
Quercitrin offers protection against brain injury in mice by inhibiting oxidative stress and inflammation
Jie-Qiong
Ma
*a,
Rong-Zhen
Luo
a,
Hai-Xia
Jiang
a and
Chan-Min
Liu
b
aSchool of Chemistry and Pharmaceutical Engineering, Sichuan University of Science and Engineering, No. 180, Huixing Road, 643000 Zigong City, Sichuan Province, PR China. E-mail: a1032042419@126.com; Fax: +86 516 83500171; Tel: +86 013700950121
bSchool of Life Science, Jiangsu Normal University, No. 101, Shanghai Road, Tongshan New Area, Xuzhou City 221116, Xuzhou City, Jiangsu Province, PR China
First published on 14th October 2015
Abstract
Quercitrin is one of the primary flavonoid compounds present in vegetables and fruits. The aim of the present study was to evaluate the effects of quercitrin against carbon tetrachloride (CCl4) induced brain injury and further to elucidate its probable mechanisms. ICR mice received CCl4 intraperitoneally with or without quercitrin co-administration for 4 weeks. Our data showed that quercitrin significantly suppressed the elevation of reactive oxygen species (ROS) production and malondialdehyde (MDA) content, reduced tissue plasminogen activator (t-PA) activity, enhanced the antioxidant enzyme activities and abrogated cytochrome P450 2E1 (CYP2E1) induction in mouse brains. Quercitrin also prevented CCl4 induced cerebral function disorders associated with its ability to inhibit the activities of monoamine oxidase (MAO), acetylcholine esterase (AChE) and the N-methyl-D-aspartate receptor 2B subunit (NR2B). In addition, western blot analysis showed that quercitrin suppressed the release of pro-inflammatory cytokines such as tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6). Taken together, our findings suggested that quercitrin may be a potential candidate to be developed as a neuroprotective agent.
1 Introduction
Flavonoids possess antioxidant and anti-inflammatory activities, readily permeate the blood–brain barrier and afford neuroprotection in a wide array of cellular and animal models of neurological diseases.1,2 Flavonoids are ubiquitous plant secondary products, which are produced in response to diverse environmental cues. In plants, naringenin was converted into dihydroflavonols. Dihydroflavonols were converted by flavonol synthase into flavonols. Flavonols were further glucosylated to flavonol glycosides.3 Quercetin is the most abundant bioflavonoid found in vegetables and fruits, which exists primarily in its glycoside form as quercitrin (3-rhamnoside). The sugar portion bound to the aglycone portion in quercitrin (QE) increases its solubility in polar solvents and consequently improves absorption (Fig. 1).1,2 Quercitrin is known to have biological effects such as anti-oxidative, anti-inflammatory, and anti-apoptosis effects.4,5 Quercitrin might be a more potent antioxidant and a neuroprotective agent than quercetin due to its high bioavailability in the digestive track.6–9 Previous reports indicated that quercitrin significantly reduced acute systemic inflammation in LPS-induced models as well as attenuated inflammation in the livers of mice.7 Quercitrin could also reduce inflammation and apoptosis in cytokine-induced models.8 In streptozotocin (STZ)-induced diabetic models, quercitrin displays organ protective properties in the pancreas, liver and kidney by improving the antioxidant status.5 | ||
| Fig. 1 Chemical structure of quercitrin (QE). | ||
Carbon tetrachloride (CCl4) is a well-known toxic compound. Administration of CCl4 to an animal is an accepted experimental model to produce damage to hepatic and other tissues.10 CCl4 can cause damage to the brain through lipid peroxidation, resulting in an up-regulation of pro-inflammatory pathways and the alterations of the neurotransmission system.11,12 Recent studies from our laboratory showed that puerarin (a natural flavonoid) could attenuate CCl4-induced oxidative stress and inflammation in the kidney.12 However, the molecular mechanisms of CCl4-induced brain injury and neuroprotective effects of quercitrin have not yet been completely understood. In the present study, we aimed to determine whether the dietary uptake of quercitrin can protect mouse brains from CCl4-induced injury by inhibiting oxidative stress and inflammation. In addition, we investigated whether the protective mechanism of quercitrin is associated with the activities of tissue plasminogen activator (t-PA), monoamine oxidase (MAO), acetylcholine esterase (AChE) and the N-methyl-D-aspartate receptor 2B subunit (NR2B).
2 Materials and methods
2.1 Chemicals and reagents
Quercitrin and CCl4 were obtained from Sigma Chemical Co. (St Louis, MO, USA). Anti-IL-6 antibody, anti-TNF-α antibody and anti-NR2B antibody are from Santa Cruz Biotechnology (Santa Cruz, CA, USA) or Cell Signaling Technology (Beverly, MA, USA). All other reagents unless indicated were obtained from Sigma Chemical Co. (St Louis, MO, USA).2.2 Animals and treatment
Male ICR mice (20–25 g) were obtained from the Branch of National Breeder Center of Rodents (Beijing). The animals were maintained in an animal facility at 20–22 °C under 40–60% relative humidity and a 12/12 h (light/dark) cycle.After acclimation for 1 week, forty mice were randomly divided into four groups. Brain injury was induced by an intraperitoneal (i.p.) injection of 2 ml of CCl4 in olive oil (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) per kg body weight twice weekly for up to 4 weeks.13 Mice in Group 1 were given two weekly injections of peanut oil (vehicle control) and received water containing 0.5% carboxymethylcellulose sodium by oral gavage; mice in Group 2 were injected with CCl4 and received water containing 0.5% carboxymethylcellulose sodium by oral gavage; mice in Group 3 and Group 4 were injected with CCl4, as in group 2, and administered with quercitrin (50 and 100 mg per kg body-weight, suspended in 0.5% carboxymethylcellulose sodium daily) by oral gavage, respectively. The choice of quercitrin dose is based on previous findings.14
:1, v/v) per kg body weight twice weekly for up to 4 weeks.13 Mice in Group 1 were given two weekly injections of peanut oil (vehicle control) and received water containing 0.5% carboxymethylcellulose sodium by oral gavage; mice in Group 2 were injected with CCl4 and received water containing 0.5% carboxymethylcellulose sodium by oral gavage; mice in Group 3 and Group 4 were injected with CCl4, as in group 2, and administered with quercitrin (50 and 100 mg per kg body-weight, suspended in 0.5% carboxymethylcellulose sodium daily) by oral gavage, respectively. The choice of quercitrin dose is based on previous findings.14
At the end of the treatment, the mouse brains were used for a biochemical analysis and western-blot evaluations. The brain tissues were quickly collected for experiments and placed in ice-cold 0.9% NaCl solution, perfused with a physiological saline solution to remove blood cells, blotted on filter paper. Then, the removed brains were immediately collected for experiments or stored at −70 °C for later use.
