to the 10 port VICI valve to account for the large differences in the CO concentration in seawater and air. Being critical to determine the atmospheric and dissolved concentrations of CO accurately, the volumes of sample loops were accurately calibrated and tested using standard gases in the laboratory.18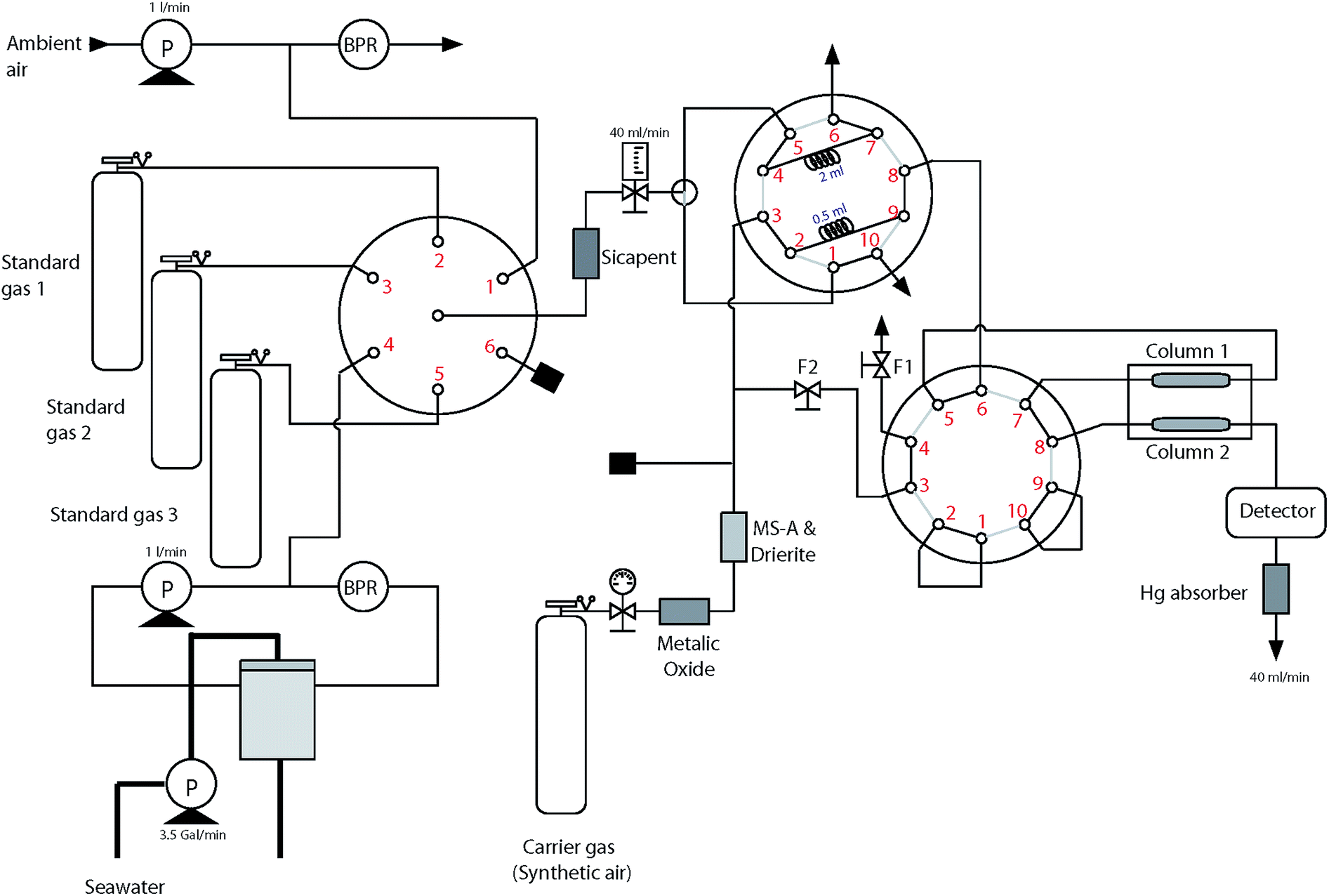 | ||
| Fig. 2 Schematic diagram of the instrumental system for analyzing atmospheric and dissolved CO. P and BPR stand for the air-cadet membrane pump and back-pressure regulator, respectively, and F1 and F2 indicate the needle valve which controls the flow rate of back-flushing the column 1. | ||
The RGA-3 gas chromatographic system consisted of a carrier gas purifying train, two CO separating columns, and a hot mercuric oxide reduction detector. The carrier gas purifying train consisted of metallic oxide, molecular sieve A, and drierite. By switching the position of the VICI valve, the CO-free synthetic air carried the sample to the RGA-3 gas chromatograph at 40 mL min−1. The two columns were made of Unibeads® 1S and Molecular Sieve 5A, and they were coiled on the aluminum mandrel in the oven, whose temperature remained constant at 130°C. When entering the detector, CO molecules reacted with mercuric oxide (HgO) on the heated bed producing mercury vapor,19 which absorbs at the specific ultraviolet (UV) line of 254 nm. The analyzing system was calibrated using three standard gases which were prepared by mixing CO molecules in synthetic air and scaled to NOAA primary standards20 (see ESI†).
2.3. Data treatment and reduction
Peak areas of the chromatograms were stored in a storage module during the campaign. As samples from the air and the equilibrator and standard gases were alternately analyzed, mole fractions of the samples were calculated using the linear regression curve obtained from the measurement of standards which bracketed the samples (see ESI for the linearity of the calibration gases†). Since gas solubility in seawater depends on the temperature and salinity, the mole fraction of dissolved gas which was equilibrated at the equilibrator temperature was corrected to the in situ seawater temperature using the ratio of the solubilities at two temperatures.15 We used the empirical solubility equations of Wiesenburg and Guinasso.21Abnormally high concentrations in the atmosphere were often encountered when the ship was stopped on station or near port. In particular, the influence of exhaust from the ship was clearly observed by the dramatic increase of the CO mole fraction. These data were visually eliminated.
