
Molecular devices based on reversible singlet oxygen binding in optical and photomedical applications
Mikhail A.
Filatov
* and
Mathias O.
Senge
School of Chemistry, SFI Tetrapyrrole Laboratory, Trinity Biomedical Science Institute, Trinity College Dublin, the University of Dublin, 152-160 Pearse Street, Dublin 2, Ireland. E-mail: filatovm@tcd.ie
First published on 31st May 2016
Abstract
Polycyclic aromatic hydrocarbons are known to bind singlet oxygen with formation of endoperoxides, which can be released upon heating. This process is now capturing broad attention due to its potential in photomedicine. Singlet oxygen generation upon decomposition of endoperoxides at ambient temperatures represents an alternative to classical photodynamic therapy (PDT), which is limited by hypoxia and reduced light penetration into cancer tissue. Moreover, endoperoxide formation with chromophoric molecules allows for a straightforward modification of the optical properties of the photoactive materials, e.g., shifting of the absorption/emission wavelength which is usually achieved by chemical tuning. This review provides a summary of the latest advances in this area of research along with a discussion of the basic properties of organic endoperoxides.
 Mikhail A. Filatov and Mathias O. Senge | Mikhail A. Filatov received his PhD degree at Moscow State University in 2008 in the field of synthesis of near-infrared phosphorescent dyes. Following he conducted postdoctoral research at the University of Burgundy, the Max Planck Institute for Polymer Research and the Bulgarian Academy of Sciences, working on molecular systems for optical and biomedical applications. He is currently a Marie Skłodowska-Curie research fellow at Trinity College Dublin where he works on new light sensitizers and sensors for photomedical treatments. He is interested in applying molecules with advanced optical properties for the construction of functional materials for healthcare. |
Mathias O. Senge, born in Silbach, Germany, graduated as Diplom-Chemiker from the Philipps Universität Marburg in 1986. After a PhD thesis in plant biochemistry with Prof. Horst Senger in Marburg (1989) and a postdoctoral fellowship with Prof. Kevin M. Smith at UC Davis, he received his habilitation in Organic Chemistry in 1996 at the Freie Universität Berlin. Next he was a Heisenberg fellow at the Freie Universität Berlin and UC Davis and held visiting professorships at Greifswald and Potsdam. In 2002 he was appointed Professor of Organic Chemistry at the Universität Potsdam and since 2005 holds the Chair of Organic Chemistry at Trinity College Dublin. His main interests are synthetic organic chemistry, the (bio)chemistry of tetrapyrroles, photochemistry, -biology and -medicine, structural chemistry, and history of science. |
Design, System, ApplicationSinglet oxygen, a metastable higher energy state of the dioxygen molecule, has been the subject of active studied for several decades. A common method to generate singlet oxygen employs the interaction between oxygen and photo-activated dye molecules. This found broad application in photomedicine due to extremely high reactivity and toxicity of singlet oxygen generated within cells. The search for novel therapeutic agents has led to organic endoperoxides – compounds being formed upon the reaction between singlet oxygen and aromatic hydrocarbon molecules. These compounds have been recognized as advanced carriers, able to store singlet oxygen at low temperature for prolonged periods of time and releasing it by decomposition upon heating. Importantly, the stability of endoperoxides and the timescale of singlet oxygen release can be readily tuned from several hours to several days by means of chemical modification. Moreover, due to reversibility of endoperoxide formation, the process can also be applied to control the optical properties of conjugated aromatic systems, which can be switched by light irradiation or heating. In recent years an increasing number of works applying this process in biomedical and optics research have appeared. In this review we summarize the recent key developments in this area. |
Introduction
The reversible formation of organic endoperoxides has been known for almost a century. In 1926 Dufraise and Moureu for the first time observed the reaction between oxygen and tetraphenyltetracene (rubrene) under light irradiation, forming endoperoxide which almost quantitatively released oxygen upon heating to 120 °C.1 Following this initial report a number of works described studies of this process with polycyclic aromatic hydrocarbons.2 In 1967, Wasserman and Scheffer showed that the oxygen released during decomposition of endoperoxides is in the singlet excited state (1Δg) form. Due to the important functions of singlet oxygen in biological processes,3 this discovery attracted significant attention with respect to the design of new pharmaceuticals. The best studied examples of such systems are alkyl-substituted naphthalene and anthracene derivatives, which can be chemically tuned to release singlet oxygen in biological media.Mechanistically, the interaction of such molecules with singlet oxygen involves a [4+2] cycloaddition of 1O2 with the electron-rich carbon atoms yielding an endoperoxide (EPO) of variable stability (Scheme 1).
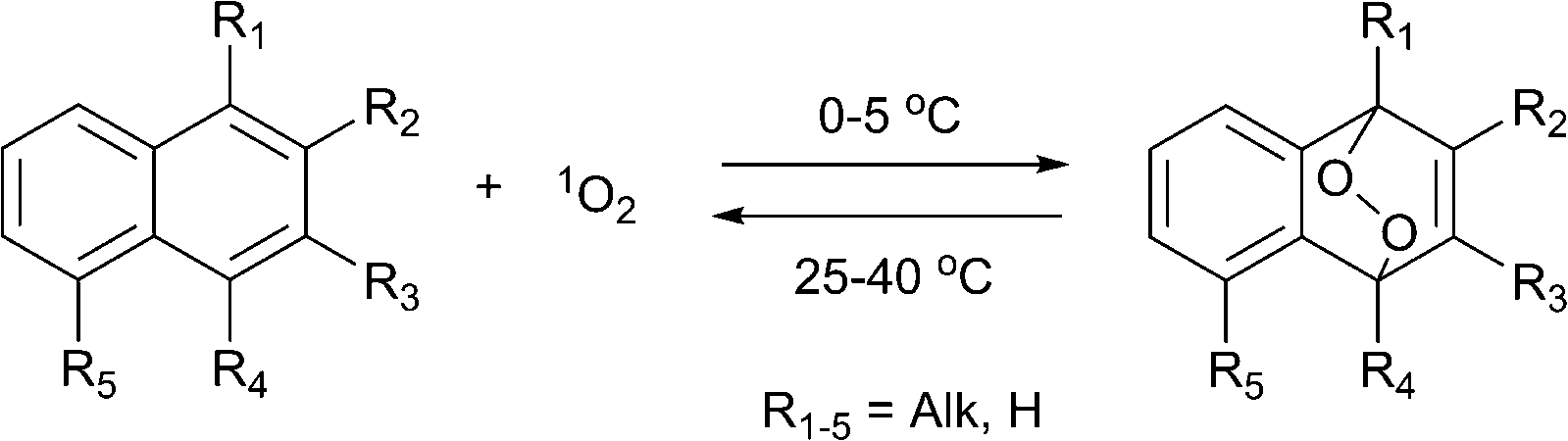 | ||
| Scheme 1 Reversible addition of singlet oxygen to naphthalenes. | ||
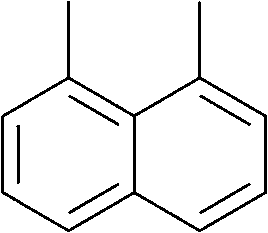
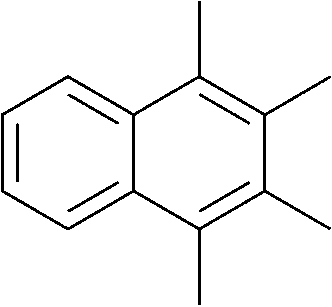
The lifetime of the endoperoxide strongly depends on the temperature. Thus, singlet oxygen can be ‘stored’ at low temperature for prolonged periods of time and then ‘released’ by decomposition upon heating. Decomposition reforms the parent organic molecule and thus, the binding-release cycle can be repeated. The lifetime can be readily tuned from several hours to several days by means of chemical modification of the parent compound (Table 1). The main structural effect that determines EPO stability is the number of fused rings in the aromatic system. Introduction of electron-rich substituents to the aromatic system increases the EPO stability in the following order H < C6H5 < CH3 < OCH3 < NR2.4 Additionally, steric effects play an important role and modulate the reactivity of the aromatic system towards 1O2. For example, 1,8-dimethylnaphthalene can form EPO of much higher stability compared to the 1,4-isomer (Table 1). This is a result of changes in geometry during conversion of naphthalene into the endoperoxide and vice versa. 1,8-Dimethyl naphthalene has a higher energy due to methyl–methyl repulsion and thus EPO decomposition is more endothermic than for 1,4-naphthalene EPOs.5
| Naphthalene | Endoperoxide | t 1/2, h |
|---|---|---|
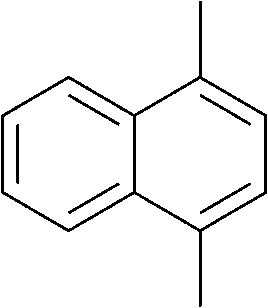
|
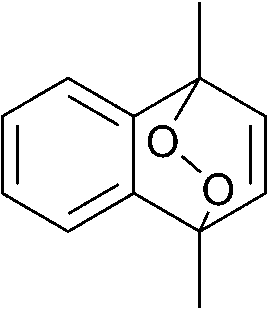
|
5 |
|
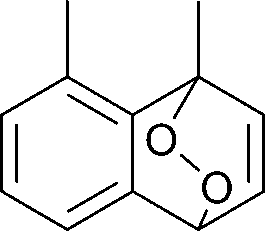
|
30 |
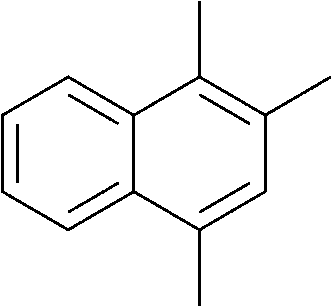
|
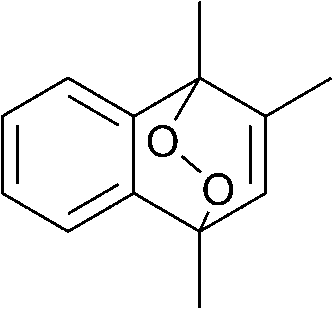
|
70 |
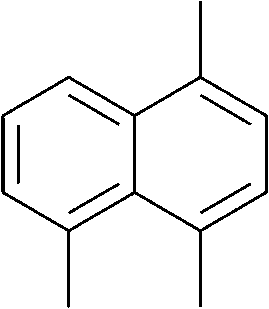
|
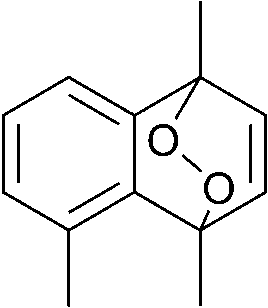
|
290 |
|
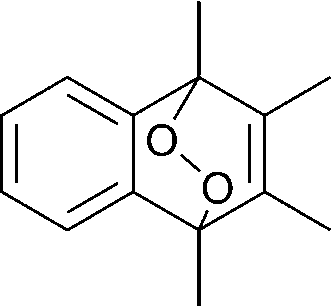
|
∞ |
The cycloreversion of the endoperoxide competes with a homolytic cleavage of the peroxide bond leading to biradical species and furthermore to rearrangements to epoxides or quinones.6 While this process must be taken into account for anthracene and higher acenes, the cycloreversion of alkyl-substituted naphthalenes is highly selective and not accompanied by rearrangement side-reactions.7
The main driving force for current studies on EPOs is the activity of 1O2 against bacteria, viruses, and malignant cells.8 Singlet oxygen is extremely reactive and toxic: under physiological conditions it only diffuses a distance of about 2 μm before reacting or relaxing to its ground state. Thus, biomolecules in solution are oxidized if they react with singlet oxygen before it deactivates. Due to this property singlet oxygen is widely used as the medically active agent in photodynamic therapy (PDT).9 First described by von Tappeiner at the beginning of the 20th century,10 it is nowadays recognized as one of the most potent anticancer treatments. The process involves light excitation of a tissue-localized photosensitizer (PS), usually resulting in a triplet excited state, and further energy transfer from PS to triplet oxygen, dissolved in the tissue, yielding 1O2. Formation of singlet oxygen and other reactive oxygen species results in oxidative damage of cells. Although PDT shows great promise for clinical application, it is limited in many aspects, e.g., low transparency of human tissue11 and hypoxia of cancer cells.12 An attractive alternative is the thermal release of 1O2 from EPOs that are selectively delivered into malignant tissue.
On the other hand, formation of EPOs can also be exploited in photochromic systems for reversible changes of optical properties, which can be induced by light or heating. Pioneering works by Schmidt and co-workers established the guidelines for the structural design of highly reversible photochromic EPO systems,13 and were followed by reports on the photooxidation of acenes resulting in a modulation of the spectral features.14 Due to the growing interest in new chromophores for optoelectronic applications EPOs formation has attracted attention as a tool to modify the HOMO–LUMO gap and emission properties.15
In recent years an increasing number of works describing reversible binding of oxygen for the development of highly reversible photochromic systems and specific sources or traps of 1O2 have appeared. This is partly driven by the discovery of new classes of molecules capable of reversible singlet oxygen binding such as dimethyldihydropyrenes16 and 2-pyridones.17 The latter provides a scaffold that allows for straightforward chemical modification and, notably, ensures reasonable solubility in water, in contrast to naphthalene and anthracene derivatives.
Some earlier review articles covered the synthesis, mechanistic aspects and applications of reversible singlet oxygen binding systems.2 Here, we provide an overview of recently reported molecular systems exploiting reversible singlet oxygen binding relevant to optical and biomedical applications. The latter are illustrated in Fig. 1. Our main focus will be on two perspectives of the reaction of singlet oxygen with aromatic systems: (1) for the modification of optical properties, e.g., shifting of absorption and emission bands; on and off switching of photo-induced electron transfer; and (2) for biomedical applications relying on the cytotoxic effect of 1O2, e.g., photodynamic therapy used for anti-cancer and anti-bacterial therapies.