This research was conducted in accordance with the Chinese laws on the use and care of laboratory animals. The committees for animal experiments of Jiangsu Normal University and Sichuan University of Science and Engineering approved these experiments.
2.3 Biochemical analysis
ROS was measured as described previously, based on the oxidation of 2′7′-dichlorodihydrofluorescein diacetate to 2′7′-dichloro-fluorescein.13 ROS formation was quantified from a DCF-standard curve and data are expressed as pmol DCF formed per min per mg protein. The activities of superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GPx), monoamine oxidase (MAO) and acetylcholine esterase (AChE) in the hippocampus homogenates were measured using the commercial kits (Jiancheng Institute of Biotechnology, Nanjing, China).The malondialdehyde (MDA) concentration in the hippocampus homogenates was measured by the thiobarbituric acid reactive substances (TBARS) assay in accordance with the manufacturer's instructions (MDA: A003-1, Jiancheng Biochemical Institute, Nanjing, China) and the absorbance was measured at a wavelength of 532 nm.
2.4 Functionally active t-PA activity assay
Functionally active t-PA was determined using the active t-PA ELISA kit according to the manufacturer's instructions.15 The amount of color development is directly proportional to the concentration of active t-PA in the sample.2.5 Western blot analysis
Western blot analysis was performed according to our previous method.16,17 Nuclear and cytoplasmic extracts for western blotting were obtained by using a nuclear/cytoplasmic isolation kit (Beyotime Institute of Biotechnology, Beijing, China). Protein levels in the hippocampus homogenates were determined using the BCA assay kit (Pierce Biotechnology, Inc., Rockford, IL, USA).2.6 Statistical analysis
All statistical analyses were performed using the SPSS software, version 11.5. A one-way analysis of variance (ANOVA; P < 0.05) was used to determine significant differences between groups and the individual comparisons were obtained by Tukey's HSD post hoc test. Statistical significance was set at P ≤ 0.05.3 Results
3.1 Quercitrin inhibited CCl4-induced oxidative stress in mouse brains
To investigate whether quercitrin can attenuate the oxidative stress damage in the brains of CCl4-treated mice, we measured the concentrations of ROS and MDA (Fig. 2). The results showed that the levels of ROS and MDA were highly increased by 107.28% and 80.04% in the brains of the CCl4-treated group compared with the control group, respectively (P < 0.01). However, treatment with quercitrin dose-dependently decreased the levels of ROS and MDA (by 30.78%, 40.03% and 27.24%, 43.15%, respectively) compared with CCl4 treatment (P < 0.01).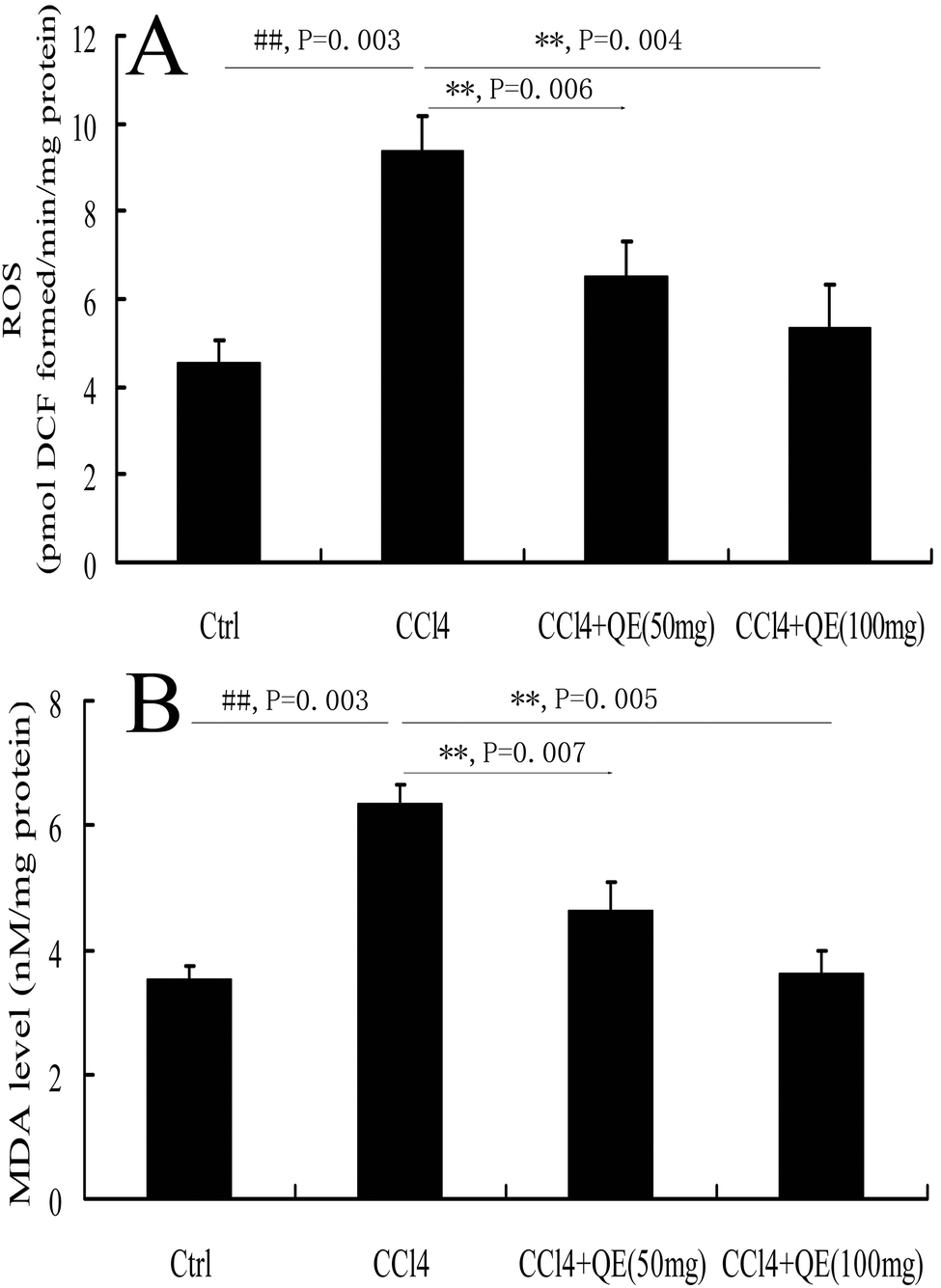 | ||
| Fig. 2 Effect of quercitrin (QE) on the oxidative stress markers in CCl4-treated mouse brains. (A) Level of ROS; (B) level of MDA. Each value is expressed as mean ± S.E.M. (n = 7). ##P < 0.01, compared with the control group; **P < 0.01, vs. the CCl4-treated group. | ||