3. Results and discussion
3.1. CO in the marine boundary layer
Along the cruise track, the mole fractions in the northern hemisphere (131 (±1, 1se)) were greater than those in the southern hemisphere by 46 ppb on average, with the transition occurring in the intertropical convergence zone (ITCZ) located at 8–12°N (Table 1). The ITCZ was not so distinctive in the spatial distribution of CO mole fractions (Fig. 3(a)) apart from the sharp decrease in the CH4 mole fraction and from the sudden change in the wind direction from north to south.15| Air masses | Latitude | CO | ||
|---|---|---|---|---|
| Mean | se | n | ||
| a se denotes the standard error (or standard deviation of the mean = sd/√N). | ||||
| Arctic polar | 53°N–50.2°N | 178 | 3 | 33 |
| Polar front | 50.2°N–49°N | 143 | 1 | 25 |
| N. Atlantic subtropical | 49°N–39°N | 154 | 1 | 86 |
| Subtropical front | 39°N–36.5°N | 135 | 3 | 67 |
| N. Atlantic subtropical | 36.5°N–24°N | 139 | 1 | 150 |
| Subtropical high | 24°N–21°N | 103 | 2 | 23 |
| N.W. African tropical | 21°N–12°N | 97 | 1 | 128 |
| ITCZ | 12°N–8°N | 105 | 1 | 33 |
| Eq. Atlantic tropical | 8°N–3.5°S | 108 | 1 | 102 |
| Subtropical high | 3.5°S–5.5°S | 75 | 1 | 19 |
| S. Atlantic tropical | 5.5°S–22°S | 78 | 0.4 | 139 |
| Subtropical high | 22°S–23°S | 83 | 1 | 8 |
| S. Atlantic subtropical | 23°S–33°S | 79 | 1 | 113 |
| Subtropical front | 33°S–34°S | 71 | 0.4 | 9 |
| S. American continent | 34°S–41°S | 90 | 1 | 150 |
| Polar front | 41°S–43°S | 79 | 4 | 17 |
| Antarctic polar | 43°S–53°S | 70 | 1 | 83 |
| N. Hemisphere | 131 | 1 | 529 | |
| S. Hemisphere | 85 | 1 | 656 | |
| Global | 106 | 1 | 1185 | |
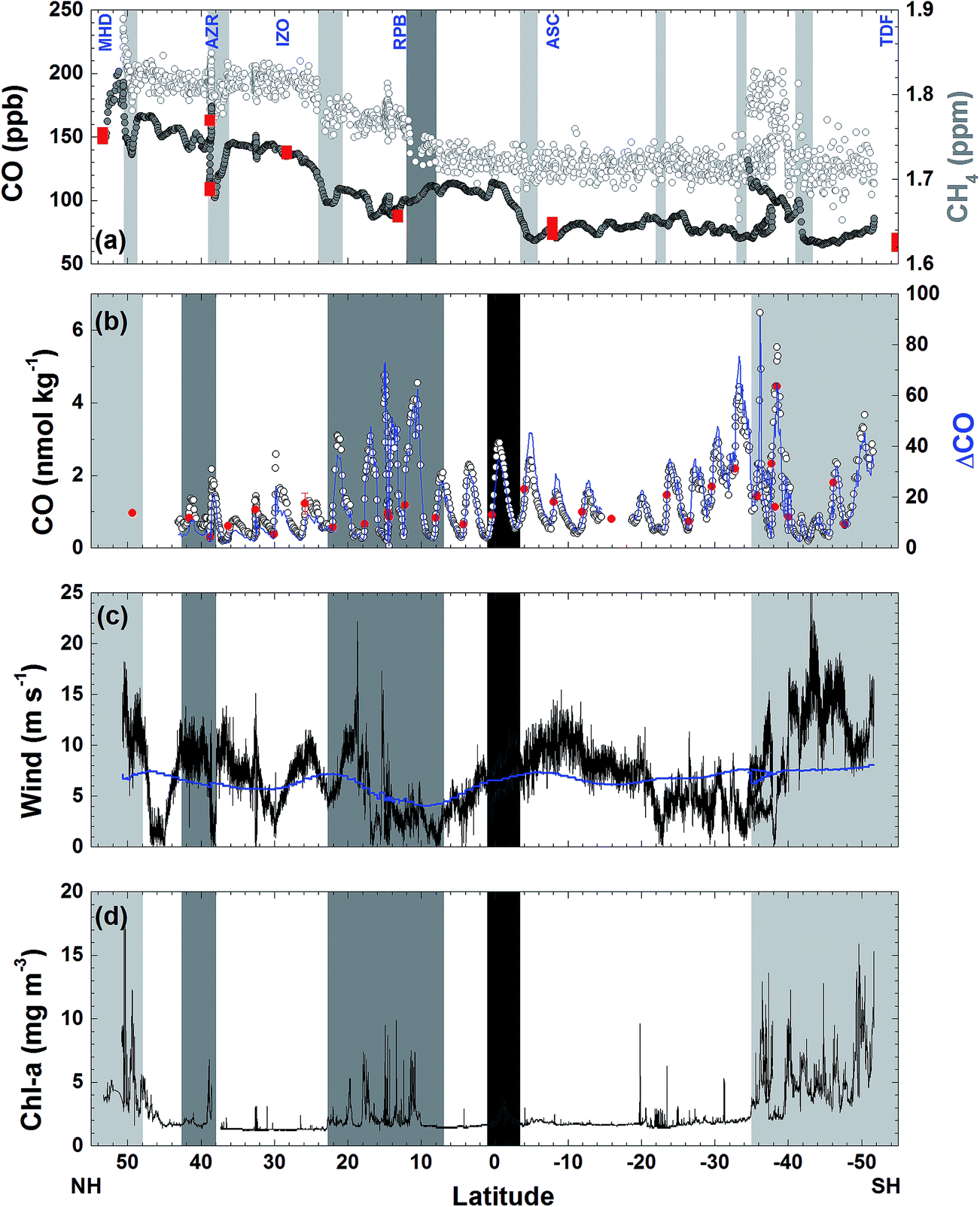 | ||
| Fig. 3 Atmospheric (a) and dissolved (b) CO concentrations, shipboard and climatological wind speeds (c), and chlorophyll a concentration (d) along the AMT-7 cruise track. In panel (a) the atmospheric CO and CH4 concentrations measured during the same period15 are shown with gray and open circles, respectively, for comparison, and red squares indicate the CO concentration at the NOAA/ESRL stations noted on the top of the panel. Light and dark gray bars indicate the frontal region of air masses and the intertropical convergence zone (ITCZ), respectively (see Table 1 for different air masses). In panel (b) open circles designate dissolved CO concentration measured underway, blue solid lines indicate running mean ΔCO, and red circles for discrete measurements of dissolved CO at the surface waters. In panel (c) black line indicates the shipboard wind speed and blue one for the climatological wind speed in COADS/CMR5. In panels (b)–(d), light gray, gray, and dark gray shades indicate coastal, coastal upwelling, and equatorial upwelling regions, respectively, and the remainder for the open ocean. | ||
The highest mole fraction reaching 229 ppb of CO was detected near the Strait of Dover, which can be attributed to anthropogenic sources coming from populated areas in the U.K. as the CH4 and N2O were high at the same period.15 As indicated in Fig. 3(a), anthropogenic point sources were also clearly detected by a sharp increase of CO mole fractions near Lisbon (38.8°N), the Madeira Islands (32.5°N), Dakar (14.6°N), and around the mouth of Rio de la Plata (34.5°S) which is encompassed by high population areas such as Buenos Aires and Montevideo.
On the other hand, several episodic decreases in the mole fractions of CO were observed at 50°N, 38°N, and 22°N, in which low CH4 concentrations were also detected parallel to the sudden change in the wind direction (Fig. 3(a)).15 These dips in the atmospheric CO are likely to result from subsidence of the air masses from the clean free troposphere where either CO is predominantly destructed by the photochemical oxidation or an air mass with low CO could be transported from the Southern Hemisphere.15,22 This air mass variation in the synoptic scale is confirmed by the coherent variation of CO observed by NOAA/ESRL network stations in the time window of ∼3 days with respect to the in situ measurements onboard (Fig. 3(a)). In particular, a rapid increase of CO concentration by 54 ppb at the Azores station (AZR) within 6 days coincides with the onboard measurements. The zonal mean mole fractions of CO between the episodic events gradually decreased southward to the ITCZ in the NH (Table 1).