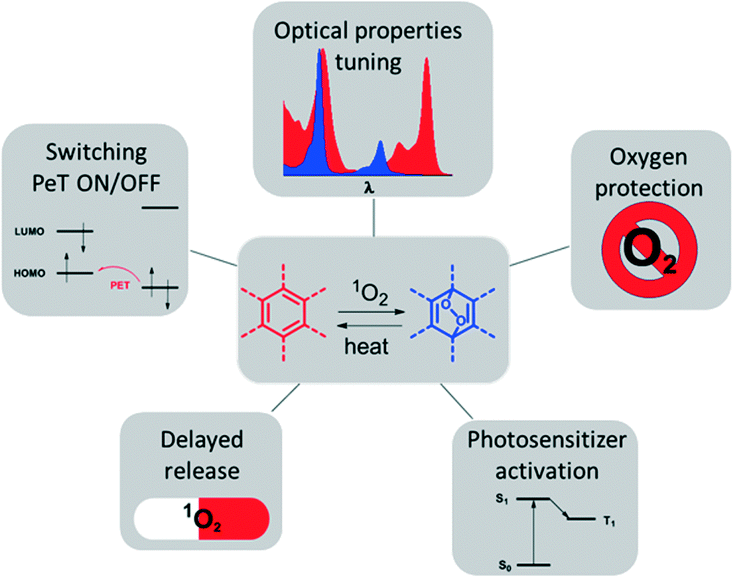 | ||
| Fig. 1 Diagram illustrating potential applications of reversible binding of singlet oxygen. | ||
Endoperoxide formation as a tool to switch optical properties
Photochromic systems
Binding of singlet oxygen to aromatic system leads to a loss of conjugation and consequently affects the HOMO–LUMO gap. This, in turn, results in a blue-shift of endoperoxide absorption and emission bands with respect to the parent aromatic hydrocarbon. Taking into account the reversible character of this reaction, it offers particular potential for the development of photochromic switches.EPOs as components of photochromic systems have been reported in a number of studies.2 9,10-Diphenylanthracene and its derivatives are particularly well suited for the target application due to the enhanced thermal stability of the endoperoxide, which decomposes only above 100 °C. Particularly, compounds 1 and 2 and similar derivatives were found to be useful as switches due to a striking difference in the fluorescence intensity caused by oxygen binding.18 Due to the large spectral shifts this has led to their use in chemical actinometry19 and an ultra-sensitive radiation dosimetry system was developed (Fig. 2).20
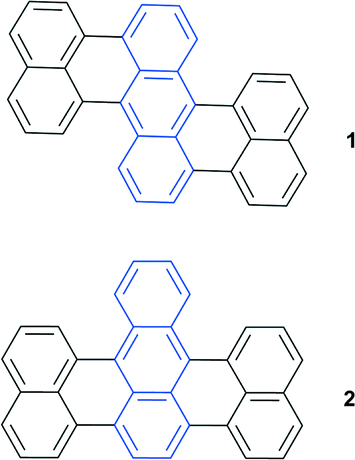 | ||
| Fig. 2 Photochromic switches based on anthracene derivatives. | ||
The formation of endoperoxides from alkyloxy–phenylanthracene 3 (Scheme 2) has been used in regenerative photolithography.21 Due to the long alkyl chain 3 can form transparent films on glass substrates. This allows ‘write’ by applying laser irradiation, ‘erase’ by heating, and ‘re-write’ by irradiation.22
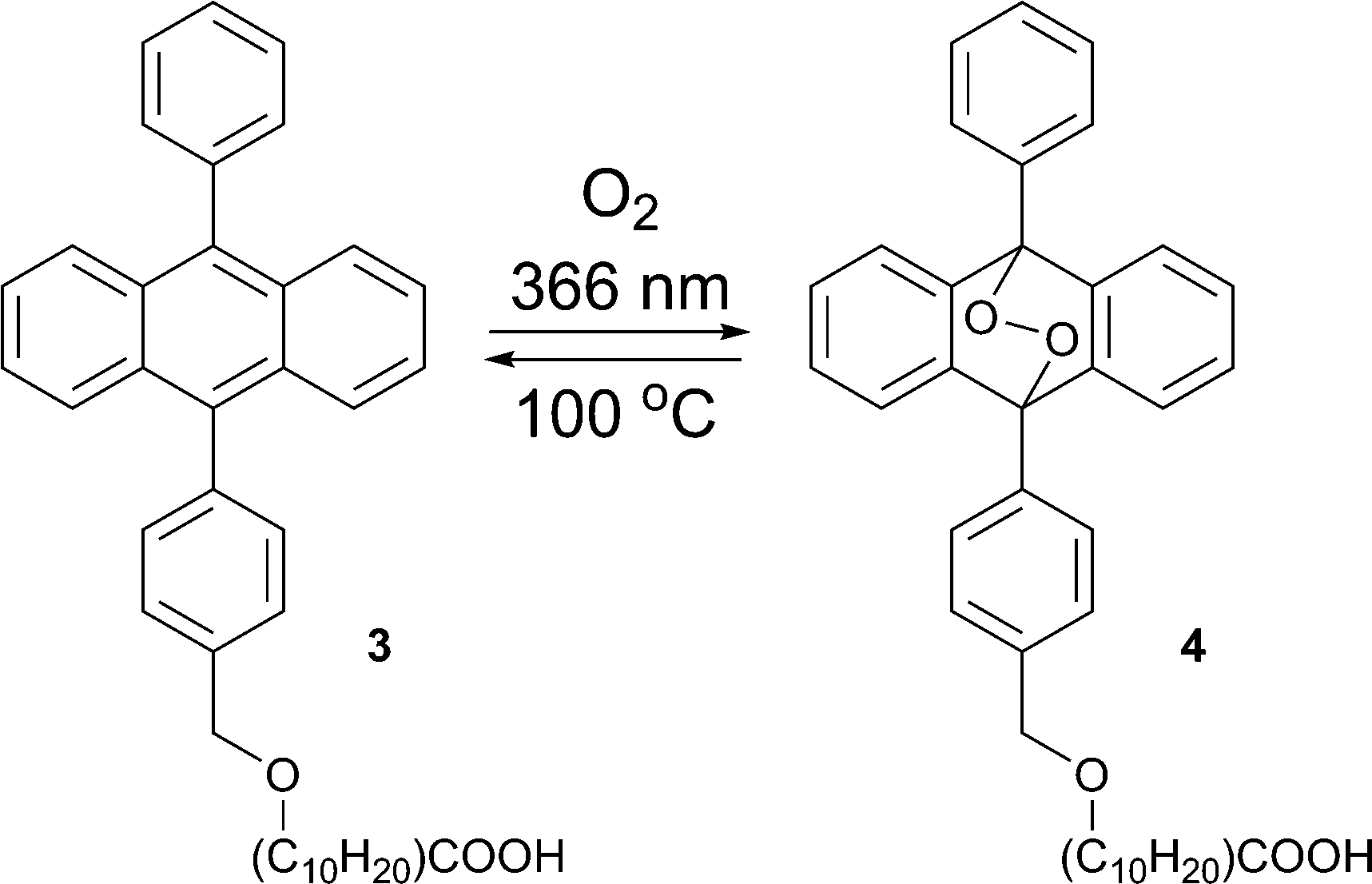 | ||
| Scheme 2 Formation of endoperoxide 4 from the anthracene derivative 3. | ||
Endoperoxides are usually prepared via sensitized photooxygenation23 or peroxidation (with the chemical source of singlet oxygen based on the H2O2–Na2MoO4 system).24 The latter is especially useful for large scale synthesis and the preparation of water-soluble derivatives.25 In both methods the yields are higher in deuterated solvents, as the lifetime of singlet oxygen is 10–1000 times longer compared to protonated solvents.23 However, this makes endoperoxide synthesis costly. Moreover, the endoperoxides have to be isolated and purified under cooling, an inconvenient method especially when column chromatography is required.26 An alternative approach uses self-sensitized singlet oxygen addition.27
Due to low values of intersystem crossing (ISC) of most aromatic hydrocarbons, or, in other words, inefficient triplet excited state formation upon light absorption, their illumination does not result in selective [4+2] oxygen addition, but rather delivers a mixture of products. Introduction of a heavy atom, e.g., Br, I or a metal into the molecule would increase ISC, thus providing self-sensitization of singlet oxygen and securing selective endoperoxide formation. Similarly, this can be achieved by incorporation of endoperoxide forming hydrocarbons into dyads with conventional 1O2 sensitizers, such as porphyrins or phthalocyanines.
The formation of such self-sensitized endoperoxides was first noticed by Rodgers and co-workers with 1,6,10,15,19,24,28,33-octabutoxynaphthalocyanine 5.28 Compound 5 reacted with oxygen in solution during steady-state photolysis with an Ar ion laser and formed an endoperoxide 6 where the oxygen had added to the benzene ring bearing butoxy substituents (Scheme 3). Product formation involved singlet oxygen generated by triplet energy transfer from the photoexcited tetrapyrrole chromophore. The product was found to decompose back to starting material upon heating or under light irradiation.
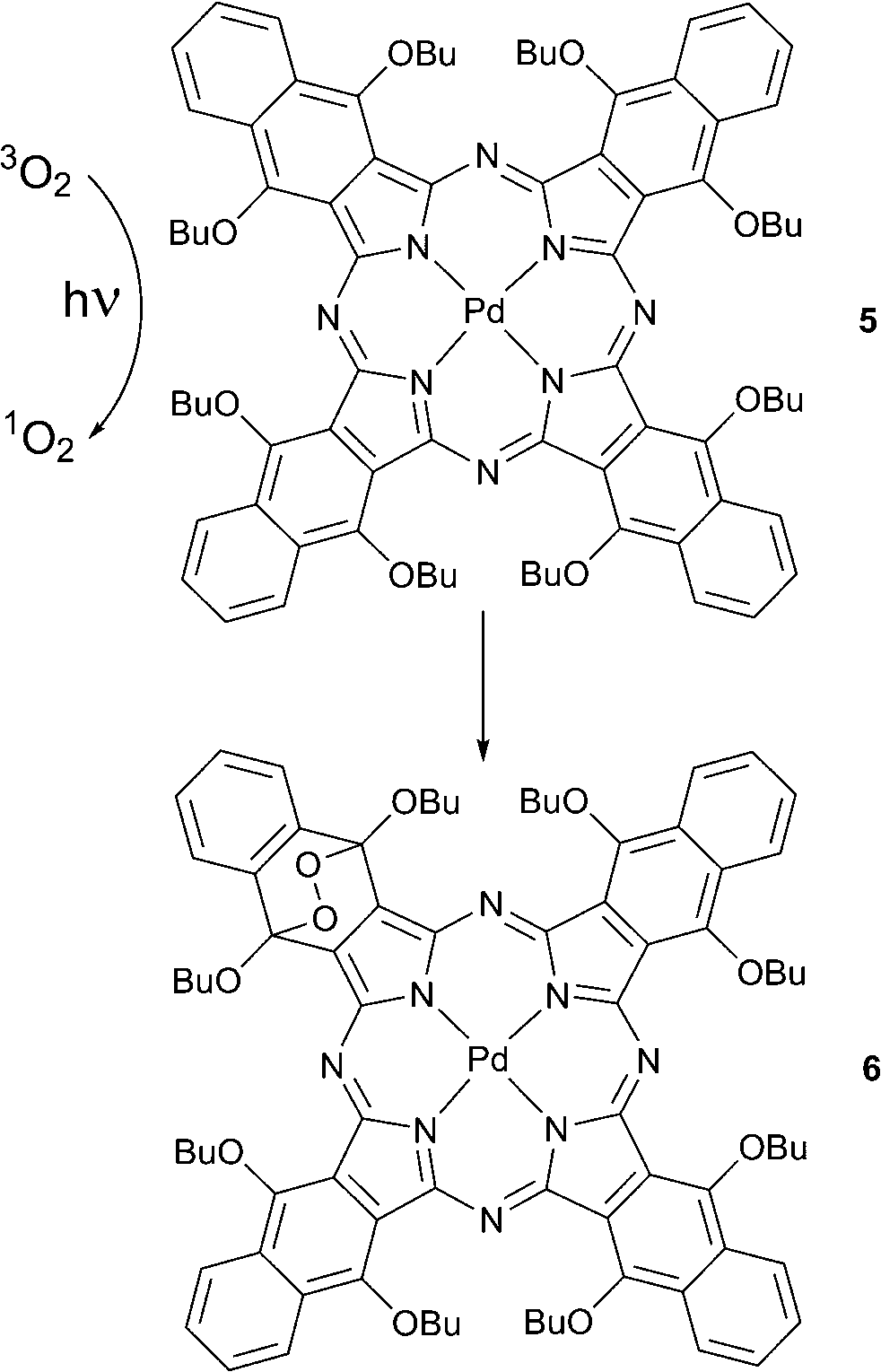 | ||
| Scheme 3 Endoperoxide of octabutoxynaphthalocyanine. | ||
Later Freyer and co-workers reported (octaphenyltetraanthraporphyrazinato)palladium(II) 7 that underwent a self-sensitized photoreaction in the presence of oxygen to form the substituted palladium phthalocyanine 8 with four endoperoxide bridges (Scheme 4).29 The product possessed photophysical properties similar to standard palladium phthalocyanines. The complex bearing four endoperoxide bridges releases singlet oxygen upon excitation by two-photon absorption (TPA) at 662 nm. The photocycloreversion can occur at all four endoperoxide subunits recovering the starting porphyrazine 7. This system is of special interest for applications, e.g., for photodynamic therapy in tissue, because in this case the penetration depth of radiation is greater due so so-called ‘transparency window of tissue’. Molecule 8 may also serve as an internal source of singlet oxygen a crucial aspect for PDT of hypoxic tumors.
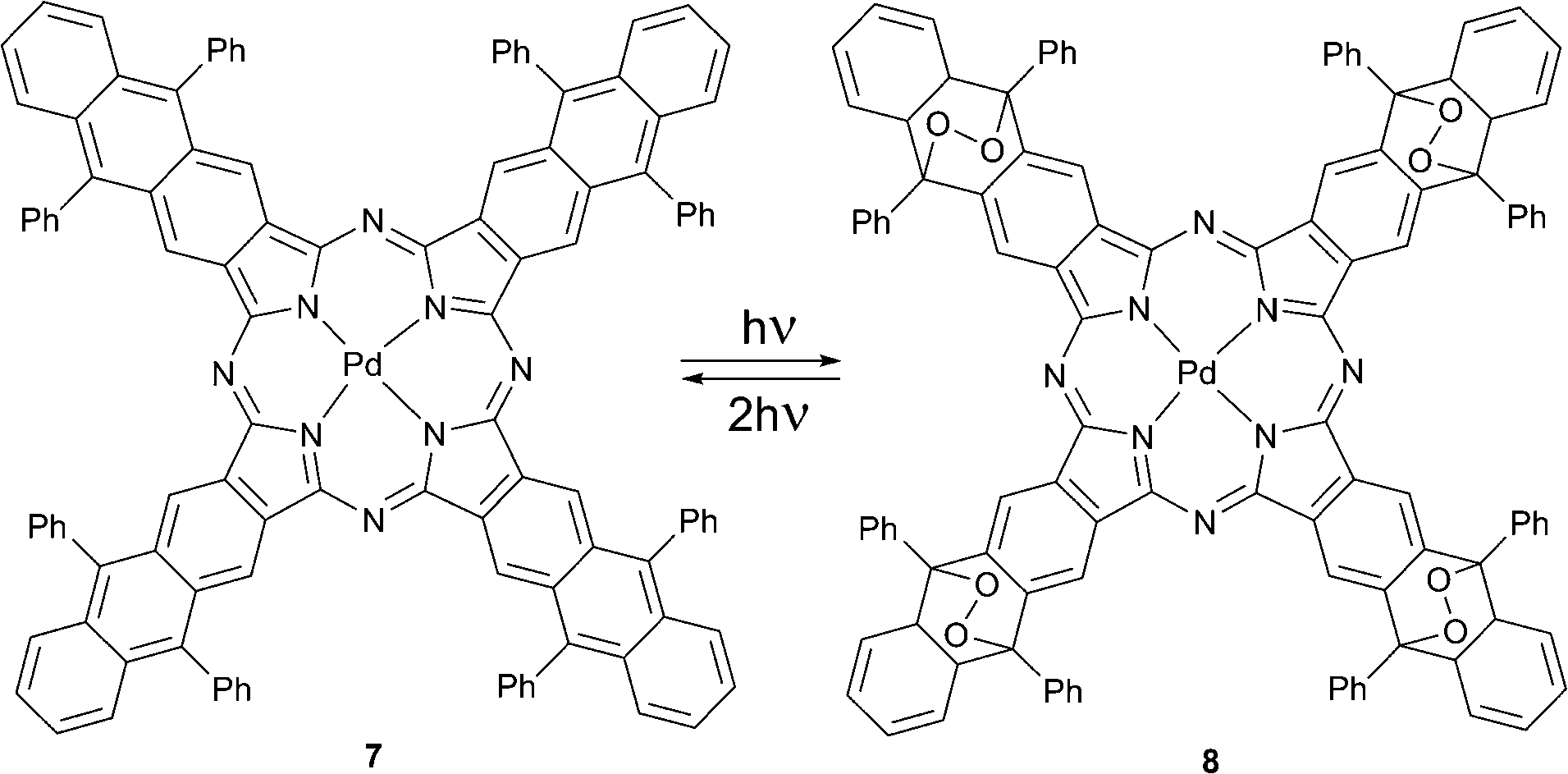 | ||
| Scheme 4 Formation of EPO 8 and its two-photon absorption induced oxygen release. | ||
A self-sensitized process resulting in binding four oxygen molecules was recently described for tetraanthraporphyrins (TAP), a newly explored class of near-IR light sensitizers relevant to triplet–triplet annihilation photon up-conversion (TTA-UC) applications.30 Upon interaction with molecular oxygen the excited state of Pd(II) TAP 9 produced singlet oxygen, followed by formation of endoperoxide (Scheme 5). The reaction was found to be irreversible, showing no regeneration of starting material upon heating. Notably, the absorption and emission bands of the product showed a blue-shift of about 200 nm compared to 9 (Fig. 3). Thus, oxygen addition of TAP sensitizer can be applied for tuning the excitation wavelength of TTA-UC systems based on rubrene as an emitter.31
 | ||
| Scheme 5 Self-sensitized formation of endoperoxides on tetraanthraporphyrin 9. | ||
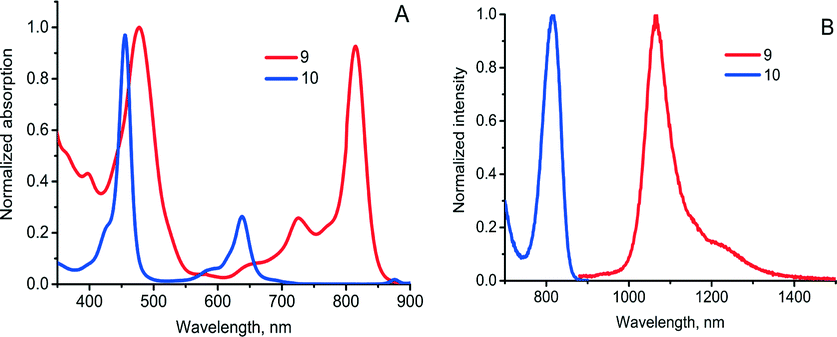 | ||
| Fig. 3 Optical properties of TAP 9 and its endo-peroxide 10. (A) absorption and (B) phosphorescence in deoxygenated toluene solution. | ||
Recently, Cobo, Royal and co-workers reported a new photochromic self-sensitized 1O2 binding system based on dimethyldihydropyrene–cyclophanediene (DHP–CPD) couple.32 Under optical and thermal stimuli this system can act as an efficient oxygen carrier and singlet oxygen generator. The colored DHP (“closed” form) can be converted into the colorless CPD isomer (“opened” form) under visible-light irradiation. Back conversion can then be accomplished either by UV-irradiation or heating.