3.2 Quercitrin inhibited the CYP2E1 level in the brains of CCl4-treated mice
In order to determine whether quercitrin can inhibit CCl4-induced brain injury, we examined the CYP2E1 level in mouse brains. As shown in Fig. 3, CCl4 treatment caused a significant increase in the CYP2E1 level by 363.21% in mouse brains as compared with that of the control group (P < 0.01). Interestingly, treatment with quercitrin dose-dependently decreased the CYP2E1 level by 43.23% and 72.19% compared with CCl4 treatment, respectively (P < 0.01).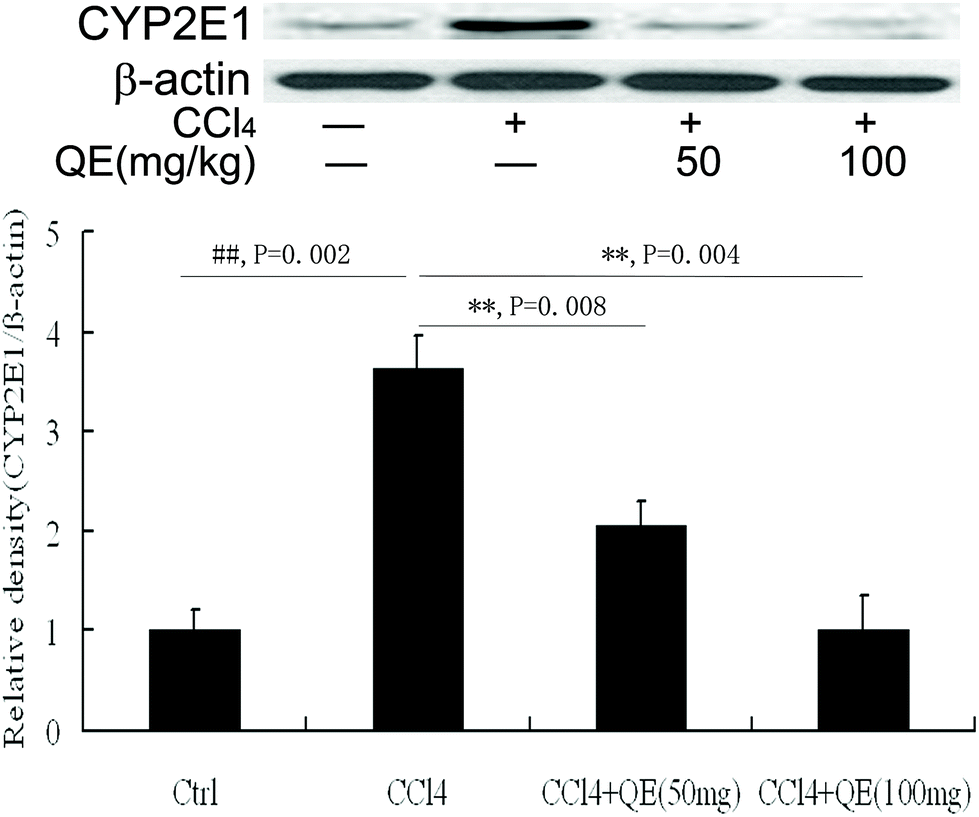 | ||
| Fig. 3 Quercitrin (QE) reduced the CAP2E1 level in the brains of mice exposed to CCl4. β-Actin was probed as an internal control in relative density analysis. The vehicle control is set as 1.0. Each value is expressed as mean ± S.E.M. **P < 0.01, compared with the control group; ##P < 0.01, vs. the CCl4-treated group. | ||
3.3 Quercitrin increased the activities of SOD, CAT and GPX in mouse brains
During the pathological process of CCl4-induced brain injury, reductions in antioxidant enzyme activities play important roles.10 CCl4 administration significantly decreased the activities of SOD, CAT and GPX in mouse brains by 24.26%, 43.57% and 24.98%, respectively, when compared to those in the control group (P < 0.01). However, pretreatment with low doses and high doses of quercitrin remarkably ameliorated the depletion of the SOD, CAT and GPX activities (Fig. 4).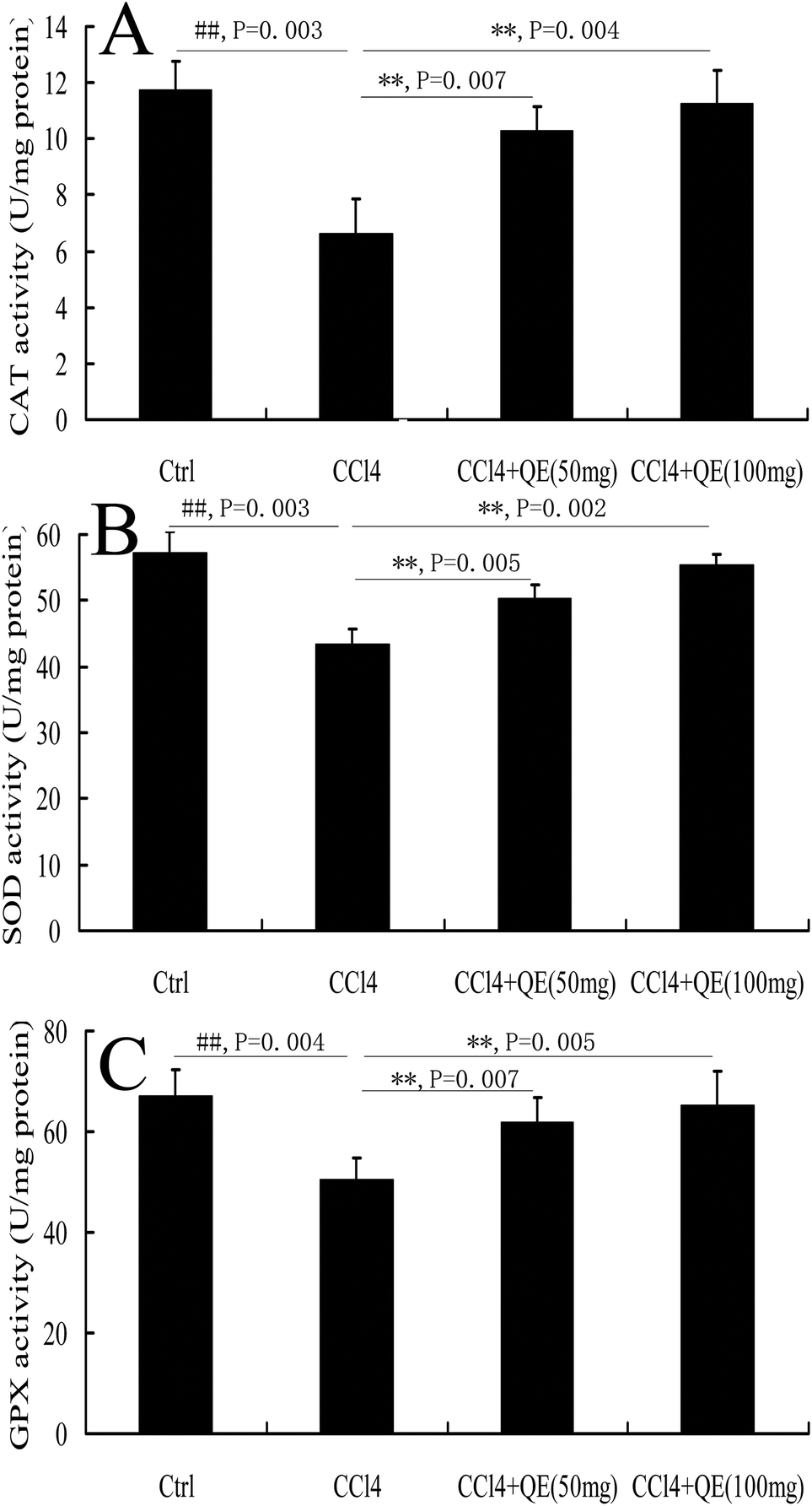 | ||
| Fig. 4 Effect of quercitrin (QE) on the antioxidant enzyme activities in the brains of mice exposed to CCl4. (A) CAT activity; (B) SOD activity; (C) GPx activity. All values are expressed as mean ± S.E.M. (n = 7). ##P < 0.01, compared with the control group; **P < 0.01, vs. the CCl4-treated group. | ||
3.4 Quercitrin decreased the t-PA activity in the brains of CCl4-treated mice
As shown in Fig. 5A, quercitrin also prevented the increase of the t-PA activity in the brain homogenates of the mice treated with CCl4. The t-PA activity in the CCl4-treated group was significantly higher (by 40.47%) than the control group (P < 0.01). However, treatment with low doses and high doses of quercitrin alongside CCl4 reduced the t-PA activity by 17.68% and 25.96% in the brains of the mice, respectively.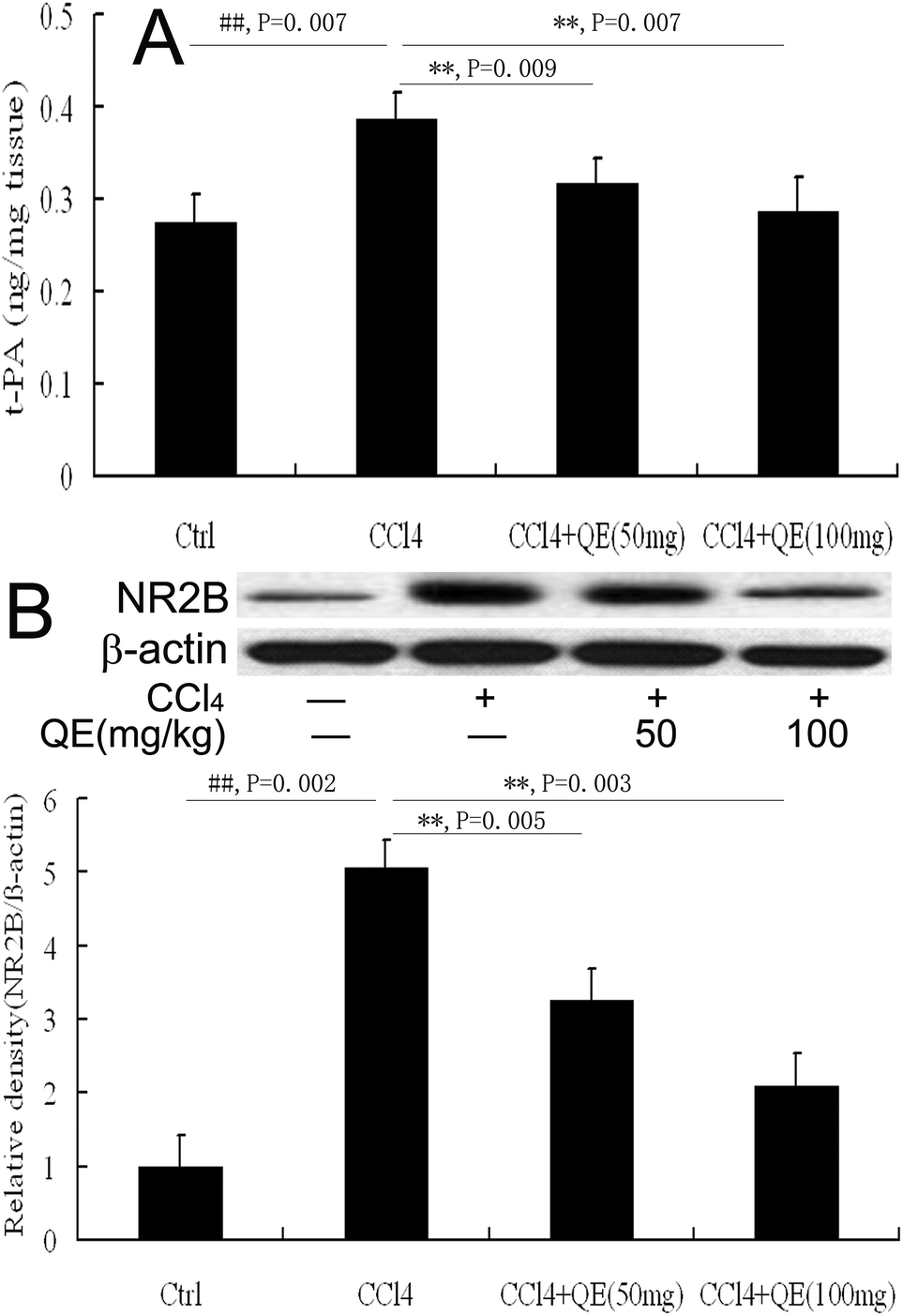 | ||
| Fig. 5 Effect of quercitrin (QE) on the activities of t-PA and NR2B in the brains of mice exposed to CCl4. (A) t-PA activity; (B) The level of NR2B. β-Actin was probed as an internal control in relative density analysis. The vehicle control is set as 1.0. Each value is expressed as mean ± S.E.M. **P < 0.01, compared with the control group; ##P < 0.01, vs. the CCl4-treated group. | ||
3.5 Quercitrin inhibited the NR2B level in the brains of CCl4-treated mice
NR2B plays critical roles in brain functions and diseases. In this study, the NR2B protein immunocontent was examined by western blot analysis on the hippocampus homogenates from mice. The results showed that the NR2B level in the brain was notably increased in the CCl4-treated mice as compared with that of the control group (Fig. 5B). Interestingly, treatment with low and high doses of quercitrin significantly down-regulated the NR2B level (by 35.53% and 58.89%, respectively) compared with CCl4 treatment (P < 0.01).3.6 Quercitrin decreased the activities of MAO and AChE in the brains of CCl4-treated mice