In the southern hemisphere, mole fractions of CO varied little (7 ppb) throughout the cruise track except for the strong enhancement near the Rio de la Plata, indicating the fairly homogeneous distribution of CO in the southern hemisphere over the Atlantic. Exception is south to the ITCZ where CO mole fractions gradually increased up to 150 ppb until the equator. This is due likely to strong emission of CO from biomass burning taking place in the south to the ITCZ which did not disclose in the atmospheric CH4 and N2O measured at the same time.15 Biomass burning produces CH4 and N2O molecules, but the emitted contents would be 11% and 0.1% of the amount of CO, respectively.15,23 Assuming that the zonal mean values of 3.5–34°S are the background concentrations in the southern hemisphere and the difference of the mean CO mole fractions between 8°N and 2°S and 3.5–34°S is due to biomass burning, and the additions of CH4 and N2O due to biomass burning are merely around 40% and 4% of the zonal difference between 8°N and 2°S and 3.5°S–34°S (Table 1).
3.2. Dissolved CO in the surface mixed layer
Concentrations of the dissolved CO and saturation anomaly (ΔCO) along the transect of the cruise track together with variations of the absorption of fluorescence (which indicates the amount of chlorophyll a (Chl-a)) are shown in Fig. 3. ΔCO is defined as the departure of the saturated mole fraction of a gas in seawater from the mole fraction in the overlying air, so that it is one lower than the saturation ratio, SR, which is the ratio of the dry mole fraction of a gas in seawater (xCO) to that in the overlying air (yCO):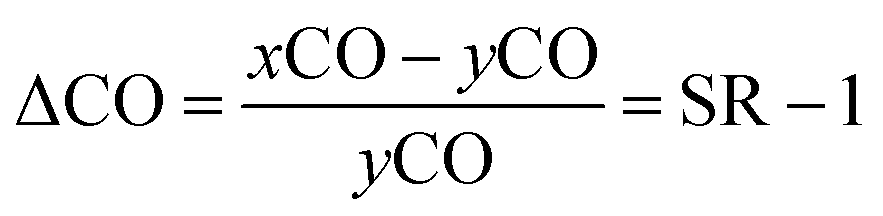 | (1) |
The mean CO concentration throughout the transect was 1.3 (±1.0, 1σ) nmol kg−1 for 978 data points, ranging from 0.19 to 6.5 nmol kg−1. These values are slightly greater than those which Bates et al.10 reported from observations in the Pacific Ocean (which ranged from 0.1 to 5.8 nM and their mean was probably between 1.1 and 1.4 nM based on the text and Table 2 in their paper). In this work, the dissolved CO was supersaturated up to 91 times (Fig. 3(b)), with the area-weighted mean value of the ΔCO being 16, which is the same as the value Bates et al.10 reported, but approximately 1.8 times as large as that Conrad et al.24 estimated based on measurements in the Atlantic about 20 years ago (ΔCO = 9) and ∼1.5 times larger on average than the value Stubbins et al.11 observed in April, 2000 (ΔCO = 11). Nevertheless, all these basin-wide observations appear to be the same magnitude of saturation anomaly within observational uncertainty.
Dissolved CO oscillated in a diurnal pattern throughout the cruise track, and the amplitude of the sinusoidal curves was larger in the region of high Chl-a content such as the coastal region and coastal upwelling area (Fig. 3(b) and 4). The largest amplitude of the diurnal cycle was 4.5 nmol kg−1 in the west African upwelling region (30 September) and the smallest was ∼0.58 nmol kg−1 near 36°N (23 September) and 43°S (23 October) in the open ocean. The high CO concentrations caused by African coastal upwelling and equatorial upwelling have been reported by Conrad et al.24 These authors mentioned that the saturation ratio had a linear relationship with the chlorophyll-a content. However, we did not find such a good correlation between photosynthetically available-radiation-normalized difference of maximal and minimal CO (ΔCO/PAR) and Chl-a (Fig. 5(a)). Although excluding the data from the coastal region gives the tendency of a linear relationship (R2 = 0.4), ΔCO/PAR appears sensitive to small change in Chl-a. This