The singlet oxygen binding and release processes in this case is critically dependent on the substituents in the DHP core. Generation of the endoperoxide from the simple dimethyldihydropyrene takes place with low yields and is not suitable for practical use.33 In contrast, the pyridinium substituted DHP 11 showed a high efficiency of the cycloaddition reaction. This was interpreted as modulation of the energy states by pyridinium groups: the electronic excited state initiating the DHP → CPD conversion becomes the lowest excited state in molecule 11.34 This significantly lowers the energy required for the conversion (from green to red light).
Irradiation of 11 with light (λ > 630 nm) in solution led to quantitative formation of the thermodynamically stable, colorless cyclophanediene 12 (Scheme 6). Irradiation in air-saturated solutions resulted in further formation of the endoperoxide 13. In this case the DHP derivative 11 plays the role of 1O2 sensitizer. Thus, photogenerated 1O2 reacts with CPD 12 to yield the corresponding EPO. Singlet oxygen can then be thermally released upon heating 13 at 35 °C for several hours. UV-spectroscopy indicated the quantitative recovery of DHP 11 within several hours, proving the absence of degradation or formation of rearrangement products. The singlet oxygen yield was found to be >85% (measured using the reaction of 13 with 2,3-dimethyl-2-butene).
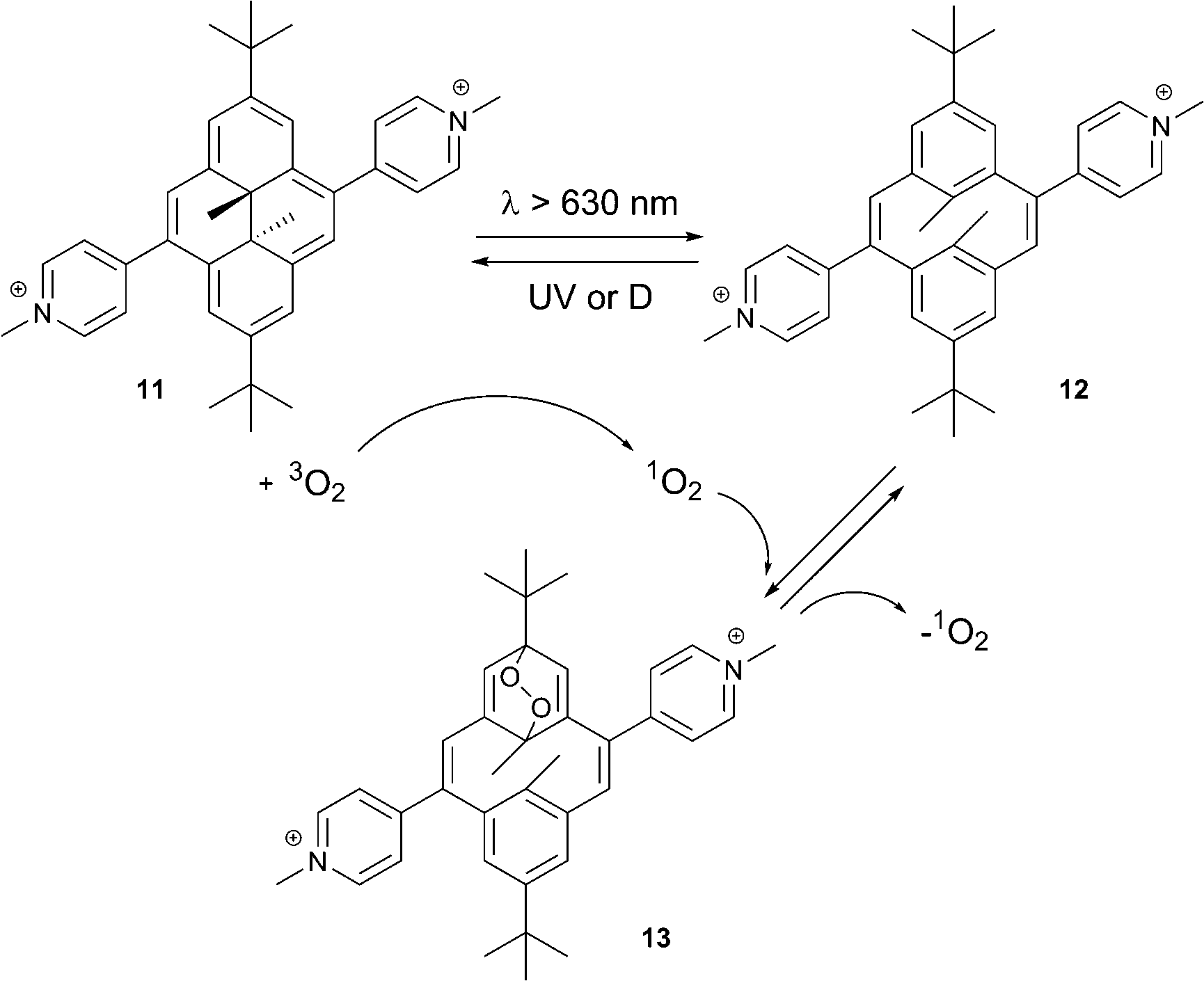 | ||
| Scheme 6 Conversion processes between the dimethyldihydropyrene–cyclophanediene (DHP–CPD) photochromic couple.32 | ||
DHP derivatives were shown to generate and release 1O2 on indium tin oxide surfaces.35 This process potentially allows the application of DHP as photoresistor and in photolithography. The required DHP derivative 14, functionalized with a silane subunit was prepared as outlined in Scheme 7. Visible light irradiation (λ > 630 nm) of the surface under air at room temperature resulted in the formation of the immobilized endoperoxide, as indicated from UV-visible spectroscopy by the disappearance of compound 14’s absorption bands. The back reaction took place with high yield either thermally (5 h at 45 °C) or upon UV light irradiation. However, after 6 cycles a slight degradation was observed as the absorption of 14 decreased by 30% and thus the system requires further optimization.
 | ||
| Scheme 7 Preparation of DHP 14 for anchoring to ITO surfaces. | ||
Protection of excited states from deactivation
Photonic applications based on triplet excited state generation36 are seriously limited due to susceptibility of triplet states towards quenching by molecular oxygen. The utility of corresponding applications is critically dependent on protection of corresponding materials from molecular oxygen.37 A “passive protection” involves barrier materials for packaging, sealing, or encapsulation of the active substances, which prevent oxygen penetration, e.g., encapsulation into polymer films38 or nano-39 and microcarriers.40 An alternative approach is “active protection”, which is based on the use of oxygen scavenging species which react either with triplet or singlet oxygen and thus prevent quenching of excited states.41Similarly, we reported a new strategy for the protection of a phosphorescent palladium(II) tetrabenzoporphyrin from quenching based on its chemical modification with endoperoxide forming groups which do not affect the photophysical properties of the porphyrin macrocycle.42 Owing to the deoxygenation ability, 15 acts as a “self-healing” triplet sensitizer.
When irradiated with a laser or kept under daylight in air-saturated solution, 15 formed a mixture of endoperoxides or ultimately, compound 16 upon complete addition (Scheme 8). Absorption and emission spectra of 15 and 16 (Fig. 4A) showed that binding of oxygen did not affect optical properties, particularly the phosphorescence quantum yields. Moreover, during short time laser irradiation of the sample (minutes or less, depending on excitation intensity), dissolved oxygen was bound to the sensitizer, performing real-time “deoxygenation” of the sample. Due to the decrease of the quencher concentration, the phosphorescence efficiency increased.
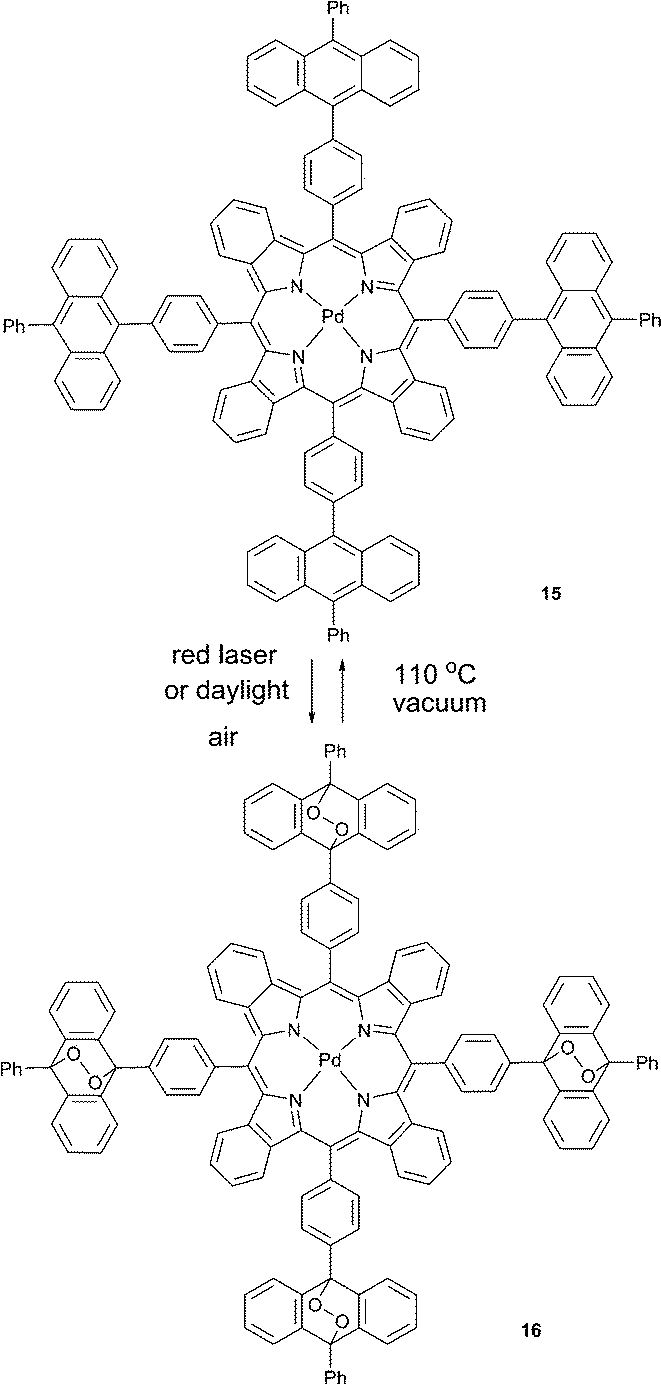 | ||
| Scheme 8 Reversible oxygen binding on the palladium benzoporphyrin sensitizer 15. | ||
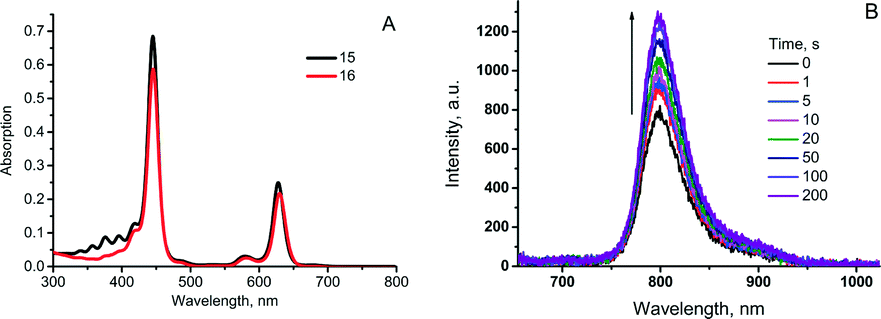 | ||
| Fig. 4 (A) Change of absorption upon transformation of 15 into EPO 16. (B) Changes in phosphorescence intensity at 798 nm for toluene solution of 15 prepared in oxygenated atmosphere during continuous irradiation by red laser (633 nm, 100 μW cm−2). | ||
Enhancement of porphyrin 15 phosphorescence could be achieved in a local area (a spot) of excitation. In the course of the measurement, the same laser beam being used for the excitation of phosphorescence can simultaneously cause local deoxygenation (Fig. 4B). Release of bound oxygen molecules could be achieved upon heating 16 in solid form at 100–110 °C in vacuum for 3–5 h. Complete recovery of the anthracene absorption and mass-spectrometry data proved the reversible oxygen binding-release cycle.
Compound 15 is a prospective material for application in optoelectronic devices. Many of such devices (e.g., OLEDs) have to be fabricated under almost O2-free conditions and must be equipped with protective plastic layers to prevent O2 and water penetration in order to maximize their lifetime. Much research and effort has been put into improved fabrication methods and appropriate device encapsulation to mitigate these environmental effects. Here, using the proposed triplet sensitizer types becomes feasible, as it provides additional protection of the excited states population, complementary to the decrease of oxygen permeability by the plastic layer. Although such a protection strategy is limited by the capacity of the sensitizer to bind no more than four oxygen molecules, it may have potential in applications which require very low oxygen concentration levels.
Intramolecular oxygen transfer
An intriguing, but yet almost unexplored, approach towards modifying the optical properties of molecules via interaction with singlet oxygen is its intramolecular transfer between different aromatic subunits. Broadly studied FRET, PET and proton transfer processes allow selective switching of optical properties and are tuned by chemical modification of donor and acceptor building blocks. If one regards singlet oxygen as a species being transferred, then the donor and acceptor should represent two aromatic hydrocarbons with low and high endoperoxide stability, respectively. An ultimate benefit of using such an approach would be the precise thermal control over absorption and emission intensities, which is interesting from a viewpoint of optical temperature sensing.43Recently, the first example of such an intramolecular 1O2 transfer between two acenes was reported by Linker's group in Potsdam.44 They prepared the ester-linked naphthalene-anthracene dyad 17, wherein the naphthalene endoperoxide moiety plays the role of donor, transferring singlet oxygen to the anthracene acceptor upon temperature increase (Scheme 9).