AChE and MAO play an important role in the central nervous system (CNS). In order to determine whether quercitrin can attenuate the neurotransmitter system damage in the brains of CCl4-treated mice, we measured the activities of AChE and MAO. The results are shown in Fig. 6. CCl4 markedly increased the activities of AChE and MAO in the mouse brain hippocampus by 58.17% and 72.29% in comparison with the control group (P < 0.01), respectively. However, treatment with quercitrin dose-dependently decreased the activities of AChE and MAO (by 30.25%, 37.66% and 25.31%, 34.33%, respectively) compared with CCl4 treatment (P < 0.01). | ||
| Fig. 6 Effect of quercitrin (QE) on the activities of AChE and MAO in the brains of mice exposed to CCl4. (A) AChE activity; (B) MAO activity. All values are expressed as mean ± S.E.M. (n = 7). ##P < 0.01, compared with the control group; *P < 0.05, **P < 0.01, vs. the CCl4-treated group. | ||
3.7 Quercitrin reduced the production of brain inflammatory cytokines of CCl4-treated mice
To examine the effect of quercitrin treatment on the CCl4-induced inflammatory cytokines, the hippocampus protein immunocontents of TNF-α and IL-6 were measured using western blot analysis. As shown in Fig. 7, the production of both TNF-α and IL-6 was significantly increased in the brains of the CCl4-treated group, compared to those in the control group. However, these increases were attenuated by quercitrin treatment (P < 0.01).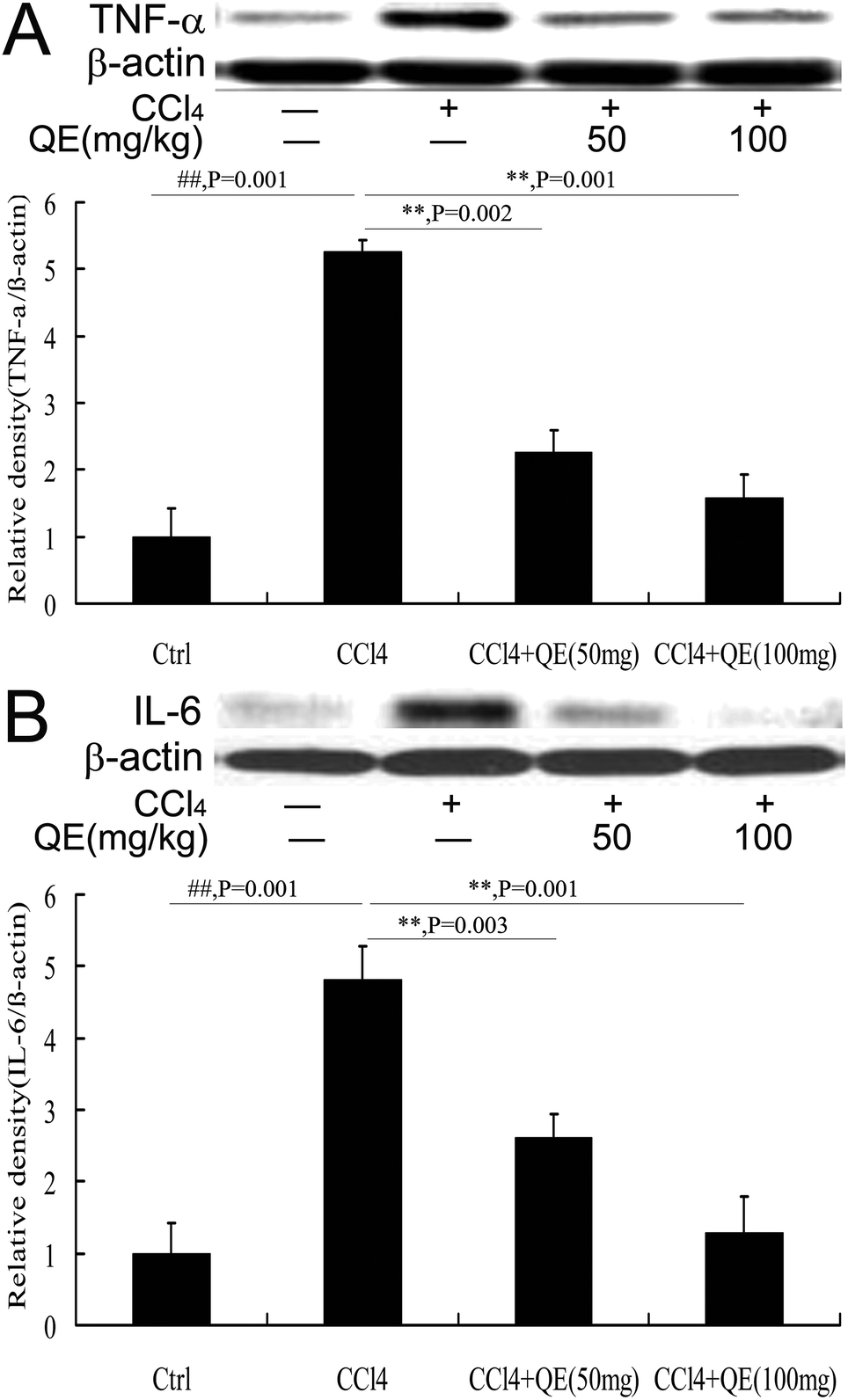 | ||
| Fig. 7 Western blot analysis of the protein immunocontent in association with inflammation in the mouse brains. (A) The level of TNF-α; (B) the level of IL-6. β-Actin was probed as an internal control in relative density analysis. The vehicle control is set as 1.0. Each value is expressed as mean ± S.E.M. **P < 0.01, compared with the control group; ##P < 0.01, vs. the CCl4-treated group. | ||
4 Discussion
The brain is an organ with a high metabolic rate that relies mainly on aerobic metabolism, which inevitably produces ROS.15 CCl4 has been extensively used in experimental models to elucidate the cellular mechanisms behind oxidative damage and inflammation.18,19 In the present study, the effect of quercitrin treatment on CCl4 induced brain injury was investigated using a mouse model. Our results demonstrated that treatment with quercitrin significantly ameliorated brain injury induced by CCl4. This protective effect was associated with its ability to inhibit oxidative stress, inflammation and neurotransmitter system damage.The brain is especially sensitive to oxidative stress. Moreover, neuronal cells are rich in polyunsaturated fatty acids, which are prone to lipid peroxidation by ROS.15 Cytochrome P450, in particular, CYP2E1, is characterized as a free enzyme