suggests that parameters other than Chl-a should also play a role in governing the amplitude of the diurnal cycle of CO. The major source of CO in the ocean is the photodegradation of chromophoric (or colored) dissolved organic matter (CDOM) by ultraviolet light.25–27 Major sinks are microbial consumption,28 the dilution by mixed layer thickening, and the emission to the atmosphere by gas exchange.29–31 Thus, the large amplitude in the high Chl-a regions could be associated with the relatively large amount of CDOM and high density of microorganisms as well. Setser et al.32 also observed a good relationship between the daily amplitude of dissolved CO and in vivo fluorescence off Baja California in the east Pacific. In spite of the high chlorophyll content, the low amplitude between 40°S and 50°S is probably related to both high wind speed, which can drive the dilution of the dissolved CO content by deepening the mixed layer and the emission by the intensive gas exchange, and the overcast clouds, which prohibited UV light from reaching the ocean surface (Fig. 3 and 4). Indeed, ΔCO/PAR in the coastal region shows a tendency of reverse relationship with wind speed (R2 = 0.4), which becomes lesser in the coastal upwelling region (R2 = 0.3) and the open ocean (R2 = 0.1) (Fig. 5(b)).
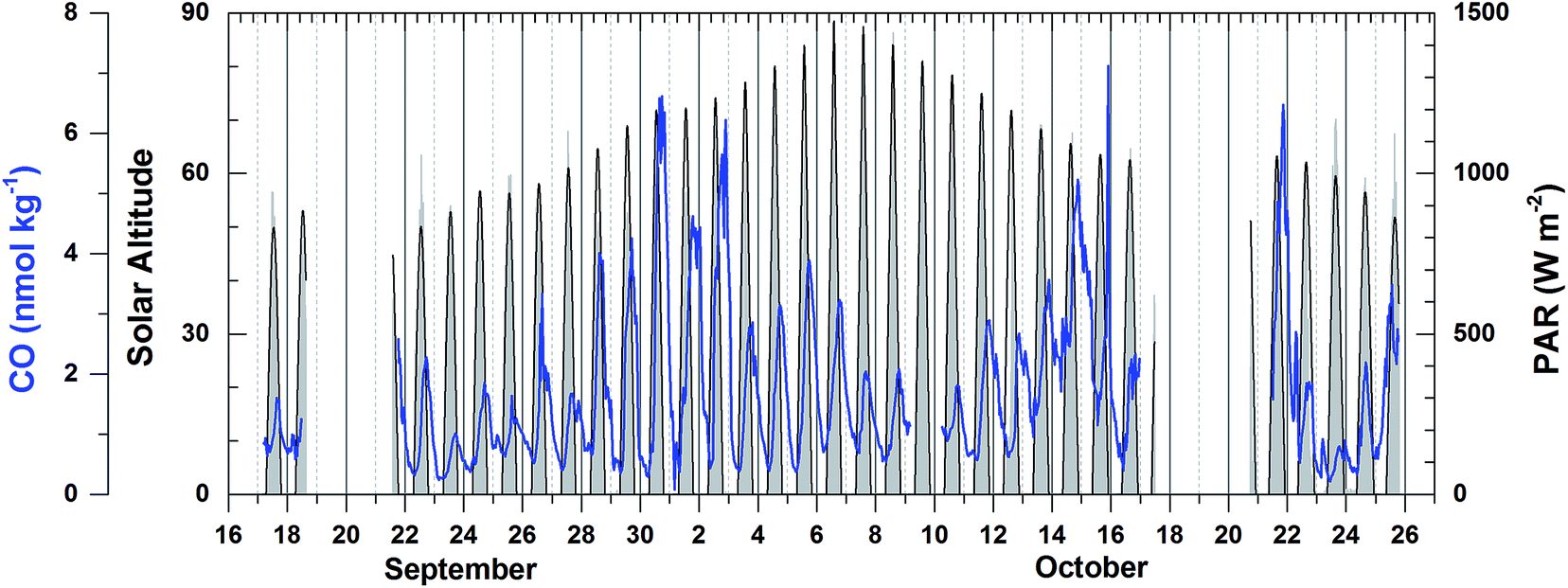 | ||
| Fig. 4 Dissolved CO concentration, solar altitude, and photosynthetically available radiation (PAR) along the cruise track in the frame of Greenwich Mean Time (GMT). | ||
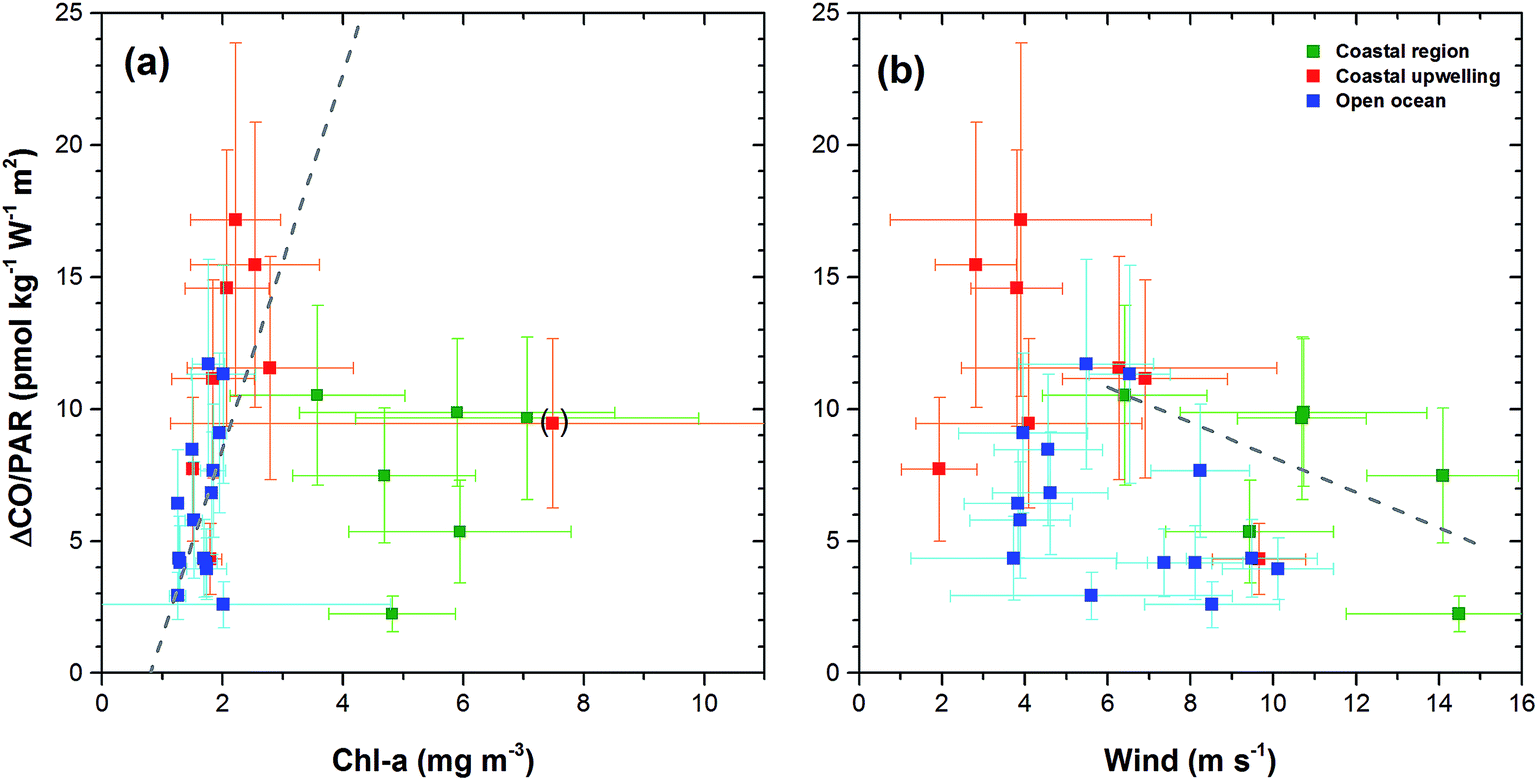 | ||
| Fig. 5 Photosynthetically available-radiation-normalized difference between maximal and minimal CO values (ΔCO/PAR) calculated from the daily harmonic fitting function of eqn (3) in text against daily mean chlorophyll a (a) and wind speed (b). Linear regression fitting for the data of the open ocean and the coastal upwelling region in (a) is shown with gray broken lines and that for the coastal region is shown in (b). | ||
A smooth fit curve of measured dissolved CO is compared with the solar zenith angle to analyze the time lag between the diurnal cycle of dissolved CO and solar radiation. To derive a smooth fit curve of the daily dissolved CO cycle, each measurement is normalized by daily mean 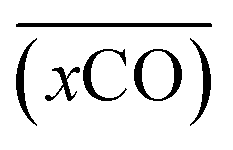 and standard deviation
and standard deviation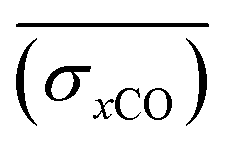 :
:
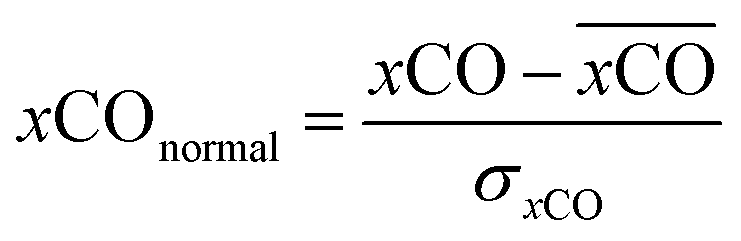 | (2) |
A third harmonic function was fitted to the normalized dissolved CO:
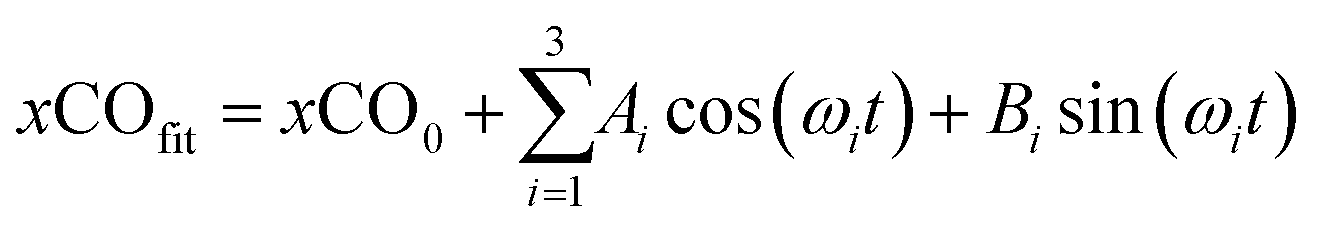 | (3) |
Then, the time lag between the minimal solar zenith angle and the maximal value of xCOfit was determined. This time lag depends on the relative strengths of the sinks and sources of dissolved CO such that it is smaller when the sink strengths are superior to the source or vice versa. As shown in Fig. 6, the time lag is on average shorter in the open ocean (Δt = 3.4 (±0.9) h) than that in the coastal upwelling (Δt = 4.0 (±1.6) h) and the coastal regions (Δt = 7.64 (±7.0) h). This suggests that the sink strengths of CO in the open ocean are relatively stronger than sources. As mentioned earlier, the major sink of dissolved CO is microbial oxidation and the major source is photochemical degradation of CDOM.27 Microbes are ubiquitous and their density would vary depending on the vital substance while photoproduction depends on the amount of CDOM. In general the CDOM content becomes larger approaching the continental coast since most of them originate from the continent. Our results abide by such common sense that the CDOM content is rather small in the open ocean.