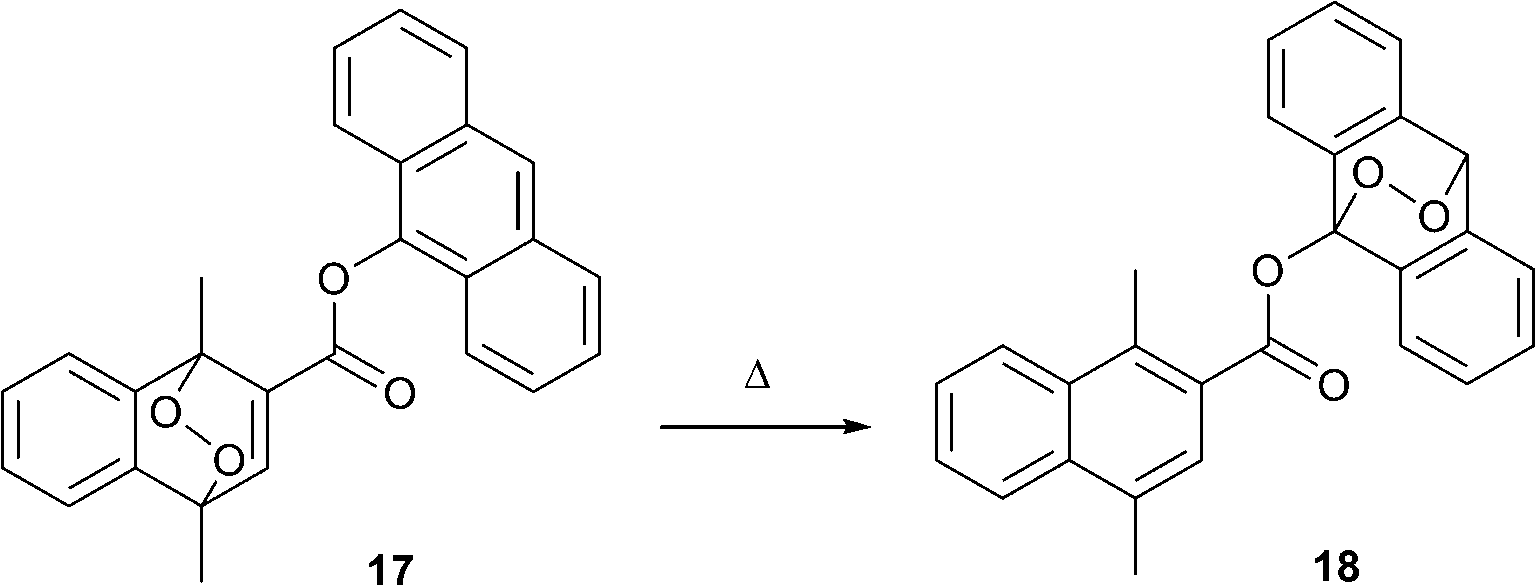 | ||
| Scheme 9 Intramolecular transfer of singlet oxygen in dyad 17. | ||
The process can be monitored by the decrease of the anthracene absorption at 380 nm and the simultaneous increase of the naphthalene absorption band at 280 nm. The efficiency of the transfer is critically dependent on the mutual orientation and distance between donor and acceptor. Thus, when the anthracene and naphthalene endoperoxide subunits were separated by a longer spacer, thermolysis resulted in release of singlet oxygen without transfer to the acceptor.45 At higher temperatures the system tends to evolve free 1O2 instead of intramolecular transfer.
Controlled release of singlet oxygen
Endoperoxides as a source of oxygen for tissue
In regenerative medicine, necrosis of engineered tissue as a result of oxygen depletion is a challenging problem. Necrosis and apoptosis of cells under oxygen-deprived conditions impair the healing process significantly, resulting in failure of vital functions of tissue. Efforts have been dedicated to the development of techniques for supplying hypoxic and anoxic tissue with oxygen.46Jessen and co-workers reported the controlled release of oxygen from water-soluble methylated pyridone endoperoxides as a method to rescue cells from death under anoxic conditions.47 These molecules, combined with ascorbic acid as singlet oxygen quencher, allowed survival of 3T3 fibroblasts (FBs) and rat smooth muscle cells (RSM) under oxygen-depleted conditions.
In order to study the rate and efficiency of oxygen release from pyridone-derived endoperoxides, a series of substituted 2-pyridones was synthesized and then subjected to photooxygenation in the presence of 5,10,15,20-tetraphenylporphyrin. Tri(ethylene glycol) ether moieties were attached to the pyridone via N-alkylation to increase the pyridine endoperoxide stability and solubility in water and the resulting endoperoxides were found to be stable when stored at −20 °C.
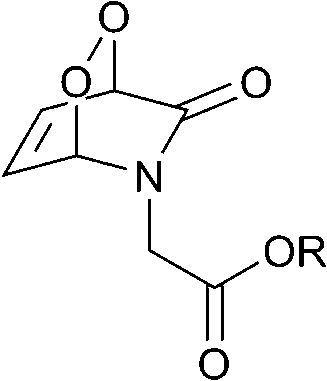
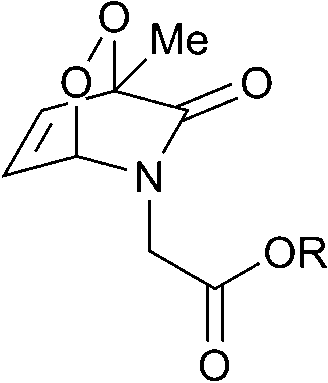
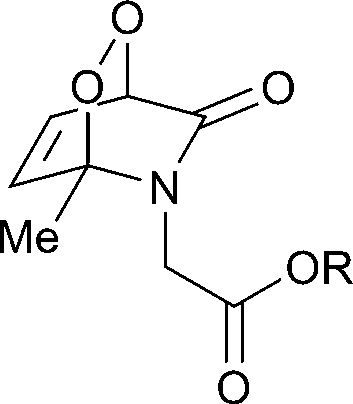
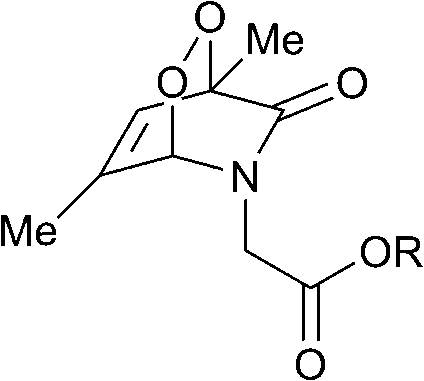
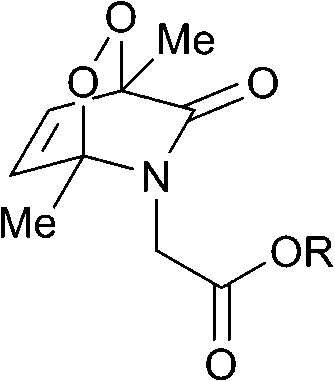
The half-lives of the endoperoxides were measured by 1H NMR spectroscopy at 37 °C in H2O/D2O (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and found to vary from 0.5 to 15 h, depending on the substitution pattern (Table 2). In most cases, thermolysis yielded the starting 2-pyridone and singlet oxygen. However, in certain cases the retro-Diels–Alder reaction was accompanied by a 2-pyridone rearrangement and consequently gave low yields of singlet oxygen. For example, non-methylated 2-pyridone endoperoxides decomposed quickly and unspecifically within 30 min at 37 °C. 3-Methyl-2-pyridone endoperoxide 19 (Scheme 10), the compound with the most favorable properties, i.e. a long half-life combined with a good yield of oxygen, was examined in cell assays.
:1) and found to vary from 0.5 to 15 h, depending on the substitution pattern (Table 2). In most cases, thermolysis yielded the starting 2-pyridone and singlet oxygen. However, in certain cases the retro-Diels–Alder reaction was accompanied by a 2-pyridone rearrangement and consequently gave low yields of singlet oxygen. For example, non-methylated 2-pyridone endoperoxides decomposed quickly and unspecifically within 30 min at 37 °C. 3-Methyl-2-pyridone endoperoxide 19 (Scheme 10), the compound with the most favorable properties, i.e. a long half-life combined with a good yield of oxygen, was examined in cell assays.
| Endoperoxide | t 1/2, h | 1O2 yield, % |
|---|---|---|
|
0.5 | 18 |
|
8.5 | 78 |
|
4 | 50 |
|
13 | 47 |
|
4 | 50 |
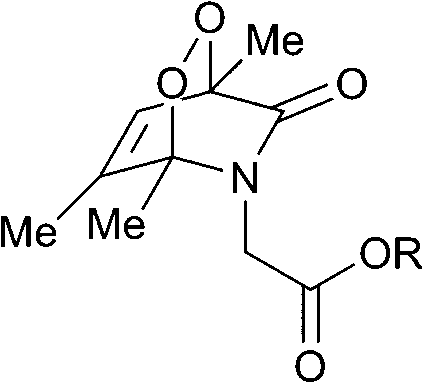
|
15 | 10 |
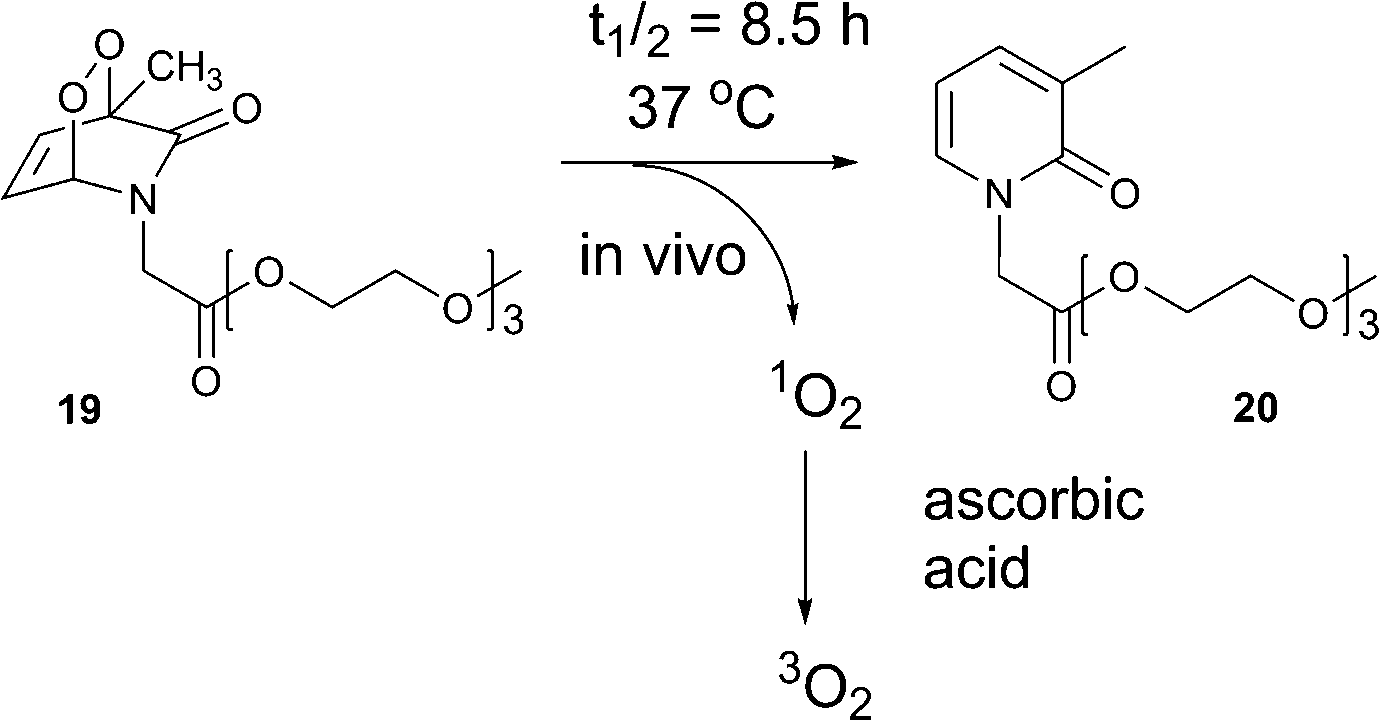 | ||
| Scheme 10 Generation of triplet oxygen upon decomposition of 2-pyridone endoperoxide 19 in the presence of ascorbic acid. | ||
The addition of vitamin C as a physical and chemical quencher was used to convert cytotoxic singlet oxygen produced upon endoperoxide thermolysis into triplet oxygen. Efficient quenching of singlet oxygen by vitamin C was demonstrated through use of the singlet oxygen sensor green assay.
Compound 19, at 50 μg mL−1, was found to be cytotoxic in FBs in the presence of vitamin C under normoxic conditions. However, reducing the endoperoxide concentration to 10 μg mL−1 in the presence of 100 μg mL−1 vitamin C, resulted in loss of any cytotoxicity and significant beneficial effect on cell viabilities under anoxic conditions in FBs and SMC was observed. Vitamin C alone (100 and 200 μg mL−1) had no effect on cell growth, but it enhanced cell survival. These results were compared with the parent pyridone 20, being formed upon thermal decomposition, which also showed no effect on cell growth. Although the half-life time of 19 is 8.5 h, SMCs showed increased cell growth under anoxic conditions for over 4 days in the presence of the endoperoxide (10 μg mL−1) and vitamin C (100 μg mL−1). Thus, the compound efficiently releases singlet oxygen within cells, followed by quenching by vitamin C, giving triplet oxygen that rescued cell growth under strictly anoxic conditions.
Clearly, 2-pyridone endoperoxides, particularly those attached to polymers, represent a promising class of materials for a broad range of medical treatments, particularly those aimed at supporting tissue with oxygen.