with a high pro-oxidant activity, and as a potential source of oxidative stress. CCl4 can be activated by the cytochrome P450 enzyme to form the trichloromethyl radical CCl3˙ and the trichloromethyl peroxy radical CCl3OO˙, which lead to lipid peroxidation and subsequent tissue damage.11 Our previous studies showed that CCl4 induced oxidative stress in the liver and kidney.13,20 In this research, the results revealed the role of lipid peroxidation in the brain hippocampus accompanied by stimulation and induction of CYP2E1 with a severe oxidative stress status in CCl4-treated mice, which is characterized by high levels of ROS and MDA (Fig. 2). Reports have shown that quercitrin could suppress oxidative stress.2,5 The present study showed that quercitrin significantly decreased the CYP2E1 level and the peroxidation product MDA confirmed that treatment with quercitrin could effectively protect against the brain oxidative stress induced by CCl4 (Fig. 2 and 3).
Primary antioxidant enzymes involved in the direct elimination of free radicals are superoxide dismutase (SOD) which removes O2˙, catalase (CAT) which converts H2O2 to water (H2O) and O2, and glutathione peroxidase (Gpx) which helps in the removal of H2O2, thereby preventing hydroxyl radical (OH−˙) formation.10,21 A reduction in the activity of these enzymes is associated with the accumulation of excess ROS, leading to deleterious effects such as loss of integrity and function of cell membranes.10,19,22 In this study, the increase in ROS and MDA levels in brains induced by CCl4 suggests enhanced lipid peroxidation leading to tissue damage and failure of antioxidant defense mechanisms to prevent the formation of excessive free radicals.10 Interestingly, treatment with quercitrin restored the activities of these antioxidant enzymes to near-normal levels in CCl4-intoxicated mice. So, quercitrin possibly confers its protective effect by restoring the activities of SOD, CAT and GPx enzymes to near-normal levels, or by preventing decreases in the activities of these enzymes (Fig. 4).
Tissue-type plasminogen activator (t-PA), a serine-protease that activates plasmin, is endogenously produced in many types of cells.23,24 t-PA can convert plasminogen into plasmin to dissolve clots and restore blood flow. t-PA also plays important roles in maintaining the function of the brain–blood barrier and has been approved for clinical use.25,26 Several research studies indicated broader and more complex functions of the t-PA/plasmin system, an intriguing one being its role in the central nervous system (CNS).15,26 The oxidative damage of neuronal cells has been causatively implicated in multiple neurodegenerative diseases, in several of which the PA/plasmin system has been involved.15,27 The PA/plasmin system has been implicated in localized extracellular matrix remodeling in several pathophysiological events, including CNS diseases, where oxidative stress plays a pivotal role.27,28 Moreover, t-PA potentiates signaling mediated by glutamatergic receptors by modifying the properties of the NMDA receptor.15 t-PA