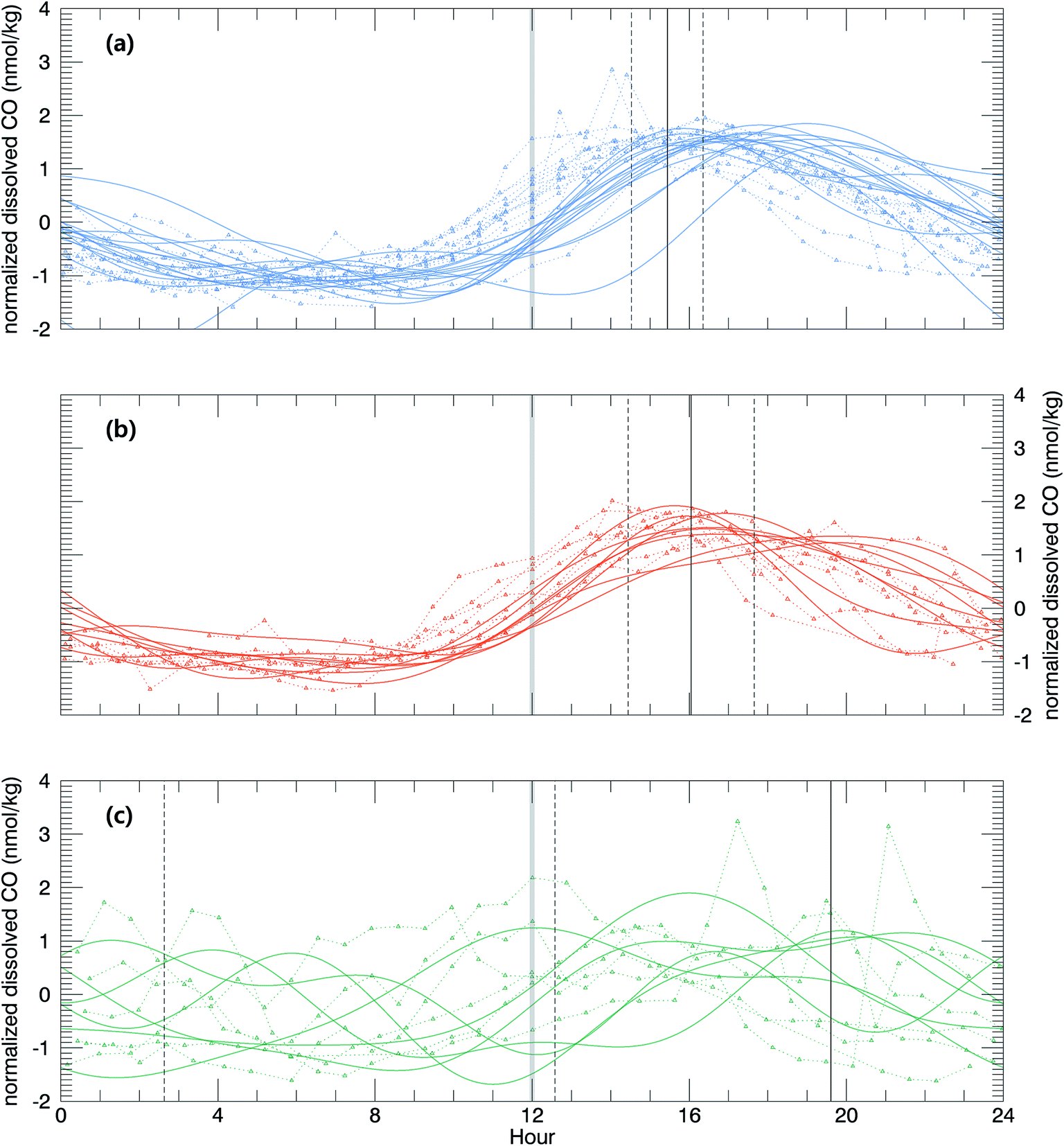 | ||
| Fig. 6 Diurnal variation of normalized dissolved CO in the open ocean (a), the coastal upwelling zone (b), and the coastal region (c). For the sake of comparison, the observation time was adjusted such that the minimal solar zenith angle (and thus maximal solar altitude) was always at noon. Vertical black solid and dotted lines indicate mean high CO time and 1 standard deviation, respectively, and gray bar for noon. | ||
3.3. CO emission from the ocean
The CO flux from the ocean was calculated following the procedure described in detail by Rhee et al.15 In brief, flux density was determined by multiplying gas transfer velocity and difference of CO concentrations between the surface seawater and overlying atmosphere. Since the concentrations are interchangeable with partial pressure by Henry's law, the flux density can be given by:| j = k(Cw − LCa) = kK0paΔCO | (4) |
We chose parameterizations for gas transfer velocity k reported by Liss and Merlivat33 (LM86), Wanninkhof34 (W92), Erickson35 (E93), and Nightingale et al.36 (N00) as they have been widely used to estimate gas flux. In general, k is a function of wind as a proxy of momentum flux across the sea surface. The shipboard wind speeds were measured along the cruise track using an anemometer which was mounted on the foremast at ∼20 m above sea level. These apparent wind speeds were corrected to true wind speeds according to Smith et al.37 and then adjusted to a 10 m height assuming a logarithmic profile of the wind speed.38 For the sake of comparison, the Compressed Marine Reports – Product 5 of the Comprehensive Ocean-Atmosphere Data Set (COADS/CMR-5)39 was chosen to obtain climatological wind speeds, sea surface temperature and salinity, surface air temperature, and relative humidity. These are mean values of 45 years of climatological data covering 1945 through 1989, and were binned as 1° × 1° monthly means. Since the wind speeds in COADS/CMR-5 are averages at the height of the anemometer on the shipboard (approximately 19.5 m), it was assumed that they are representative of 20 m above sea level. The COADS/CMR-5 wind speed did not reproduce the variability of our shipboard wind speed (Fig. 3(c)). Nonetheless the mean wind speeds are similar to each other; the mean values from shipboard observation and COADS/CMR-5 are 6.8 (±3.7, 1σ) m s−1 and 6.4 (±0.9, 1σ) m s−1, respectively.