Porphyrin sensitizers that trap, store, and release singlet oxygen
Connors and co-workers presented a composite molecule 21 with four 2-pyridone subunits attached at the meso (5,10,15,20) positions of a porphyrin macrocycle. In the presence of oxygen and upon irradiation with light this compound generated, stored and released singlet oxygen bound directly to a porphyrin photosensitizer dye (Scheme 11).481H NMR spectroscopy revealed that this cycle can be repeated a number of times without detectable degradation or formation of side products. Unlike conventional photosensitization methods to produce singlet oxygen, the reversible storage capacity of compound 21 ensures that singlet oxygen will continue to be generated for hours after the excitation light source has been removed.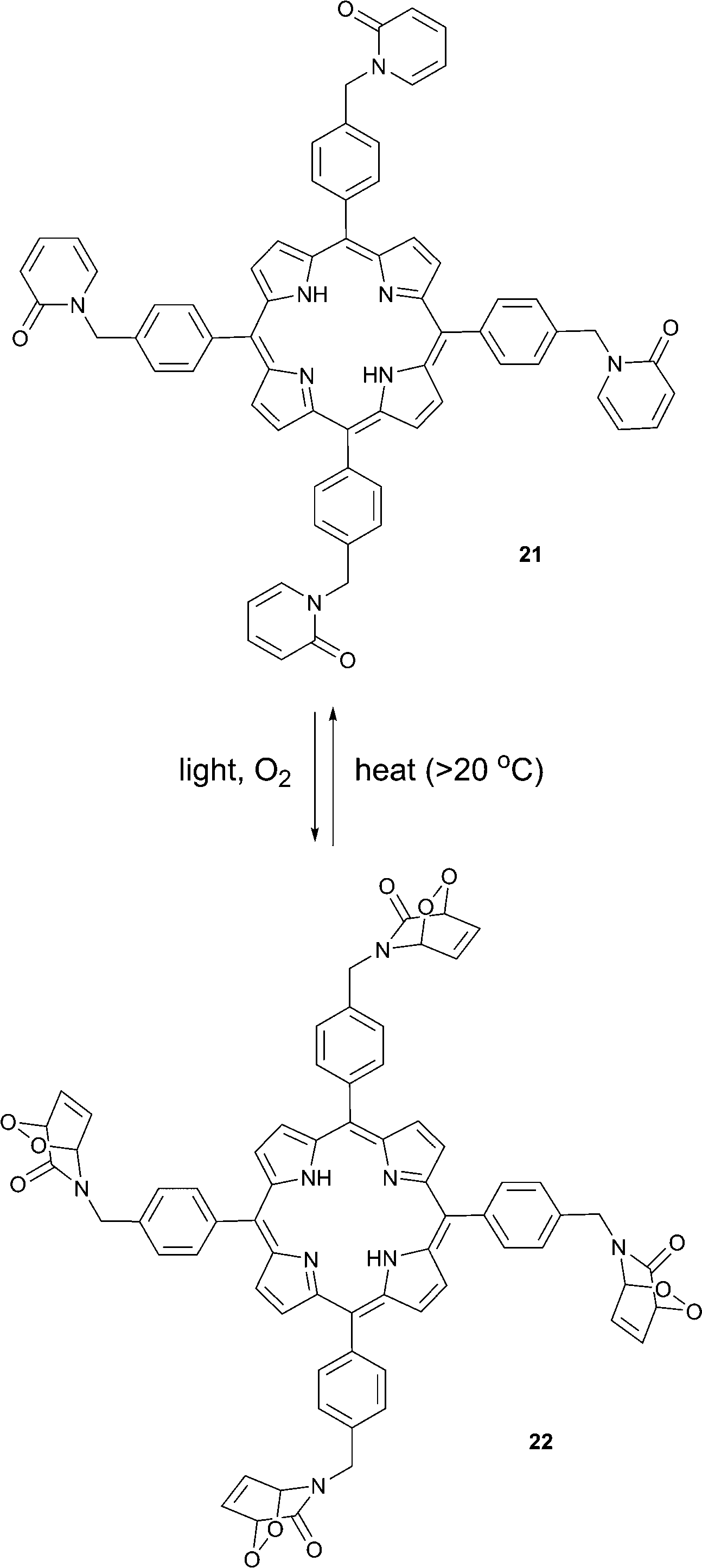 | ||
| Scheme 11 Reversible generation, trapping, and release of singlet oxygen by porphyrin 21. | ||
The corresponding endoperoxide 22 could be obtained upon irradiation of porphyrin 21 in a deuterated solvent under a stream of O2 gas at room temperature. The reaction proceeded without significant formation of by-products, as evidenced by 1H NMR studies. Further heating of 22 in solution to 40 °C resulted in decomposition, recovering the parent compound. Interestingly, here endoperoxide thermolysis was not accompanied by ring rearrangement, particularly the Kornblum–Delamare reaction,49 as opposed to the 2-pyridone derivatives studied by Jessen and co-workers.47
Oxidation of the mustard gas simulant, 2-chloroethyl ethyl sulfide (CEES), by singlet oxygen released in the dark from the molecule 22 was demonstrated, too. Obviously, molecules such as 22, possess significant potential for photomedical applications. Conventional PDT sensitizers produce singlet oxygen only in the presence of light and thus any therapeutic effect is limited to the time of tissue treatment with light. In contrast, when the sensitizer is ‘equipped’ with singlet oxygen traps, such as 2-pyridone moieties, a part of the singlet oxygen formed upon photosensitization is bound to the trapping molecule. A fraction of singlet oxygen formed directly upon sensitization is then responsible for an immediate therapeutic PDT response. Following this initial response, a “slow” phase of the therapeutic effect could occur via singlet oxygen evolution from the trapping moieties. Ultimately, this may result in an increased therapeutic effect of the treatment.
Photothermal release
Metal nanoparticles (NPs) are known to absorb visible light at their surface plasmon resonance and convert it to heat localized near the surfaces of the nanoparticles.50 Heat generated from optically stimulated NPs have been used to kill cancer cells in a localized, noninvasive manner, the so-called photothermal therapy.51Anthracene 9,10-endoperoxides possess half-live times on the scale of years at room temperature and normally release singlet oxygen only when being heated above 100 °C.52 For this reason, their application in biomedical treatments is very limited. However, if a local heating in the desired region of tissue is applied, anthracene EPOs could deliver singlet oxygen in biological systems.
Branda and co-workers were the first to demonstrate that the heat generated near the surfaces of gold nanoparticles can cause endoperoxide decomposition with the formation of singlet oxygen.53 Their approach for the light-induced release of 1O2 is shown in Scheme 6 and uses the NPs system 23 with anthracene anchored to a gold nanoparticle surface through Au–S bonds (Fig. 5). This required preparation of a thiolated anthracene derivative and anchoring to the NPs. The average number of anthracene ligand molecules per nanoparticle was estimated to be approximately 4300. The anthracene endoperoxides formation on the surface could be monitored by UV-vis spectroscopy, as the endoperoxides do not absorb in the visible region, while anthracene possesses characteristic bands in the 350–425 nm region. Upon heating to 95 °C in a solution of tetrachloroethane the endoperoxide moieties released oxygen on a timescale of several days. This property is crucial for applications under biological conditions, as the stability of the endoperoxides ensures no 1O2 release during the delivery period to the tissue. However, when exposed to green laser light (532 nm, 10 Hz, 5 mJ per pulse, 10 ns), the nanoparticles 23 produce heat, sufficient to trigger thermolysis of the anthracene endoperoxide, resulting in release of 1O2. Notably, the process did not result in a significant increase of the temperature of the bulk sample. Conversion of the endoperoxide to the anthracene is rapid and complete within 30 seconds after laser excitation. The release of singlet oxygen was confirmed by reaction with 1,3-diphenylisobenzofuran (DPBF) as an indicator, which showed a decrease in absorption.
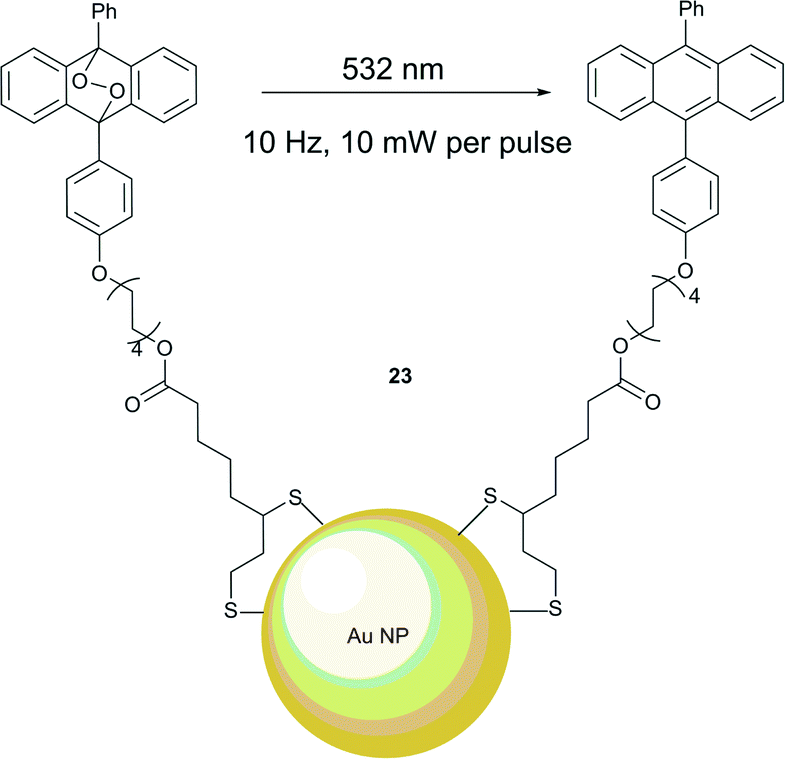 | ||
| Fig. 5 Photothermal release of singlet oxygen from endoperoxide ligands attached to gold nanoparticles. | ||
Photodynamic therapy
Photodynamic therapy (PDT) is a broadly used technique for killing diseased cells by incorporating a combination of light and a light-activated drug, known as a photosensitizer (PS).9 The singlet excited state of the PS formed upon absorption of light undergoes intersystem crossing to populate a triplet excited state. The latter transfers energy to ground-state triplet molecular oxygen, generating singlet oxygen. Singlet oxygen is extremely reactive and toxic for cells. PDT is currently used to treat a wide range of diseases characterized by neoplastic growth, such as various cancers, actinic keratosis, age-related macular degeneration and is used for disinfection and sterilization.54A critical limitation of PDT therapy is associated with the sensitivity of skin to strong light exposure. Systemic PDT causes photosensitivity in patients' skin for up to three months55 following treatment, dependent upon the photosensitizing drug used. Side effects may include pain, burning/stinging sensation, and itchiness. These sensations usually decrease rapidly once the illumination is paused or exposure is terminated. A local anesthetic may be applied to the treated area before or during PDT to relieve a pain. Due to these side-effects and depending on the condition of the skin, PDT light treatment of the patient usually lasts no longer than 30–60 min and requires repetitive procedures, the scheduling of which must take into account the time required for the recovery of irradiated skin zones.
The need for repetitive irradiation procedures is, in part, associated with the short lifetime of 1O2 in biological media, which is susceptible to physical quenching by water molecules. This process leads to ground-state triplet oxygen instead of oxidation reactions with biomolecules within malignant cells. Depending on the solvent, singlet oxygen decays back to the ground state with a half-life time (τ1/2) in the range of 10−6–10−2 s.56 Thus, the therapeutic effect of PDT is limited to the actual time of light treatment, since photosensitized singlet oxygen deactivates instantly. An extension of the singlet oxygen generation timescale without increasing the light irradiation time, which causes side-effects, would allow for a profound improvement of the PDT method. As a solution, a process whereby singlet oxygen is gradually generated during an extended period of time after the light treatment could be applied.
Another limiting factor stems from most tumors developing hypoxic regions, which are highly resistant to chemotherapy and radiotherapy.57 In such regions PDT cannot provide therapeutic action, as the singlet oxygen concentration achieved upon irradiation is too low to induce cytotoxic effects. Moreover, PDT itself causes hypoxia in tissue due to fast consumption of the cellular oxygen.58 A possible solution to overcome hypoxia in PDT is fractional irradiation of the tissue with time periods in between, so that the intracellular oxygen concentration can recover to a normal level.59 Again, effective PDT under hypoxic conditions can be achieved by using chemically generated singlet oxygen during the dark period of fractional PDT, where photosensitized generation of 1O2 is not possible. Thus, a combination of a photosensitizer and a chemical source of singlet oxygen in a single molecule is highly desirable.
From this viewpoint an application of organic endoperoxides has obvious prospects. The decomposition of naphthalene endoperoxides has already been shown to be suitable as a source of singlet oxygen for biomedical applications. Examples include the induction of cellular damage in malignant tissue and the potential utilization of biomimetic endoperoxide-bond cleavage processes for the controlled deactivation of bacteria, viruses, and parasites.60 Singlet oxygen releasing naphthalene endoperoxide derivatives embedded in nanoparticles were shown to induce a very strong cytocidal effect on the proliferation of human breast cancer cells.61 However, the use of endoperoxides in these biomedical applications is still in its infancy due to lack of carrier systems which can ensure: 1) selective targeting of diseased cells and, particularly, 2) prevent endoperoxide decomposition until the drug is delivered into cells. This contrasts with conventional PDT treatments, wherein a photosensitizer drug selectively accumulates in tumor tissue and generates singlet oxygen only if an external stimulus (light) is applied.
Endoperoxides as a source of singlet oxygen for hypoxic tissue PDT
Recently, Yoon, Akkaya and co-workers demonstrated for the first time that photothermal singlet oxygen release from anthracene EPOs on the gold nanorods (GNR), discussed above, can induce apoptosis and lead to cell death.62 The target molecular system involved 9,10-diphenylanthracene EPO attached to the surface of gold nanorods through thiol linkers. Spectroscopic data revealed that each GNR carried approximately 6.5 × 109 endoperoxide molecules. Upon irradiation of the resulting GNR solution at r.t. with 830 nm laser light, singlet oxygen formation took place, as evidenced through trapping with 1,3-diphenylisobenzofuran.The cytotoxic effect of the GNR–endoperoxide generated 1O2 was demonstrated with HeLa cells incubated with GNR for 24 h. After redistribution in the cells the GNRs were found to be located within vacuoles. The cells were then irradiated for 10 min with an 808 nm laser (2.0 W cm−2) followed by cell viability assays. The control compound, GNR without attached EPOs did not generate any oxidative stress in the cell culture during the irradiation period. In contrast, GNR-EPOs caused formation of reactive singlet oxygen species, as indicated by microscopy studies with oxidative stress detection reagents. Addition of sodium azide, known to quench singlet oxygen, resulted in a strong suppression of cell death. The cell viability increased concomitantly to the concentration of azide added, confirming the hypothesis that cytotoxicity is due to singlet oxygen formation from the endoperoxide.
However, it should be noted, that the light intensities required to induce a photothermal effect on gold nanorods are rather high compared to those applied in regular PDT treatments. Clinical PDT protocols are restricted to use light intensities at the lesion surface of not exceeding 200 mW cm−2 in order to avoid skin side-effects. Thus, although photothermal 1O2 is an alternative to a conventional PDT treatment, many efforts are required to optimize the properties of the system.
Fractional photodynamic therapy
Endoperoxide-based therapies promise to overcome one of the major PDT limitations – strong tumor hypoxia. It was previously assumed that a substantial therapeutic effect requires large amounts of singlet oxygen to be produced and thus stoichiometric agents, such as EPOs, would possess very limited capacity to induce cell death. However, the demonstration that even small amounts of singlet oxygen can cause an apoptotic response clearly mandates a reevaluation and opens a new chapter in photomedicinal treatments with oxygen.Akkaya and co-workers reported a bifunctional compound meeting the requirements for enhanced fractional photodynamic therapy.63 When excited with red laser light (λ = 650 nm), the BODIPY dye 24 generated singlet oxygen, which later reacted with the 2-pyridone subunit to form the corresponding endoperoxide 25 (Scheme 12). Upon cessation of irradiation the thus formed endoperoxide underwent thermal cycloreversion producing singlet oxygen in the absence of light.
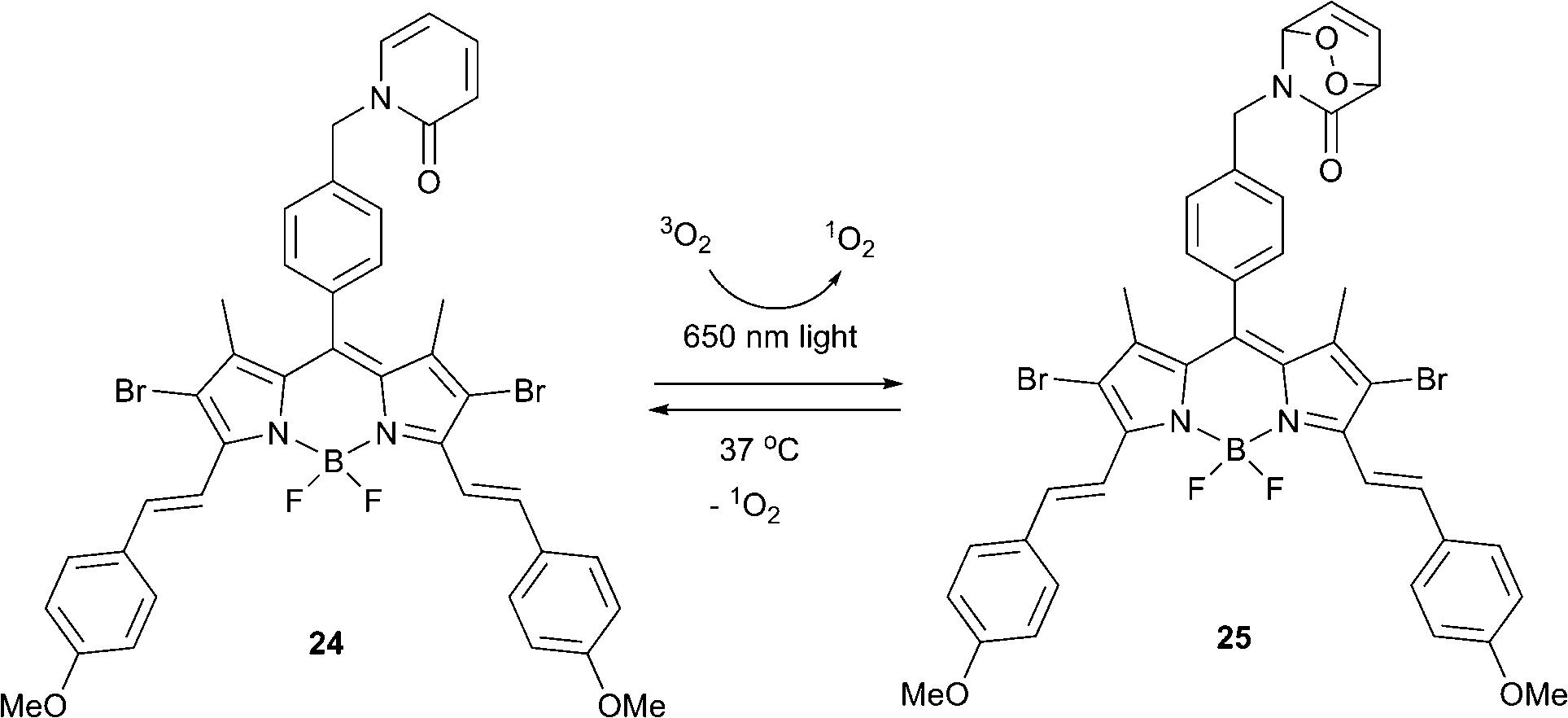 | ||
| Scheme 12 Singlet oxygen generation by the bifunctional sensitizer 24. | ||
The bifunctional photosensitizer was tested in HeLa cell cultures. The non-ionic surfactant cremophor was used to deliver the compound within a micellar structure and cells were exposed to varying concentrations of compounds 24 and 25 for comparison. Irradiation was performed for 10 min every one hour for 24 cycles. Cell viability and cytotoxicity assays showed that even low doses of compound 25 resulted in a significant decrease of cell viability. The CC50 (50% cytotoxic concentration) for EPO 25 was found to be considerably lower than that of 24 after fractional irradiation of both, with a total illumination time of 4 h. This validated that dark production of singlet oxygen by delayed release from endoperoxide fragment in a large difference in cytotoxicity.