mediates cell signaling effects through binding to the cell surface molecule NMDA receptor.15,29 NMDA receptor 2B (NR2B) subunits are essential for neurotransmission, neuronal development and synaptic plasticity, and have been shown to be involved in learning and memory abilities.26,30 Inhibition of NR2B receptors was found to reverse signaling abnormalities and spine density as well as stereotyped behavior and depression-like behavior in GluD1 KO mice.31 In the present study, the results showed that CCl4 increased the levels of active t-PA in rat brain homogenates (Fig. 5A), which were in agreement with previous reports.13 The injured neurons in CCl4 treated mice may provoke enhancement of localized t-PA activity through formation of aggregates that facilitate t-PA binding in the injured area.32 Alternatively, the induction of active t-PA in the brain was probably due to the transcriptional activation of the respective genes in neuronal or glial cells.15 This results in quercitrin, like other flavonoids, decreasing the levels of t-PA activity, which may be attributed to inhibited oxidative stress, decreased secretion of t-PA by neuronal cells, decreased transcription of the t-PA gene in neuronal and glial cells, or even to the reduction in t-PA binding sites as a result of restricted neuronal damage.15,33 The results of NR2B in the brain hippocampus of CCl4-treated mice were much higher than those in the control group, suggesting that CCl4 caused neurotransmission system dysfunction by affecting the function of NMDA receptors and some glutamate (Glu) transporters.11 While quercitrin may display the neuroprotective effects associated with decreased NR2B levels in mice (Fig. 5B), acetylcholine is considered as one of the important neurotransmitters implicated in memory function. One of the major markers for cholinergic function is the determination of AChE activity.34,35 AChE hyperactivity can cause memory deficit in animal models.10 Several studies reported that oxidative stress was related to the AChE activity in the brain.35,36 In this study, CCl4 markedly increased the AChE activity and oxidative stress in the mouse brain, which was in agreement with the previous research.10 Monoamine oxidase (MAO) plays a vital role in the inactivation of neurotransmitters. MAO dysfunction (too much/too little MAO activity) is thought to be responsible for various mental and neurodegenerative disorders.11,37 MAO, which constitutes the major pathway for inactivation of the catecholamine neurotransmitters, produces hydrogen peroxide and triggers the formation of other forms of ROS as well, which can induce neuronal damage. MAO can therefore be regarded as a key marker of the cellular antioxidant and pro-oxidant statuses.38,39 Our results revealed a significant increase in MAO activity in brain tissue after CCl4 induction, suggesting that CCl4 induced neurotransmitter dysfunction and oxidative stress. However, quercitrin treatment markedly decreased the activities of AChE and MAO in mouse brains, indicating its potent neuroprotective capacity (Fig. 6).