Another thing to consider when applying those parameterizations for the gas transfer velocity is the normalization of the Schmidt number, Sc. The Schmidt number is defined as the ratio of the kinematic viscosity of the solution to the diffusion coefficient of the dissolved gas. The LM86 and W92 models are based on the gas transfer velocities of CO2 at 20 °C for fresh water and for seawater at 35‰, respectively. The E93 model was basically based on radon (222Rn) experiments during GEOSECS, but it did not specify the temperature or salinity. Therefore, it is necessary to make a conversion of these models, so they may be applied to other gases or different conditions. This has been done by normalizing Sc with a specific value of the exponent (−2/3 or −1/2) because different thermodynamic conditions affect the diffusion coefficient of the dissolving gas and the kinematic viscosity of the solution, and different gases have different diffusion coefficients under the same physicochemical conditions. The diffusion coefficient of CO was calculated using the empirical equations from Wise and Houghton.40 The diffusion coefficient was reduced by 6% to correct for the salt effect,41 since all empirical equations were determined from experimental results in fresh water. The kinematic viscosity of the solution is defined as the ratio of the dynamic viscosity to the density of the solution. The dynamic viscosity of seawater was calculated by Korson et al.,42 correcting for the salt effect using the relative viscosity of seawater given by Millero.43 The density of seawater was calculated by Millero and Poisson.44 Zafiriou et al.'s45 parameterization of the Sc of CO does not count on salinity but temperature only, which prevents us from attempting the use of this simple parameterization.
The CO density flux was then calculated by feeding gas transfer velocity, solubility, and partial pressures measured underway from both the surface seawater and the overlying atmosphere in eqn (4). The ocean was divided into three typical oceanographic regions – the open ocean, coastal region, and coastal upwelling zone – and determined mean density fluxes in the individual regions. Assuming that these mean values of the flux densities are representative for each region, the regional and global fluxes were evaluated in Table 2. The open ocean appears to be a dominant source region for CO due to the area weighting factor and small variation of the density flux in different regions. This agrees with Bates et al.'s10 observation in the Pacific.
| Area (×106 km2) | ΔCOb | Shipboard | Climatology | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Windb (m s−1) | Fluxa (Tg(CO) a−1) | Mean flux fraction (%) | Windb (m s−1) | Fluxa (Tg(CO) a−1) | Mean flux fraction (%) | |||||||||||
| LM86 | W92 | E93 | N00 | Mean | LM86 | W92 | E93 | N00 | Mean | |||||||
| a LM86, W92, E93, and N00 indicate k parameterizations from Liss and Merlivat,33 Wanninkhof,34 Erickson,35 and Nightingale et al.,36 respectively. b Global ΔCO and wind are area-weighted mean values. | ||||||||||||||||
| Coastal region | 48.4 | 19.0 | 11 | 2.4 | 4.3 | 3.9 | 3.3 | 3.5 | 25 | 7.5 | 1.6 | 3.2 | 1.7 | 2.1 | 2.2 | 15 |
| Coastal upwelling | 0.4 | 18.4 | 4.9 | 0.006 | 0.01 | 0.02 | 0.009 | 0.01 | 0.07 | 5.4 | 0.007 | 0.02 | 0.01 | 0.01 | 0.01 | 0.08 |
| Open ocean | 313.3 | 15.7 | 6.3 | 7.2 | 12.5 | 12.3 | 10.4 | 10.6 | 75 | 6.5 | 8.6 | 16.8 | 10.7 | 11.2 | 11.8 | 85 |
| Global | 362 | 16.2b | 6.8b | 9.6 | 16.8 | 16.2 | 13.7 | 14.1 | 6.6b | 10.2 | 20.0 | 12.5 | 13.3 | 14.0 | ||
The global flux of CO depends on the model of gas transfer velocities and the wind speed applied since it was assumed that the saturation anomalies and the partial pressures were invariable temporally. Taking all the fluxes calculated by the different models and winds into account, we estimate a global oceanic CO flux of 14 Tg(CO) a−1, ranging from 9.6 to 20 Tg(CO) a−1. Regardless of the wind speed taken from either shipboard measurements or climatological data, the mean values and ranges are astonishingly similar to each other probably due to diurnal variation of the dissolved CO concentration and its order of magnitude large saturation anomaly. Adopting the method of Nevison et al.46 for evaluating the uncertainty of a flux, we assumed approximately a 70% uncertainty for the mean flux. The range of uncertainty with the most likely value is listed in Table 3 together with the previous estimates in the literature. Our estimate is very similar to that reported by Bates et al.10 and Stubbins et al.11 and is within the range reported by Conrad et al.24 Among the compiled data in Table 3, Bates et al.,10 Conrad et al.,24 and Stubbins et al.11 estimated the flux based on the basin-wide observations in the Pacific and the Atlantic. Bates et al.10 computed the global CO flux using a 6% smaller world ocean area compared with that reported by Menard and Smith.13 However, this would be compensated for by employing the uncorrected climatological wind speeds, at a height of ∼20 m; correction of this to a standard height of 10 m would decrease the wind speed by 7%. Thus, the reported data by Bates et al.10 can be compared with our estimation directly. However, the approach of Conrad et al.24 to estimate the flux is quite different from ours and Bates et al.10 They employed the stagnant film model with a film thickness of 10 to 50 μm, which corresponds to the gas transfer velocities of 16 to 83 cm h−1 following the stagnant film model which Conrad et al.24 used. When those gas transfer velocities are deconvoluted to wind speeds, they correspond to from 8 to 20 m s−1 winds using W92 parameterization. Those wind speeds are obviously higher than the mean values of our ship winds and climatological winds, as well as their observations of winds. In addition, although Conrad et al.24 came up with a saturation ratio of 10 after normalizing the values obtained in the Atlantic to the world ocean, they took a range of 30 ± 20 when calculating the flux taking into account the enhancement of dissolved CO near the seawater surface in the depth profile. Consequently, Conrad et al.24 may have overestimated the global flux of CO. Thus, accounting for the lower limit of their estimation, the emission from the ocean is likely to be within the range of 4 to 30 Tg(CO) a−1. This probably led to the overestimated oceanic emission for CO in IPCC1992 which is based on WMO1985,47 Seiler and Conrad,48 and Khalil and Rasmussen49 (Table 3). These studies again evaluated the flux using the observations in the 1970s by Linnenbom et al.50 and Seiler,51 in which the dissolved concentrations were significantly higher as outlined by Bates et al.10 Although Seiler and Conrad48 did not refer to Conrad et al.24 nor did they state any other references for the source strength of the ocean, the values seemed to come from Conrad et al.24 Therefore, it is unambiguous that the IPCC1992 report overestimated the oceanic flux of CO. The overestimation by Conrad et al.24 is also supported by another Atlantic meridional observation by Stubbins et al.11 in April, 2000. Their estimate of ocean emission of CO is 8.6 ± 6 Tg(CO) a−1, which is at the lower end of our estimates. In spite of direct observations of ocean flux of CO in the open ocean, a series of IPCC reports essentially refer to the estimation in IPCC1992 (Table 3). Although mentioning the most likely value of 50 Tg(CO) a−1 in IPCC2001, which is based on the global inverse model simulation by Bergamaschi et al.,52 the estimation is still 4–5 times larger than the value estimated on the basis of direct observations in the ocean, which Stubbins et al. also pointed out. Recent IPCC reports published in 20063 and 201353 did not mention the CO emissions from the ocean. Recent inverse model simulation,8 which investigated the atmospheric CO budget by means of not only molecular concentration but also characteristic source signatures of oxygen isotope ratios in CO, came up with the reduced source strength of the ocean of 20 Tg(CO) a−1, approaching to the value estimated by direct observations. This suggests that the previous estimates of ocean source strength are likely to be biased and we urged to count on the direct observations of ocean source strength in the model simulation and in the future IPCC reports.