Selective activation of photosensitizers by interaction with singlet oxygen
In order to minimize undesired side-effects, including damage to healthy tissue during PDT treatment, photosensitization of 1O2 may be controlled at different levels. Specific approaches towards such a control are based, for example, on PS self-quenching,64 FRET65 or electron transfer.66 Recently, the chemical activation of a photosensitizer specifically in the targeted tissue has emerged as an additional, effective method.67Cosa and co-workers described a photosensitizer that activates upon interaction with 1O2 and thus can be utilized towards the controlled delivery of 1O2 specifically in cells or tissues that are under oxidative stress associated with increased metabolic activity.68 The photosensitizer was based on a two-segment photosensitizer-trap molecule, consisting of a BODIPY dye and a chromanol α-tocopherol ring (Scheme 13). Bromo- or iodo-substituted BODIPY ensured efficient intersystem crossing to the triplet state and production of singlet oxygen, while the chromanol segment provided photo-induced electron transfer (PeT) that competes with ISC, thus effectively reducing the yield of the triplet state.
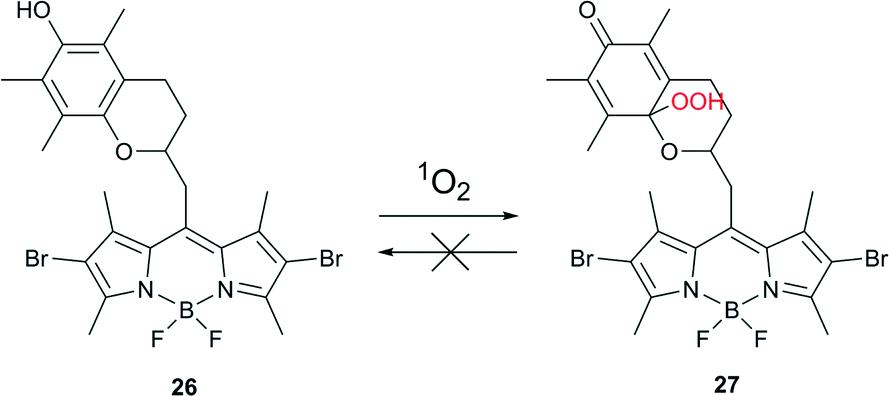 | ||
| Scheme 13 Activation of sensitizer 26via reaction with singlet oxygen. | ||
Singlet and triplet quenching of the BODIPY excited state by chromanol provided two means of prevention for 1O2 formation. Moreover, α-tocopherol, known to be an efficient physical quencher of 1O2, provided a third level of suppression of 1O2 production. Oxidation of a trap segment with 1O2 or other reactive oxygen species unblocked the depopulation of BODIPY excited states and allowed for 1O2 sensitization. Thus compound 26 is a versatile ROS-activatable sensitizer with potential application in tissues where the metabolic imbalance leads to increased ROS production, e.g., cancer cells and wound tissue. The applicability of compound 26 was validated by selective inactivation of ROS-stressed bacteria. Escherichia coli were incubated with 500 nM hydrogen peroxide, known to stimulate ROS production in cells, for 2 h at 37 °C. A drastic decrease in colony forming units was recorded following the combined action of compound 26 and irradiation of stressed cells. No activation was observed for non-stressed healthy cells.
Compound 26 exemplifies a new concept for developing PSs that will enable the controlled delivery of 1O2 into cells where metabolic imbalance leads to a large production of ROS.
Bioimaging
Sensing of singlet oxygen
Reactive singlet oxygen species (ROS), including singlet oxygen, are part of the normal cell signaling system. They play an important role in cell death and stress-response processes. Consequently, recent years have seen more interest in the development of probes for ROS.69The development of detection methods and sensors for 1O2 is of critical importance due to its relevance for antibacterial and cancer treatments.70 Singlet oxygen is best detected by its phosphorescence at 1275 nm. However, as the emission quantum yield is extremely low (<10−6) this requires specialized instrumentation.71 Indirect methods for the detection have been developed based on oxidation of chemical acceptors by 1O2, giving either colorimetric or fluorescent responses. The first fluorescent probes for singlet oxygen were based on the fluorescence decrease of anthracene derivatives, e.g., diphenylisobenzofuran and diethylamino dansyl upon reaction with 1O2. The simple 9,10-diphenylanthracene was initially reported for this,72 subsequently, water-soluble anthracene derivatives were developed.73 However, these probes displayed a lack of selectivity for 1O2. Thus, water-soluble rubrene (5,6,11,12-tetraphenyltetracene) derivatives have been employed, which show rapid reaction with and high selectivity for 1O2. However, their synthesis is rather involved.74
The most widely accepted strategy for the development of modern singlet oxygen probes is based on the formation of endoperoxides within the chromophore molecule, resulting in ON/OFF switching of the intramolecular electron transfer (PET). Such a probe design was initially suggested by Nagano's group.75 Typically, the probe is a dyad composed of a covalently-linked electron donor (an oxygen binding aromatic hydrocarbon) and an acceptor (fluorescent dye). The energy levels of the subunits should correspond to the requirement that the HOMO of the fluorophore is lower than that of the oxygen binding subunit (Fig. 6). In this case PET quenching of the fluorophore takes place in the absence of singlet oxygen. When the donor is converted to EPO the fluorophore emission increases.
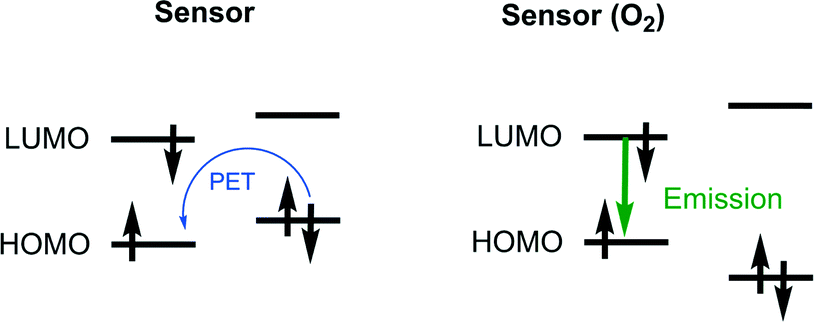 | ||
| Fig. 6 Fluorescence quenching of singlet oxygen probes via intramolecular PET. | ||
A commercially available fluorescent probe, compound 28, which is highly selective for 1O2 among other ROS, the so-called Singlet Oxygen Sensor Green (SOSG), has recently received much attention in bioimaging.76 This compound is poorly fluorescent (quantum yield ∼1.9%)77 due to quenching of the fluorescein singlet excited state by PET. Reaction with 1O2 results in the formation of an anthracene-EPO and consequently blocks PET due to lowering of the HOMO level. The fluorescence quantum yield of this product was measured to be 37%, showing a maximum emission at 525 nm. Although having some advantages over other fluorescent probes, particularly high selectivity, SOSG was shown to generate singlet oxygen itself upon irradiation77 thus limiting its use for quantitative measurements.78
Other examples of well-established fluorescent PET probes include the 9,10-diphenyl and 9,10-dimethylanthracene-conjugated xanthenes 29a–c, anthracene-substituted tetrathiafulvalenes 30a–b, and phosphorescent europium 31a–b, terdium (32) and rhenium (33) complexes (Fig. 7), which are discussed in a recent review devoted to luminescent probes.79 An obvious common limitation of these probes is the need for excitation in the visible part of the spectrum, which causes a cell autofluorescence signal. Thus, special attention is now paid to probes which can be excited in the “tissue transparency window” – a spectral range >650 nm, where natural pigments have negligible absorption.
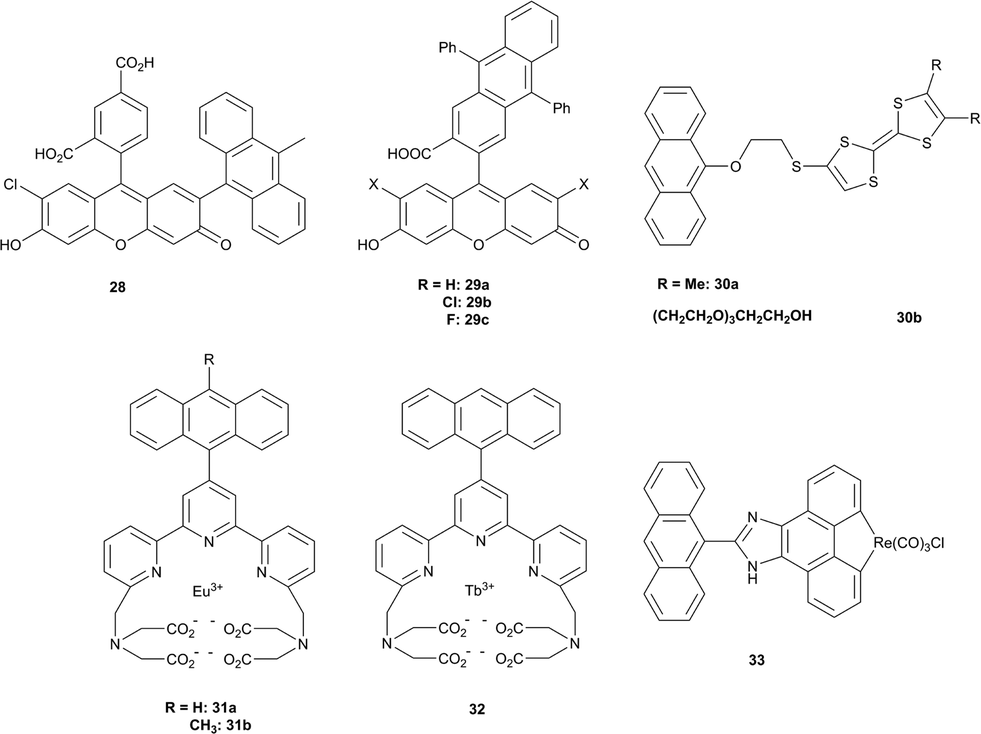 | ||
| Fig. 7 Singlet oxygen probes based on intramolecular PET. | ||
Recently, Majima and co-workers developed near-infrared fluorescent probes composed of 9,10-dimethylanthracene and silicon-containing rhodamine 34 (Fig. 8).80 Similar to xanthene–anthracene dyads, the Si-rhodamine chromophore is quenched by PET in the absence of oxygen, resulting in 1% fluorescence quantum yield. Upon reaction with 1O2 it was transformed into the bright form, showing an approximately 18-fold increase of the fluorescence intensity at 660 nm (17% quantum yield). In contrast to SOSG, compound 30 is much less prone to generate singlet oxygen itself upon irradiation (Φ1O2 = 0.02) and only negligible self-oxidation was observed.
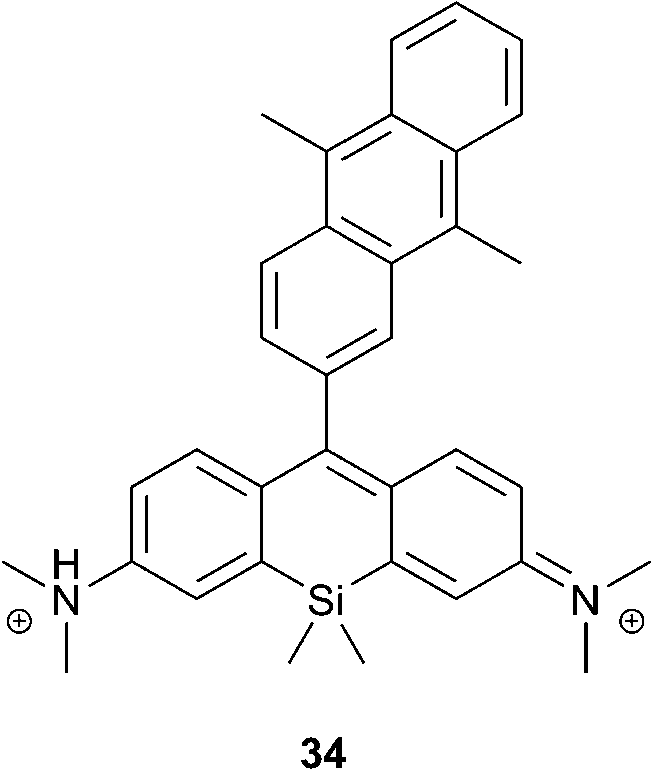 | ||
| Fig. 8 A far-red fluorescent probe for singlet oxygen. | ||
Upon incubation with cells, the probe localized selectively in mitochondria. Among different intracellular sensitizers, 34 selectively detected 1O2 generated by 5-aminolevulinic acid-derived protoporphyrin, co-localized in mitochondria. At the same time, other lysosomal porphyrins did not induce fluorescence changes of the probe. This approach is proposed for the visualization of mitochondrial 1O2 production during PDT with high spatial resolution.