Various inflammatory cytokines play a significant role in the development of an acute or chronic inflammatory response. TNF-α and IL-6 have been widely considered to be the most important inflammatory cytokines in CCl4 induced brain injury.11 It is well known that oxidative stress also stimulates the release of these cytokines and amplifies the inflammation response to cause further damage.13,17,39 Moreover, the increased CYP2E1 and produced ROS accompanied by oxidative stress can activate the inflammation process by producing TNF-α, IL-6 and other pro-inflammatory cytokines.11,40 Our study showed increased levels of CYP2E1 and proinflammatory cytokines in the CCl4-exposed mouse brain hippocampus and quercitrin treatment could effectively suppress them (Fig. 3 and 7). These results indicated that quercitrin application markedly inhibited CCl4-induced oxidative stress and inflammation in mouse brains.
In conclusion, this is the first report on the fact that quercitrin has protective effects on brain injury due to exposure to CCl4. The primary mechanisms of this effect could be due to attenuation of brain oxidative stress, suppression of inflammation, and amelioration of neurotransmitter dysfunction (Fig. 8). Quercitrin decreased the activities of ROS, CYP2E1, MDA, t-PA, NR2B, AChE and MAO, and increased the activities of antioxidant enzymes in mouse brains exposed to CCl4. Quercitrin also suppressed the levels of key pro-inflammatory cytokines triggered by an inflammatory stimulus. Overall, despite the positive effects of quercitrin on CCl4-induced brain injury, more in-depth mechanistic studies are needed to support these beneficial effects.
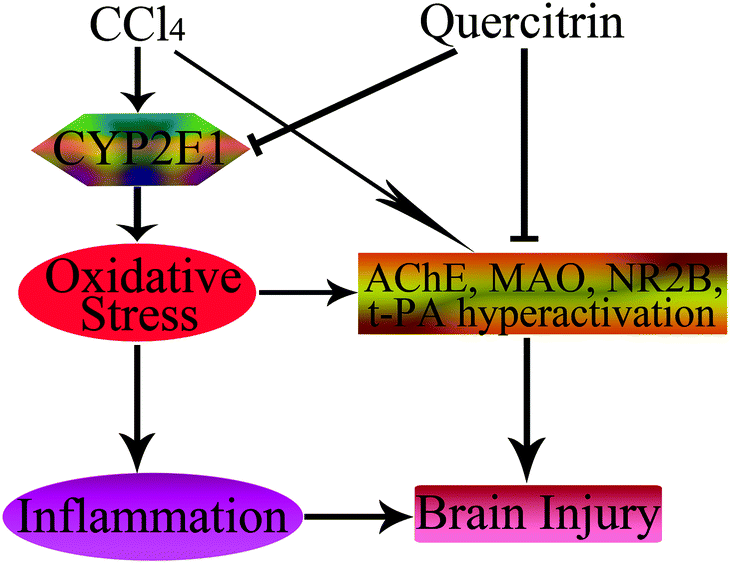 | ||
| Fig. 8 Schematic diagram showing the protective effects of quercitrin in CCl4-induced brain injury. The → indicates activation or induction, and ┤ indicates inhibition or blockade. | ||
Abbreviations
| AChE | Acetylcholine esterase |
| CAT | Catalase |
| CYP2E1 | Cytochrome P450 2E1 |
| GPX | Glutathione peroxidase |
| IL-6 | Interleukin-6 |
| MAO | Monoamine oxidase |
| MDA | Malondialdehyde |
| NR2B | N-Methyl-D-aspartate receptor 2B subunit |
| QE | Quercitrin |
| ROS | Reactive oxygen species |
| SOD | superoxide dismutase |
| t-PA | Tissue-type plasminogen activator |
| TNF-α | Tumor necrosis factor-α. |
Conflict of interest
The authors declare that there are no conflicts of interest.Acknowledgements
This work is supported by the Doctor Science Foundation of Sichuan University of Science and Engineering.Notes and references
- J. M. Gee, M. S. DuPont, M. J. Rhodes and I. T. Johnson, Free Radicals Biol. Med., 1998, 25, 19–25 CrossRef CAS.
- K. Kawabata, R. Mukai and A. Ishisaka, Food Funct., 2015, 6, 1399–1417 CAS.
- B. Winkel-Shirley, Curr. Opin. Plant Biol., 2002, 5, 218–223 CrossRef CAS.
- S. Rattanajarasroj and S. Unchern, Neurochem. Res., 2010, 35, 1196–1205 CrossRef CAS PubMed.
- R. Babujanarthanam, P. Kavitha, U. S. Mahadeva Rao and M. R. Pandian, Mol. Cell. Biochem., 2011, 358, 121–129 CrossRef CAS PubMed.
- P. C. Hollman, J. H. de Vries, S. D. Van Leeuwen, M. J. Mengelers and M. B. Katan, Am. J. Clin. Nutr., 1995, 62, 1276–1282 CAS.
- C. Morand, C. Manach, V. Crespy and C. Remesy, Free Radical Res., 2000, 33, 667–676 CrossRef CAS.
- H. Y. Jo, Y. Kim, S. Y. Nam, B. J. Lee, Y. B. Kim, Y. W. Yun and B. Ahn, J. Vet. Sci., 2008, 9, 267–272 CrossRef.