| Reference | Range | Most likely | Remark |
|---|---|---|---|
| a Emissions are in Tg (CO) a−1. b WMO198547 essentially refers to Logan et al.63 which is based on the observations by Linnenbom et al.50 and Seiler.51 c Khalil and Rasmussen49 refers to Logan et al.63 | |||
| Basin-wide observations | |||
| This study | 4–24 | 14 | Estimate based on data from the Atlantic in October, 1998 |
| Conrad et al.60 | 10–180 | 100 | Estimate based on data from the Atlantic during 1978–1980 |
| Bates et al.61 | 6–30 | 13 | Estimate based on data from the Pacific during 1987–1994 |
| Stubbins et al.11 | 2.6–15 | 8.6 | Estimate based on data from the Atlantic in April, 2000 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| IPCC reports | |||
| IPCC199257 | 20–200 | Refers to WMO1985,47b Seiler and Conrad,48 and Khalil and Rasmussen49c | |
| IPCC199458 | 20–200 | Refers to IPCC199257 | |
| IPCC199659 | 20–200 | Refers to IPCC199257 | |
| IPCC200112 | 20–200 | 50 | Refers to Bergamaschi et al.,52 WMO1999,62 IPCC199659 |
At this point, it is worthwhile to mention the possible uncertainty of the dissolved concentration of CO above the sampling depth. As demonstrated in previous investigations,24,54 the dissolved CO concentration decreases exponentially with depth, and Conrad et al.24 stated that their estimation of the CO flux might be a lower limit because the CO concentration within 1 m depth was twice that at a sampling depth of 4 m and because Seiler55 had reported a saturation ratio of 1000. Along the same lines, Springer-Young et al.56 argued that their estimated flux may represent the current oceanic flux of CO. However, such a large dissolved concentration in the upper few meters appears to be unlikely based on the measurements by Johnson17 that the dissolved CO concentrations within the upper 5 m were observed to be constant within analytical error. Diurnal variation of vertical profiles of CO in the BATS site in the Atlantic also demonstrated a well-mixed surface layer in less than 10 m deep.45 In addition, Johnson17 tested the performance of the equilibrator which was used for the underway measurements of dissolved gas by Bates et al.,10 concluding that there might be a >25% error for dissolved CO measurements primarily due to the long e-folding time (or characteristic time) of CO, the large supersaturation of seawater, and diurnal variation. Our equilibrator was 5.7 times smaller than that Bates et al.10 used; thus the e-folding time is shorter. We tested the performance of our equilibrator to see if the underway measurement was underestimated. This was done by comparing the results from the underway measurement and discrete sampling for dissolved CO. Throughout the campaign the Rosette was cast 32 times. The uppermost surface water was sampled from a depth of 4 to 12 m. As demonstrated in Fig. 3(b), there was no significant difference between the two depths. Therefore, analytical evidence of ours and Johnson17 shows that the global oceanic fluxes of CO estimated by Bates et al.,10 Stubbins et al.,11 and by this work are more reliable than those values mentioned in IPCC reports.12,57–59
4. Conclusions
The global emission of CO from the ocean was estimated to be 4–24 Tg(CO) a−1 with a central value of 14 Tg(CO) a−1 based on observations in the Atlantic during fall 1998. This agrees well with those reported by Bates et al.10 and Stubbins et al.11 Considering that the data of Bates et al.10 and Stubbins et al.11 covered different seasons and regions in the Pacific and the Atlantic, direct basin-wide observations suggest the source strength of ocean for the atmospheric CO budget as 4–30 Tg(CO) a−1, with the most likely value of 14 Tg(CO) a−1. Consequently, a series of IPCC reports12,57–59 overestimated the global oceanic emission of CO by as much as five times or more by referring to model simulations or uncorrected observational results.Acknowledgements
We gratefully acknowledge the help of Jim Aiken, Douglas Trevitt, and other crew members of the RRS James Clark Ross in collecting samples. This research, funded by the Max Planck Society, has been done while T. S. R. was at the Max Planck Institute for Chemistry supervised by M. O. Andreae. T. S. R. is pleased to acknowledge the useful comments of D. R. Schink, R. A. Duce, and M. O. Andreae on an early version of the manuscript. Funding from Korean polar research programs (PE13410, PE15061, and PM15070) allowed us to disclose this work in the public at the final stage.References
- J. S. Daniel and S. Solomon, J. Geophys. Res.: Atmos., 1998, 103, 13249–13260 CrossRef CAS.
- G. Myhre, D. Shindell, F. Bréon, W. Collins, J. Fuglestvedt, J. Huang, D. Koch, J. Lamarque, D. Lee, B. Mendoza, T. Nakajima, A. Robock, G. Stephens, T. Takemura and H. Zhang, in Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, ed. T. F. Stocker, D. Qin, G.-K. Plattner, M. Tignor, S. K. Allen, J. Boschung, A. Nauels, Y. Xia, V. Bex and P. M. Midgley, Cambridge University Press, Cambridge, U.K., New York, U.S.A, 2013 Search PubMed.
- IPCC, Climate Change 2007: The Physical Science Basis. Contribution of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge, U.K., 2007 Search PubMed.
- P. C. Novelli, K. A. Masarie, P. P. Tans and P. M. Lang, Science, 1994, 263, 1587–1590 CAS.
- P. C. Novelli, K. A. Masarie, P. M. Lang, B. D. Hall, R. C. Myers and J. W. Elkins, J. Geophys. Res.: Atmos., 2003, 108, 4464–4477 CrossRef.
- O. Pechony, D. T. Shindell and G. Faluvegi, J. Geophys. Res.: Atmos., 2013, 118, 8054–8066 CAS.
- A. F. Arellano, P. S. Kasibhatla, L. Giglio, G. R. van der Werf and J. T. Randerson, Geophys. Res. Lett., 2004, 31, L01104–L011108 Search PubMed.
- K. Park, L. K. Emmons, Z. Wang and J. E. Mak, Atmosphere, 2015, 6, 547–578 CrossRef.
- K. Park, L. K. Emmons, Z. Wang and J. E. Mak, Geophys. Res. Lett., 2013, 40, 221–226 CrossRef CAS.
- T. S. Bates, K. C. Kelly, J. E. Johnson and R. H. Gammon, J. Geophys. Res.: Atmos., 1995, 100, 23093–23101 CrossRef CAS.
- A. Stubbins, G. Uhera, V. Kitidis, C. S. Law, R. C. Upstill-Goddard and E. M. S. Woodward, Deep Sea Res., Part II, 2006, 53, 1685–1694 CrossRef.
- IPCC, Climate Change 2001: The Scientific Basis, Contribution of Working Group I to the Third Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge, U.K., 2001 Search PubMed.
- H. W. Menard and S. M. Smith, J. Geophys. Res., 1966, 71(18), 4305–4325 CrossRef.
- J. Aiken, N. Rees, S. Hooker, P. Holligan, A. Bale, D. Robins, G. Moore, R. Harris and D. Pilgrim, Prog. Oceanogr., 2000, 45, 257–312 CrossRef.
- T. S. Rhee, A. J. Kettle and M. O. Andreae, J. Geophys. Res.: Atmos., 2009, 114, D12304, DOI:10.1029/2008jd011662.
- A. J. Kettle, T. S. Rhee, M. von Hobe, A. Poulton, J. Aiken and M. O. Andreae, J. Geophys. Res.: Atmos., 2001, 106, 12193–12209 CrossRef CAS.
- J. E. Johnson, Anal. Chim. Acta, 1999, 395, 119–132 CrossRef CAS.
- T. S. Rhee, Ph.D. thesis, Texas A&M University, 2000.
- J. D. McCullough, R. A. Crane and A. O. Beckman, Ind. Eng. Chem., Anal. Ed., 1947, 19, 999–1002 CrossRef CAS.
- P. C. Novelli, J. W. Elkins and L. P. Steele, J. Geophys. Res.: Atmos., 1991, 96, 13109–13121 CrossRef CAS.
- D. A. Wiesenburg and N. L. Guinasso Jr, J. Chem. Eng. Data, 1979, 24, 356–360 CrossRef CAS.
- H. Matsueda, H. Inoue and M. Ishii, Geophys. Res. Lett., 1993, 20, 695–698 CrossRef CAS.
- M. O. Andreae and P. Merlet, Global Biogeochem. Cycles, 2001, 15, 955–966 CrossRef CAS.
- R. Conrad, W. Seiler, G. Bunse and H. Giehl, J. Geophys. Res., C: Oceans Atmos., 1982, 87, 8839–8852 CrossRef CAS.
- W. H. Pos, D. D. Reimer and R. G. Zika, Mar. Chem., 1998, 62, 89–101 CrossRef CAS.
- A. Stubbins, G. Uher, C. S. Law, K. Mopper, C. Robinson and R. C. Upstill-Goddard, Deep Sea Res., Part II, 2006, 53, 1695–1705 CrossRef.
- O. C. Zafiriou, S. S. Andrews and W. Wang, Global Biogeochem. Cycles, 2003, 17, 1015–1029 CrossRef.
- H. Xie, O. C. Zafiriou, T. P. Umile and D. J. Kieber, Mar. Ecol.: Prog. Ser., 2005, 290, 1–14 CrossRef CAS.
- A. Gnanadesikan, J. Geophys. Res.: Oceans, 1996, 101(C5), 12177–12191 CrossRef CAS.
- R. D. Jones and R. Y. Morita, Can. J. Microbiol., 1983, 29, 1545–1551 CrossRef CAS.
- F. Nakagawa, U. Tsunogai, T. Gamo and N. Yoshida, J. Geophys. Res.: Oceans, 2004, 109, D08308 CrossRef.
- P. J. Setser, J. L. Bullister, E. C. Frank, J. N. L. Guinasso and D. R. Schink, Deep-Sea Res., Part A, 1982, 29, 1203–1215 CrossRef CAS.
- P. S. Liss and L. Merlivat, Air-sea gas exchange rates: Introduction and synthesis, in The Role of Air-Sea Exchange in Geochemical Cycling, ed. P. Buat-Ménard, 1986, D. Reidel, Hingham, Mass., pp. 113–129 Search PubMed.
- R. Wanninkhof, J. Geophys. Res.: Oceans, 1992, 97, 7373–7382 CrossRef.
- D. J. I. Erickson, J. Geophys. Res.: Oceans, 1993, 98, 8471–8488 CrossRef.
- P. D. Nightingale, G. Malin, C. S. Law, A. J. Watson, P. S. Liss, M. I. Liddicoat, J. Boutin and R. C. Upstill-Goddard, Global Biogeochem. Cycles, 2000, 14, 373–387 CrossRef CAS.
- S. R. Smith, M. A. Bourassa and R. J. Sharp, J. Atmos. Ocean Tech., 1999, 17, 939–952 CrossRef.
- E. B. Kraus and J. A. Businger, Atmosphere-ocean interaction, Clarendon Press, New York, 2nd edn, 1994 Search PubMed.
- C. C. Young, A. M. da Silva and S. Levitus, New Atlas Details Surface Marine Atmosphere - Joint project between UWM and NODC to aid global climate research, 1995 Search PubMed.
- D. Wise and G. Houghton, Chem. Eng. Sci., 1968, 23, 1211–1216 CrossRef CAS.
- B. Jahne, G. Heinz and W. Dietrich, J. Geophys. Res.: Oceans, 1987, 92, 10767–10776 CrossRef.
- L. Korson, W. Drost-Hansen and F. J. Millero, J. Phys. Chem., 1969, 73, 34–39 CrossRef CAS.
- F. J. Millero, in The Sea, John Wiley, New York, 1974, vol. 5, pp. 3–80 Search PubMed.
- F. J. Millero and A. Poisson, Deep-Sea Res., Part A, 1981, 28, 625–629 CrossRef.
- O. C. Zafiriou, H. Xie, N. B. Nelson, R. G. Najjar and W. Wang, Limnol. Oceanogr., 2008, 53, 835–850 CrossRef CAS.
- C. D. Nevison, R. F. Weiss and D. J. Erickson III, J. Geophys. Res.: Oceans, 1995, 100, 15809–15820 CrossRef CAS.
- WMO, Atmospheric ozone 1985, Report 16, World Meteorological Organization, Geneva, Switzerland, 1985 Search PubMed.
- W. Seiler and R. Conrad, in The Geophysiology of Amazonia, ed. R. E. Dickinson, John Wiley & Sons, New York, 1987, pp. 133–160 Search PubMed.
- M. A. K. Khalil and R. A. Rasmussen, Chemosphere, 1990, 20, 227–242 CrossRef CAS.
- V. Linnenbom, J. Swinnerton and R. Lamontagne, J. Geophys. Res., 1973, 78, 5333–5340 CrossRef CAS.
- W. Seiler, Tellus, 1974, 26, 116–135 CrossRef CAS.
- P. Bergamaschi, R. Hein, C. A. M. Brenninkmeijer and P. J. Crutzen, J. Geophys. Res.: Atmos., 2000, 105, 1929–1945 CrossRef CAS.
- IPCC, Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge, U.K. and New York, NY, USA, 2013 Search PubMed.
- J. E. Johnson and T. S. Bates, Global Biogeochem. Cycles, 1996, 10(2), 347–359 CrossRef CAS.
- W. Seiler, in Environmental biogeochemistry and geomicrobiology, ed. W. E. Krumbein, Ann Arbor Science, Ann Arbor, 1978, pp. 773–810 Search PubMed.
- M. Springer-Young, I. D. J. Errickson and T. P. Carsey, J. Geophys. Res.: Atmos., 1996, 101, 4479–4484 CrossRef CAS.
- IPCC, Climate change 1992: The supplementary report to the IPCC scientific assessment, Cambridge University Press, 1992 Search PubMed.
- IPCC, Climate Change 1994: Radiative Forcing of Climate Change and An Evaluation of the IPCC IS92 Emission Scenarios, Cambridge University, 1995 Search PubMed.
- IPCC, Climate Change 1995-The science of climate change, Cambridge University, 1996 Search PubMed.
- R. Conrad and W. Seiler, Deep-Sea Res., Part A, 1988, 35, 1903–1917 CrossRef CAS.
- T. S. Bates, K. C. Kelly, J. E. Johnson and R. H. Gammon, J. Geophys. Res.: Atmos., 1996, 101(D3), 6953–6961 CrossRef CAS.
- WMO, Scientific Assessment of Ozone Depletion: 1998. Global Ozone Research and Monitoring Project - Report No. 44, Report 16, World Meteorological Organization, Geneva, Switzerland, 1999 Search PubMed.
- J. A. Logan, M. J. Prather, S. C. Wofsy and M. B. McElroy, J. Geophys. Res.: Oceans, 1981, 86, 7210–7254 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5em00546a |
| ‡ Currently at Inha university, Incheon, Korea |
| This journal is © The Royal Society of Chemistry 2016 |