Optical molecular imaging
There is an ongoing interest in development of chemiluminescent probes for in vivo imaging as an alternative to radioisotopes, which are known to cause ionization reactions harmful for living systems.81 For bioimaging chemiluminescent probes offer the advantage of better signal-to-noise ratio, as no excitation is required and thus autofluorescence of biomolecules is not observed.82 Although many organic molecules exhibit chemiluminescence, potential candidates for bioimaging are scarce, since visible light emission is readily absorbed and scattered by tissue.83Thermolysis of organic peroxides was observed to be followed by light emission in the early works of Dufraisse and co-workers, who showed emission at 1276 nm upon decomposition of 1,4-dimethoxy-9,10-diphenylanthracene. This corresponds to a 22.5 kcal energy difference between the singlet and triplet oxygen forms.84 Later, in reports by Krasnovsky, an emission deriving from phthalocyanine dye molecules was found to take place in the presence of naphthalene endoperoxides.85 This was interpreted based on the hypothesis of Khan and Kasha86 as an energy transfer from an excited singlet oxygen molecule to a fluorescent dye (step 1 in Fig. 9). However, the energy difference between singlet oxygen and ground-state triplet oxygen is not large enough for the excitation of the phthalocyanines and other dyes used in the corresponding experiments. Thus it was assumed, that the energy transfer takes place between a dye and two molecules of singlet oxygen, a so-called ‘dimol’ (step 2 in Fig. 9).87
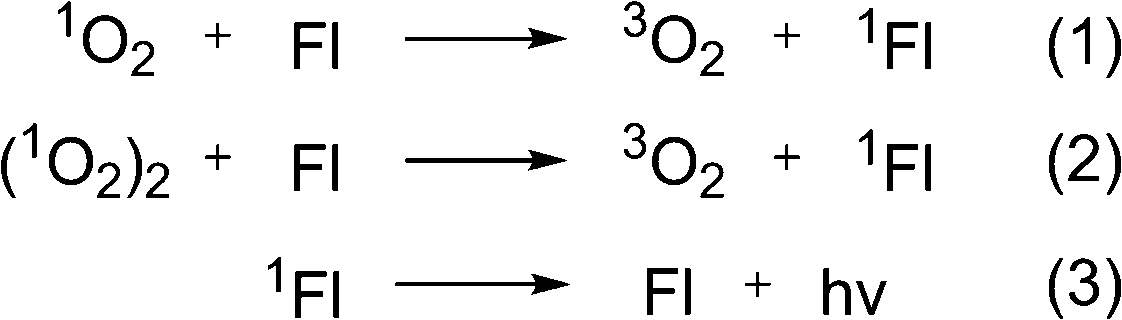 | ||
| Fig. 9 Energy transfer from singlet oxygen to a dye. | ||
Based on this processes, Smith and co-workers developed a thermally activated near-infrared chemiluminescent molecular system for in vivo optical imaging.88 An interlocked [2]-rotaxane 35, composed of a tetralactam macrocycle and a squaraine dye possesses a strong absorption in the far-red region, able of singlet oxygen generation. Thus, under irradiation with light in the presence of air it formed an endoperoxide at one of the anthracene subunits. This highly selective formation of a mono(endoperoxide) is due to the encapsulated squaraine steric effect, which prevents cycloaddition on the second anthracene subunit to take place. The reaction is not accompanied by formation of side-products, even at prolonged irradiation times.
In contrast to other anthracene endoperoxides, cycloreversion of 36 took place even at room temperature and fully regenerated the starting squaraine rotaxane (Scheme 14). The half-life time of the endoperoxide was found to be 3.2 h at 38 °C. Release of oxygen was accompanied by emission of light at 733 nm from the excited state squaraine. Further investigations showed that excitation of the squaraine chromophore takes place as an energy transfer from singlet oxygen to ground-state squaraine. This emission can be regenerated after the sample is irradiated with light in the presence of oxygen. The chemiluminescence wavelength can be tuned by introducing other squaraine dyes into the parent tetralactam macrocycle.
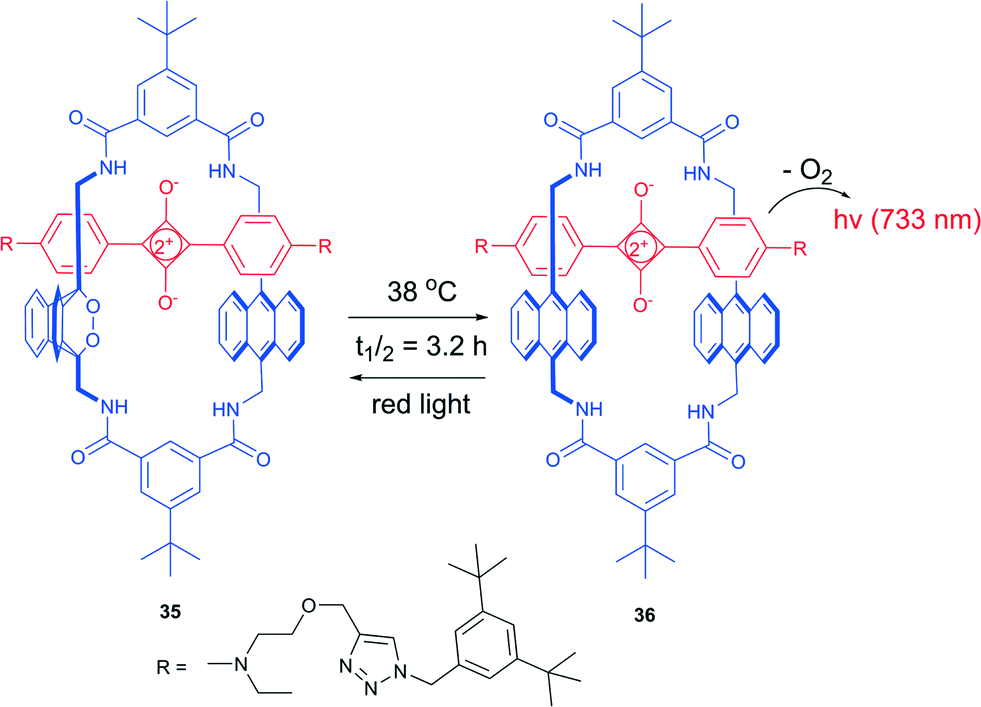 | ||
| Scheme 14 Thermal oxygen release from a rotaxane endoperoxide.87 | ||
EPO 36 was then used for optical imaging in live mice after encapsulation into carboxylate-modified polystyrene microparticles. Upon warming to body temperature, 36 emits near-infrared light that readily penetrated through the tissue and could be detected using a CCD camera (Fig. 10). The target background ratio (TBR) for chemiluminescence was found to be as high as 11.6. On the other hand, the squaraine chromophores in molecules 35 and 36 could be selectively excited and the resulting fluorescence monitored. However, the fluorescence microscopy by means of squaraine excitation from the particles gave substantially lower resolution (TBR 1.1) compared to chemiluminescence, due to light scattering and tissue autofluorescence.
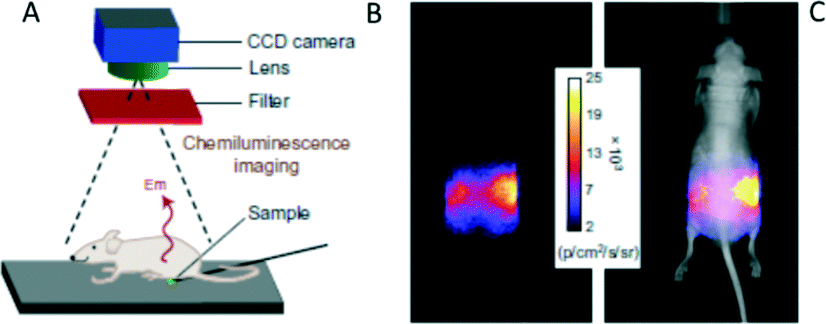 | ||
| Fig. 10 Chemiluminescence from 32 penetrates through a living mouse. (A) Experimental set-up for the measurements. (B,C) Pixel intensity map of chemiluminescence transmitted through the mouse.87 Copyright Macmillan Publishers Ltd. | ||
Although EPO 36 can release cytotoxic singlet oxygen, fast energy transfer to the squaraine dye ensure its complete deactivation prior to defusion from the local site. Indeed, standard cell toxicity experiments indicated that 36 (up to 20 mM) did not alter cell vitality in the dark.89 Thus, chemiluminescence from rotaxane EPOs represents a promising tool for in vivo molecular imaging applications. However, some technical complications need to be overcome, particularly the complicated synthesis, selective delivery to target sites, and stability at ambient temperature.
Conclusions
Clearly, the formation of organic endoperoxides upon reaction of singlet oxygen with aromatic systems, is currently garnering special attention. Their use can deliver a completely new paradigm for 1O2 carriers in photomedical treatments, particularly in photodynamic therapy.The ongoing expansion of the arsenal of reversible singlet-oxygen binding molecules and, particularly, its release at ambient temperatures opens new perspectives for biomedical applications. A combination of endoperoxide-forming molecules with conventional photosensitizers allows: 1) an increase of the actual lifetime of this highly reactive species in tissue thus prolonging the therapeutic effect, 2) its use as a source of oxygen in hypoxic tissue where conventional PDT is not effective. In addition, a combination of EPO materials with new photothermal nanomaterials provides a tool for precise control of the 1O2 production in selected regions of tissue. From this perspective a development of new target delivery methods for EPO molecules and corresponding materials is highly desirable.
On the other hand, formation of EPOs upon photooxidation of aromatic systems suggests new means to modulate a molecule's optical properties. In the past EPO formation has been regarded as an undesired complication of working with highly conjugated materials. However, as outlined here, the process can become a useful alternative to existing tools for the chemical tuning of photoactive materials. Additionally, there is a critical need to develop new biologically compatible probes for the selective detection of singlet oxygen. Currently employed fluorescent probes suffer from various limitations and here the search for photochromic systems based on EPO formation is a pending challenge.
Practical applications of reversible singlet oxygen binding molecular devices are currently still in the developmental stage and restricted by the synthetic availability of appropriate molecules, target delivery methods, and detection techniques. However, in light of the prospective benefits for photomedicine and the many ongoing studies solutions for the current limitations can be expected in the near future.
Acknowledgements
M. F. acknowledges a European Commission for Marie Curie Individual Fellowship (CONSORT, Grant No. 655142). M. O. S. acknowledges grants from Science Foundation Ireland (14/TIDA/2362 and 13/IA/1894).Notes and references
- C. S. Foote and S. Wexler, J. Am. Chem. Soc., 1964, 86, 3879 CrossRef CAS.
- (a) W. Adam and M. Prein, Acc. Chem. Res., 1996, 29, 275 CrossRef CAS; (b) J.-M. Aubry, C. Pierlot, J. Rigaudy and R. Schmidt, Acc. Chem. Res., 2003, 36, 668 CrossRef CAS PubMed.
- For example, singlet oxygen production by enzyme systems: J. R. Kanofsky, Chem.-Biol. Interact., 1989, 70, 1 CrossRef CAS PubMed participation in the metabolism of arachidonic acid: E. Cadenas and H. Sies, Methods Enzymol., 2000, 319, 67 Search PubMed DNA oxidation: H. Welfers, D. Schulte-Frohlinde and H. Sies, FEBS Lett., 1987, 211, 49 CrossRef cataract formation: J. S. Zigler and J. D. Goosey, Curr. Eye Res., 1984, 3, 59 CrossRef PubMed.
- S. Nonell and C. Flors, Singlet Oxygen: Applications in Biosciences and Nanosciences, Royal Society of Chemistry, Oxford, 2016 Search PubMed.
- H. H. Wasserman, K. B. Wiberg, D. L. Larsen and J. Parr, J. Org. Chem., 2005, 70, 105 CrossRef CAS PubMed.
- A. Lauer, A. L. Dobryakov, S. A. Kovalenko, H. Fidder and K. Heyne, Phys. Chem. Chem. Phys., 2011, 13, 8723 RSC.
- N. J. Turro and M. F. Chow, J. Am. Chem. Soc., 1981, 103, 7218 CrossRef CAS.
- (a) Z. Malik, J. Hanania and Y. Nitzan, J. Photochem. Photobiol., B, 1990, 5, 281 CrossRef CAS; (b) E. C. Ziegelhoffer and T. J. Donohue, Nat. Rev. Microbiol., 2009, 7, 856 CAS; (c) D. Arian, L. Kovbasyuk and A. Mokhir, J. Am. Chem. Soc., 2011, 133, 3972 CrossRef CAS PubMed; (d) R. Choudhury and A. Greer, Langmuir, 2014, 30, 3599 CrossRef CAS PubMed; (e) E. Clû, J. W. Snyder, P. R. Ogilby and K. V. Gothelf, ChemBioChem, 2007, 8, 475 CrossRef PubMed; (f) M. K. Kuimova, G. Yahioglu and P. R. Ogilby, J. Am. Chem. Soc., 2009, 131, 332 CrossRef CAS PubMed; (g) T. J. Dougherty, C. J. Gomer, B. W. Henderson, G. Jori, D. Kessel, M. Korbelik, J. Moan and Q. Peng, J. Natl. Cancer Inst., 1998, 90, 889 CrossRef CAS PubMed.
- (a) A. M. Asadirad, Z. Erno and N. R. Branda, Chem. Commun., 2013, 49, 5639 RSC; (b) R. Bonnett, Chem. Soc. Rev., 1995, 24, 19 RSC; (c) D. Bartusik, D. Aebisher, A. Ghogare, G. Ghosh, I. Abramova, T. Hasan and A. Greer, Photochem. Photobiol., 2013, 89, 936 CrossRef CAS PubMed; (d) T. J. Dougherty, C. J. Gomer, B. W. Henderson, G. Jori, D. Kessel, M. Korbelik, J. Moan and Q. Peng, J. Natl. Cancer Inst., 1998, 90, 889 CrossRef CAS PubMed; (e) S. B. Brown, E. A. Brown and I. Walker, Lancet Oncol., 2004, 5, 497 CrossRef CAS PubMed.
- (a) H. von Tappeiner and A. Jodlbauer, Dtsch. Arch. Klin. Med., 1904, 80, 427 Search PubMed; (b) H. von Tappeiner and H. Jesionek, Munch. Med. Wochenschr, 1903, 50, 2042 Search PubMed; (c) H. Jesionek and H. von Tappeiner, Dtsch. Arch. Klin. Med., 1905, 82, 223 Search PubMed.
- A. E. Profio and D. R. Doiron, Photochem. Photobiol., 1987, 46, 591 CrossRef CAS PubMed.
- R. Sullivan and C. H. Graham, Cancer Metastasis Rev., 2007, 26, 319 CrossRef CAS PubMed.
- R. Schmldt, W. Drews and H.-D. Brauer, J. Phys. Chem., 1982, 86, 4909 CrossRef.
- Z. Liang, W. Zhao, S. Wang, Q. Tang, S.-C. Lam and Q. Miao, Org. Lett., 2008, 10, 2661 CrossRef PubMed.
- I. Kaur, W. Jia, R. P. Kopreski, S. Selvarasah, M. R. Dokmeci, C. Pramanik, N. E. McGruer and G. P. Miller, J. Am. Chem. Soc., 2008, 130, 16274 CrossRef CAS PubMed.
- C. Bohne and R. H. Mitchell, J. Photochem. Photobiol., C, 2011, 12, 126 CrossRef CAS.
- M. Matsumoto, M. Yamada and N. Watanabe, Chem. Commun., 2005, 483 RSC.
- (a) R. Schmidt, Z. Naturforsch. A, 1984, 39, 998 CrossRef; (b) R. Schmidt, W. Drews and H.-D. Brauer, J. Phys. Chem., 1982, 86, 4909 CrossRef CAS; (c) R. Schmidt, W. Drews and H.-D. Brauer, J. Am. Chem. Soc., 1980, 102, 2791 CrossRef CAS; (d) R. Schmidt and H.-D. Brauer, J. Photochem., 1986, 34, 1 CrossRef CAS.
- H. J. Kuhn, S. E. Braslavsky and R. Schmidt, Pure Appl. Chem., 1989, 61, 187 CrossRef CAS.
- B. Stevens, US Pat., 4853548, 1989 Search PubMed.
- (a) W. Fudickar and T. Linker, Chem. – Eur. J., 2006, 12, 9276 CrossRef CAS PubMed; (b) W. Fudickar and T. Linker, Langmuir, 2010, 26, 4421 CrossRef CAS PubMed.
- W. Fudickar, A. Fery and T. Linker, J. Am. Chem. Soc., 2005, 127, 9386 CrossRef CAS PubMed.
- C. Schweitzer and R. Schmidt, Chem. Rev., 2003, 103, 1685 CrossRef CAS PubMed.
- J. M. Aubry and B. Cazin, Inorg. Chem., 1988, 27, 2013 CrossRef CAS.
- J. M. Aubry, B. Cazin and J. Duprat, J. Org. Chem., 1989, 54, 726 CrossRef CAS.
- M. Klaper and T. Linker, Chem. – Eur. J., 2015, 21, 8569 CrossRef CAS PubMed.
- E. J. Corey and W. C. Taylor, J. Am. Chem. Soc., 1964, 86, 3881 CrossRef CAS.
- B. D. Rihter, M. E. Kenney, W. E. Ford and M. A. J. Rodgers, J. Am. Chem. Soc., 1993, 115, 8146 CrossRef CAS.
- W. Freyer, H. Stiel, M. Hild, K. Teuchner and D. Leupold, Photochem. Photobiol., 1997, 66, 596 CrossRef CAS.
- M. A. Filatov, S. Baluschev, I. Z. Ilieva, V. Enkelmann, T. Miteva, K. Landfester, S. E. Aleshchnkov and A. V. Cheprakov, J. Org. Chem., 2012, 77, 11119 CrossRef CAS PubMed.
- V. Yakutkin, M. A. Filatov, I. Z. Ilieva, K. Landfester, G. Nelles, T. Miteva and S. Baluschev, Photochem. Photobiol. Sci., 2016 Search PubMed , in press.
- S. Cobo, F. Lafolet, E. Saint-Aman, C. Philouze, C. Bucher, S. Silvi, A. Credi and G. Royal, Chem. Commun., 2015, 51, 13886 RSC.
- H. Cerfontain, A. Koeberg-Telder, B. H. Bakker, R. H. Mitchell and M. Tashiro, Liebigs Ann./Recl., 1997, 873 CrossRef CAS.
- D. Roldan, S. Cobo, F. Lafolet, N. Vilà, C. Bochot, C. Bucher, E. Saint-Aman, M. Boggio-Pasqua, M. Garavelli and G. Royal, Chem. – Eur. J., 2015, 21, 455 CrossRef CAS PubMed.
- A. Bakkar, S. Cobo, F. Lafolet, E. Saint-Aman and G. Royal, J. Mater. Chem. C, 2015, 3, 12014 RSC.
- (a) J. R. Sommer, R. T. Farley, K. R. Graham, Y. Yang, J. R. Reynolds, J. Xue and K. S. Schanze, ACS Appl. Mater. Interfaces, 2009, 1, 274 CrossRef CAS PubMed; (b) T. V. Esipova, A. Karagodov, J. Miller, D. F. Wilson, T. M. Busch and S. A. Vinogradov, Anal. Chem., 2011, 83, 8756 CrossRef CAS PubMed; (c) J. F. Sun, F. F. Zhong and J. Z. Zhao, Dalton Trans., 2013, 42, 9595 RSC; (d) A. Turshatov, D. Busko, S. Baluschev, T. Miteva and K. Landfester, New J. Phys., 2011, 10, 083035 CrossRef.
- M. Filatov, I. Ilieva, K. Landfester and S. Baluschev, Exploring the IR-limit of the Triplet–Triplet Annihilation Upconversion: Tetraaryltetraanthra[2,3]porphyrin – family, Solar Power Technologies & Materials, in Nanotechnology 2013: Bio Sensors, Instruments, Medical, Environment and Energy, Nano Science and Technology Institute (NSTI), 2013, ch. 6, vol. 3, pp. 531–534 Search PubMed.
- (a) X. Song, L. Huang and B. Wu, Anal. Chem., 2008, 80, 5501 CrossRef CAS PubMed; (b) R. Islangulov, J. Lott, C. Weder and F. Castellano, J. Am. Chem. Soc., 2007, 129, 12652 CrossRef CAS PubMed; (c) F. Marsico, A. Turshatov, R. Pekoz, Y. Avlasevich, M. Wagner, K. Weber, D. Donadio, K. Landfester, S. Baluschev and F. R. Wurm, J. Am. Chem. Soc., 2014, 136, 11057 CrossRef CAS PubMed.
- (a) C. Wohnhaas, K. Friedemann, D. Busko, K. Landfester, S. Baluschev, D. Crespy and A. Turshatov, ACS Macro Lett., 2013, 2, 446 CrossRef CAS; (b) S. Ji, W. Wu, H. Guo and J. Zhao, Angew. Chem., Int. Ed., 2011, 50, 1626 CrossRef CAS PubMed.
- (a) Q. Liu, T. Yang, W. Feng and F. Li, J. Am. Chem. Soc., 2012, 11, 5390 CrossRef PubMed; (b) A. Monguzzi, M. Frigoli, C. Larpent, R. Tubino and F. Meinardi, Adv. Funct. Mater., 2012, 22, 139 CrossRef CAS.
- (a) V. Ramamurthy, J. V. Caspar, D. F. Eaton, E. W. Kuo and D. R. Corbin, J. Am. Chem. Soc., 1992, 114, 3882 CrossRef CAS; (b) A. S. Carreteroa, A. S. Castilloa and A. F. Gutiérreza, Crit. Rev. Anal. Chem., 2005, 35, 3 CrossRef; (c) M. R. Fernández de la Campa, Y. Ming Liu, M. E. Díaz García and A. Sanz Medel, Anal. Chim. Acta, 1990, 238, 297 CrossRef.
- M. A. Filatov, E. Heinrich, D. Busko, I. Z. Ilieva, K. Landfester and S. Baluschev, Phys. Chem. Chem. Phys., 2015, 17, 6501 RSC.
- A. Turshatov and S. Baluschev, Triplet–Triplet Annihilation-Assisted Upconversion: All-Optical Tools for Probing the Physical Parameter of Soft Matter Handbook of Coherent-Domain Optical Methods, Springer Science + Business Media, New York, 2013, ISBN-13: 978–1461451754 Search PubMed.
- M. Klaper and T. Linker, J. Am. Chem. Soc., 2015, 137, 13744 CrossRef CAS PubMed.
- H. H. Wasserman, T. Y. Ching, B. H. Lipshutz, H. Matsuyama, F. E. Scully and P. Wang, Bioorg. Med. Chem. Lett., 1992, 2, 1137 CrossRef CAS.
- M. Radisic, H. Park, F. Chen, J. E. Salazar-Lazzaro, Y. Wang, R. Dennis, R. Langer, L. E. Freed and G. Vunjak-Novakovic, Tissue Eng., Part B, 2010, 16, 169 CrossRef PubMed.
- S. Benz, S. Nötzli, J. S. Siegel, D. Eberli and H. J. Jessen, J. Med. Chem., 2013, 56, 10171 CrossRef CAS PubMed.
- C. Changtonga, D. W. Carneya, L. Luoa, C. A. Zotoa, J. L. Lombardib and R. E. Connors, J. Photochem. Photobiol., A, 2013, 260, 9 CrossRef.
- N. Kornblum and H. E. DeLaMare, J. Am. Chem. Soc., 1951, 73, 880 CrossRef CAS.
- A. O. Govorov and H. H. Richardson, Nano Today, 2007, 2, 30 CrossRef.
- (a) X. Huang, I. H. El-Sayed, W. Qian and M. A. El-Sayed, J. Am. Chem. Soc., 2006, 128, 2115 CrossRef CAS PubMed; (b) A. M. Gobin, D. P. O'Neal, D. M. Watkins, N. J. Halas, R. A. Drezek and J. L. West, Lasers Surg. Med., 2005, 37, 123 CrossRef PubMed.
- S. Martins, J. P. S. Farinha, C. Baleizao and M. N. Berberan-Santos, Chem. Commun., 2014, 50, 3317 RSC.
- A. M. Asadirad, Z. Erno and N. R. Branda, Chem. Commun., 2013, 49, 5639 RSC.
- (a) P. J. Rosenfeld, D. M. Brown, J. S. Heier, D. S. Boyer, P. K. Kaiser, C. Y. Chung and R. Y. Kim, N. Engl. J. Med., 2006, 355, 1419 CrossRef CAS PubMed; (b) M. Ferrari, Nat. Rev. Cancer, 2005, 5, 161 CrossRef CAS PubMed.
- W. G. Roberts, K. M. Smith, J. L. McCullough and M. W. Berns, Photochem. Photobiol., 1989, 49, 431 CrossRef CAS PubMed.
- K. I. Salokhiddinov, I. M. Byteva and G. P. Gurinovich, J. Appl. Spectrosc., 1981, 34, 561 CrossRef.
- J. A. Bertout, S. A. Patel and M. C. Simon, Nat. Rev. Cancer, 2008, 8, 967 CrossRef CAS PubMed.
- T. M. Busch, S. M. Hahn, S. M. Evans and C. J. Koch, Cancer Res., 2000, 60, 2636 CAS.
- Z. Xiao, S. Halls, D. Dickey, J. Tulip and R. B. Moore, Clin. Cancer Res., 2007, 13, 7496 CrossRef CAS PubMed.
- (a) C. Pierlot, J.-M. Aubry, K. Briviba, H. Sies and P. di Mascio, Methods Enzymol., 2000, 319, 188 Search PubMed; (b) K. Otsu, K. Sato, Y. Ikeda, H. Imami, Y. Nakagawa, Y. Ohba and J. Fujii, Biochem. J., 2005, 389, 197 CrossRef CAS PubMed; (c) K. Otsu, K. Sato, M. Sato, H. Ono, Y. Ohba and Y. Katagata, Cell Biol. Int., 2008, 32, 1380 CrossRef CAS PubMed.
- D. Posavec, M. Zabel, U. Bogner, G. Bernhardt and G. Knör, Org. Biomol. Chem., 2012, 10, 7062 CAS.
- S. Kolemen, T. Ozdemir, D. Lee, G. Mi Kim, T. Karatas, J. Yoon and E. U. Akkaya, Angew. Chem., Int. Ed., 2016, 55, 1 CrossRef PubMed.
- I. S. Turan, D. Yildiz, A. Turksoy, G. Gunaydin and E. U. Akkaya, Angew. Chem., 2016, 128, 1 CrossRef.
- K. K. Ng, J. F. Lovell, A. Vedadi, T. Hajian and G. Zheng, ACS Nano, 2013, 7, 3484 CrossRef CAS PubMed.
- S. A. E. Marras, F. Russell-Kramer and S. Tyagi, Nucleic Acids Res., 2002, 30, e122 CrossRef PubMed.
- M. R. Wasielewski, Chem. Rev., 1992, 92, 435 CrossRef CAS.
- (a) G. Zheng, J. Chen, K. Stefflova, M. Jarvi, H. Li and B. C. Wilson, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8989 CrossRef CAS PubMed; (b) J. F. Lovell, T. W. Liu, J. Chen and G. Zheng, Chem. Rev., 2010, 110, 2839 CrossRef CAS PubMed; (c) A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung and K. Burgess, Chem. Soc. Rev., 2013, 42, 77 RSC; (d) L. Huang, W. Yang and J. Zhao, J. Org. Chem., 2014, 79, 10240 CrossRef CAS PubMed; (e) T. Yogo, Y. Urano, A. Mizushima, H. Sunahara, T. Inoue, K. Hirose, M. Iino, K. Kikuchi and T. Nagano, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 28 CrossRef CAS PubMed.
- A. M. Durantini, L. E. Greene, R. Lincoln, S. R. Martínez and G. Cosa, J. Am. Chem. Soc., 2016, 138, 1215 CrossRef CAS PubMed.
- (a) T. Nagano, J. Clin. Biochem. Nutr., 2009, 45, 111 CrossRef CAS PubMed; (b) N. Soh, Anal. Bioanal. Chem., 2006, 386, 532 CrossRef CAS PubMed; (c) A. Gomes, E. Fernandes and J. L. F. C. Lima, J. Biochem. Biophys. Methods, 2005, 65, 45 CrossRef CAS PubMed; (d) K. Setsukinai, Y. Urano, K. Kakinuma, H. J. Majima and R. Nagano, J. Biol. Chem., 2003, 278, 3170 CrossRef CAS PubMed; (e) V. S. Lin, B. C. Dickinson and C. Chang, Methods Enzymol., 2013, 526, 19 CAS.
- P. R. Ogilby, Chem. Soc. Rev., 2010, 39, 3181 RSC.
- S. Hackbarth and B. Röder, Photochem. Photobiol. Sci., 2015, 14, 329 CAS.
- M. J. Steinbeck, A. U. Khan and M. J. Karnovsky, J. Biol. Chem., 1992, 267, 13425 CAS.
- V. Nardello and J.-M. Aubry, Methods Enzymol., 2000, 319, 50 CAS.
- J. Rigaudy and N. K. Cuong, C. R. Acad. Sci., 1962, 254, 4184 CAS.
- N. Umezawa, K. Tanaka, Y. Urano, K. Kikuchi, T. Higuchi and T. Nagano, Angew. Chem., Int. Ed., 1999, 38, 2899 CrossRef CAS.
- (a) Y. Osakada, K. Kawai, T. Tachikawa, M. Fujitsuka, K. Tainaka, S. Tero-Kubota and T. Majima, Chem. – Eur. J., 2012, 18, 1060 CrossRef CAS PubMed; (b) C. Flors, M. J. Fryer, J. Waring, B. Reeder, U. Bechtold, P. M. Mullineaux, S. Nonell, M. T. Wilson and N. R. Baker, J. Exp. Bot., 2006, 57, 1725 CrossRef CAS PubMed; (c) Y. Shen, H. Lin, Z. Huang, L. Xiao, D. Chen, B. Li and S. Xie, Proc. SPIE, 2010, 7845, 78451F CrossRef; (d) L. Xiao, L. Gu, S. B. Howell and M. J. Sailor, ACS Nano, 2011, 5, 3651 CrossRef CAS PubMed; (e) X. Ragas, L. P. Cooper, J. H. White, S. Nonell and C. Flors, ChemPhysChem, 2011, 12, 161 CrossRef CAS PubMed; (f) A. Gollmer, J. Arnbjerg, F. Blaikie, B. Pedersen, T. Breitenbach, K. Daasbjerg, M. Glasius and P. Ogilby, Photochem. Photobiol., 2011, 87, 671 CrossRef CAS PubMed.
- S. Kim, M. Fujitsuka and T. Majima, J. Phys. Chem. B, 2013, 117, 13985 CrossRef CAS PubMed.
- H. Lin, Y. Shen, D. Chen, L. Lin, B. C. Wilson, B. Li and S. Xie, J. Fluoresc., 2012, 23, 41 CrossRef PubMed.
- (a) X. Chen, X. Tian, I. Shin and J. Yoon, Chem. Soc. Rev., 2011, 40, 4783 RSC; (b) X. Li, X. Gao, W. Shi and H. Ma, Chem. Rev., 2014, 114, 590 CrossRef CAS PubMed.
- S. Kim, T. Tachikawa, M. Fujitsuka and T. Majima, J. Am. Chem. Soc., 2014, 136, 11707 CrossRef CAS PubMed.
- J. G. Mancini and M. N. Ferrandino, Curr. Opin. Urol., 2010, 20, 163 CrossRef PubMed.
- J. V. Frangioni, Mol. Imaging, 2009, 8, 303 Search PubMed.
- (a) J. A. Prescher and C. H. Contag, Curr. Opin. Chem. Biol., 2010, 14, 80 CrossRef CAS PubMed; (b) A. Roda, M. Guardigli, E. Michelini and M. Mirasoli, TrAC, Trends Anal. Chem., 2009, 28, 307 CrossRef CAS.
- (a) C. Moureu, C. Dufraisse and P. M. Dean, C. R. Acad. Sci., 1926, 182, 1584 Search PubMed; (b) C. Dufraisse and L. Velluz, Bull. Soc. Chim. Fr., 1942, 9, 171 CAS; (c) C. Dufraisse, J. Rigaudy, J. J. Basselier and N. K. Cuong, C. R. Acad. Sci., 1965, 260, 5031 CAS.
- Y. Fu, A. A. Krasnovsky and C. S. Foote, J. Am. Chem. Soc., 1993, 115, 10282 CrossRef CAS.
- A. U. Khan and M. Kasha, J. Am. Chem. Soc., 1966, 88, 1574 CrossRef CAS.
- W. Adam, D. V. Kazakov and V. P. Kazakov, Chem. Rev., 2005, 105, 3371 CrossRef CAS PubMed.
- (a) C. G. Collins, J. M. Baumes and B. D. Smith, Chem. Commun., 2011, 47, 12352 RSC; (b) J. M. Baumes, J. J. Gassensmith, J. Giblin, J.-J. Lee, A. G. White, W. J. Culligan, W. M. Leevy, M. Kuno and B. D. Smith, Nat. Chem., 2010, 2, 1025 CrossRef CAS PubMed.
- J.-J. Lee, A. Goncalves, B. A. Smith, R. Palumbo, A. G. White and B. D. Smith, Aust. J. Chem., 2011, 64, 604 CAS.
| This journal is © The Royal Society of Chemistry 2016 |