- X. Dai, Y. Ding, Z. Zhang, X. Cai and Y. Li, Int. J. Mol. Med., 2013, 31, 265–271 CAS.
- T. Annadurai, S. Vigneshwari, R. Thirukumaran, P. A. Thomas and P. Geraldine, J. Physiol. Biochem., 2011, 67, 519–530 CrossRef CAS PubMed.
- D. A. Ghareeb, H. S. Hafez, H. M. Hussien and N. F. Kabapy, Metab. Brain Dis., 2011, 26, 253–267 CrossRef CAS PubMed.
- C. Soria Fregozo, M. L. Miranda Beltrán, M. E. Flores Soto, M. I. Pérez Vega, C. Beas Zárate and L. Huacuja Ruiz, Neurologia, 2012, 27, 261–267 CrossRef CAS PubMed.
- J. Q. Ma, J. Ding, Z. H. Xiao and C. M. Liu, Food Chem. Toxicol., 2014, 71, 264–271 CrossRef CAS PubMed.
- J. H. Lee, J. Ahn, J. W. Kim, S. G. Lee and H. P. Kim, Arch. Pharmacal Res., 2015, 38, 1304–1311 CrossRef CAS PubMed.
- S. N. Lavrentiadou, M. P. Tsantarliotou, I. A. Zervos, E. Nikolaidis, M. P. Georgiadis and I. A. Taitzoglou, Food Chem. Toxicol., 2013, 61, 196–202 CrossRef CAS PubMed.
- C. M. Liu, G. H. Zheng, Q. L. Ming, C. Cheng and J. M. Sun, J. Agric. Food Chem., 2013, 61, 1146–1154 CrossRef CAS PubMed.
- C. M. Liu, Y. Z. Sun, J. M. Sun, J. Q. Ma and C. Cheng, Biochim. Biophys. Acta, 2012, 1820, 1693–1703 CrossRef CAS PubMed.
- S. Basu, Toxicology, 2003, 189, 113–127 CrossRef CAS.
- T. Jayakumar, M. Sakthivel, P. A. Thomas and P. Geraldine, Chem. Biol. Interface, 2008, 176, 108–120 CrossRef CAS PubMed.
- J. Q. Ma, J. Ding, L. Zhang and C. M. Liu, Food Chem. Toxicol., 2014, 64, 41–48 CrossRef CAS PubMed.
- B. Halliwell and J. M. Gutteridge, J. Lab. Clin. Med., 1992, 19, 598–620 Search PubMed.
- A. C. Reddy and B. R. Lokesh, Mol. Cell. Biochem., 1992, 111, 117–124 CAS.
- C. Rosnoblet, U. M. Vischer, R. D. Gerard, J. C. Irminger, P. A. Halban and E. K. O. Kruithof, Arterioscler., Thromb., Vasc. Biol., 1999, 19, 1796–1803 CrossRef CAS.
- Z. Zhang, X. Chen, L. Li, K. Zhang, S. Tian, H. Gao and H. Li, Neurol. Sci., 2013, 34, 1605–1611 CrossRef PubMed.
- B. Engelhardt, Cell Tissue Res., 2003, 314, 119–129 CrossRef CAS PubMed.
- X. Zhang, X. Li, M. Li, J. Ren, K. Yun, Y. An, L. Lin and H. Zhang, Eur. J. Pharmacol., 2015, 755, 58–65 CrossRef CAS PubMed.
- J. P. Melchor and S. Strickland, Thromb. Haemostasis, 2005, 93, 655–660 CAS.
- M. P. Tsantarliotou, N. A. Kokolis and A. Smokovitis, Theriogenology, 2008, 69, 458–465 CrossRef CAS PubMed.
- O. Nicole, F. Docagne, C. Ali, I. Margaill, P. Carmeliet, E. T. Mackenzie, D. Vivien and A. Buisson, Nat. Med., 2001, 7, 59–64 CrossRef CAS PubMed.
- P. V. Massey, B. E. Johnson, P. R. Moult, Y. P. Auberson, M. W. Brown, E. Molnar, G. L. Collingridge and Z. I. Bashir, J. Neurosc., 2004, 24, 7821–7828 CrossRef CAS PubMed.
- S. C. Gupta, R. Yadav, R. Pavuluri, B. J. Morley, D. J. Stairs and S. M. Dravid, Neuropharmacology, 2015, 93, 274–284 CrossRef CAS PubMed.
- A. L. Samson, R. J. Borg, B. Niego, C. H. Y. Wong, P. J. Crack, Y. Tang and R. L. Medcalf, Blood, 2009, 114, 1937–1946 CrossRef CAS PubMed.
- I. A. Zervos, E. Nikolaidis, S. N. Lavrentiadou, M. P. Tsantarliotou, E. K. Eleftheriadou, E. P. Papapanagiotou, D. J. Fletouris, M. Georgiadis and I. A. Taitzoglou, Toxicol. Sci., 2011, 36, 423–433 CrossRef CAS.
- M. A. Papandreou, M. Tsachaki, S. Efthimiopoulos, P. Cordopatis, F. N. Lamari and M. Margarity, Behav. Brain Res., 2011, 219, 197–204 CrossRef CAS PubMed.
- J. B. Melo, P. Agostinho and C. R. Oliveira, Neurosci. Res., 2003, 45, 117–127 CrossRef CAS.
- O. Cioanca, M. Hancianu, M. Mihasan and L. Hritcu, Neurochem. Res., 2015, 40, 952–960 CrossRef CAS PubMed.
- J. H. Meyer, N. Ginovart, A. Boovariwala, S. Sagrati, D. Hussey, A. Garcia, T. Young, N. Praschak-Rieder, A. A. Wilson and S. Houle, Arch. Gen. Psychiatry, 2006, 63, 1209–1216 CrossRef CAS PubMed.
- M. Bortolato, K. Chen and J. C. Shih, Adv. Drug Delivery Rev., 2008, 60, 1527–1533 CrossRef CAS PubMed.
- L. L. Guo, Z. Z. Guan, Y. Huang, W. Y. L. Ang and J. S. Shi, Exp. Toxicol. Pathol., 2013, 65, 579–584 CrossRef CAS PubMed.
- H. Yu, L. Zheng, L. Yin, L. Xu, Y. Qi, X. Han, Y. Xu, K. Liu and J. Peng, Int. Immunopharmacol., 2014, 19, 233–244 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |