
Engineering affinity agents for the detection of hemi-methylated CpG sites in DNA†
B. E.
Tam
,
K.
Sung
and
H. D.
Sikes
*
Department of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA. E-mail: sikes@mit.edu
First published on 7th September 2016
Abstract
Wild-type methyl-CpG-binding domain (MBD) proteins specifically bind symmetrically methylated DNA sequences, and assays have been developed that use these proteins for profiling DNA methylation. Here, we use directed evolution in the yeast surface display format to identify a new protein variant that binds hemi-methylated CpG dinucleotides.
Design, System, ApplicationDirected evolution is a valuable technique for developing proteins with desirable catalytic or binding properties. Used in conjunction with a yeast surface display platform, a large number of proteins with random mutations can be quickly screened to select those with the desired properties. Here, a protein that binds to hemi-methylated DNA with high affinity and excellent specificity with respect to unmethylated DNA is desired. Current affinity agent-based methylation detection assays require symmetrically methylated DNA sequences for a positive readout. With a protein that binds hemi-methylated DNA, we can alter these assays to, instead, use a simple, unmethylated DNA probe to capture a DNA sequence of interest and profile its methylation status. This allows for the development of simpler, less expensive assays that can more easily be developed into clinical methylation tests to aid in the detection and treatment of several types of cancers. |
Introduction
DNA methylation patterns, and predominantly the methylation of cytosine bases in CpG dinucleotides, have a strong correlation with numerous cancer subtypes. Currently, CpG methylation of the MGMT gene promoter is used to guide treatment decisions for patients with glioblastoma,1 and MLH1 promoter methylation is used to determine whether patients with certain types of colorectal cancer have a sporadic or familial form of the disease.2–4 The utility of promoter methylation as a biomarker for cancer diagnostics is demonstrated by the commercially available tests Cologuard® and Epi ProColon® for colorectal cancer screening.5–7 While there are only a few gene promoters that are routinely evaluated for methylation at this time,8,9 studies have shown tumor-associated promoter methylation of more than 45 genes in various types of cancers.10Current clinical methylation detection assays involve bisulfite conversion of unmethylated cytosines to uracil while leaving the methylated cytosines unchanged, producing modified DNA sequences that can be distinguished by sequencing or PCR.9,11 Such methods have numerous drawbacks. For example, large quantities of DNA are required because 90% can be degraded during the conversion step, and without careful optimization and control of the reaction conditions, false positives and negatives are common.12,13 One of the methods proposed to eliminate the need for bisulfite treatment is to use an affinity agent, such as a methyl-CpG binding domain protein (MBD), to detect methylation of cytosine bases. Such affinity agents are commonly used in assays such as MethylCap-Seq, where genomic DNA is enriched for fragments containing methylated cytosines and then sequenced to identify the methylated regions.14 In addition, several assays based on DNA microarrays have been developed to profile methylation patterns of sequences of interest.
Early methylation-specific microarrays involved either bisulfite conversion followed by hybridization on an array of DNA probes specific to the sequences of the converted unmethylated or methylated states15 or enrichment of methylated sequences in genomic DNA via proteins that bind methylated CpGs followed by hybridization with DNA arrays to compare the prevalence of a specific sequence in the enriched and unenriched samples.16 More recently, researchers have used DNA probes to capture a specific DNA sequence followed by MBD binding as a simple method to determine the sequence-specific methylation status of the gene or region of interest. Yu et al.17 hybridized target sequences to probes on a chip and detected MBD binding with surface plasmon resonance. This method, however, required a 1 μM concentration of target DNA.17 Heimer et al.18 performed the hybridization and MBD binding steps on an agarose-coated surface and reduced the limit of detection to 1 nM while detecting bound MBD directly with fluorescence labeling. This method eliminates the need for bisulfite conversion, PCR, sequencing, and enrichment steps, providing many advantages over conventional methylation detection assays.
Wild-type MBD proteins bind specifically to symmetrically methylated DNA, in which the cytosines on both DNA strands are methylated. Therefore, the capture probes must contain methylated CpGs in order for these assays to work. Such probes cannot be produced by standard enzymatic methods, such as PCR; instead, they require chemical synthesis. The incorporation of modified phosphoramidites into synthetic oligos yields probes that are much more expensive than those that only require unmodified bases. Developing a protein to recognize hemi-methylated DNA, where the cytosine bases are only methylated on one of the two DNA strands, would allow for the detection of a methylated sequence from a patient's sample bound to an unmethylated capture probe. This offers a key advantage in assay development. In other applications, where researchers are developing DNA microarrays to detect specific genetic mutations19 or viral RNA,20 several probes for each target sequence must often be tested experimentally, even after using specialized software to design the probes. This is also an important problem when designing probes for CpG islands, where high GC content and repeat sequences add additional challenges to probe design. If probes for the methylation assay need not be methylated, a larger number of potential probes could be screened more easily, making the process of advancing microarray assays to the clinics much more feasible and efficient.
Here, we report the development of an MBD variant that binds to hemi-methylated DNA with high affinity. A library created from human MBD2, previously shown to have the highest affinity for methylated DNA of the MBD family,21 was used as a starting point. Variants with improved affinity for hemi-methylated DNA were isolated and analyzed in a yeast surface display construct.22 The top performing variant was produced as a GFP fusion protein23 and tested in an array-based binding assay on a microscope slide to demonstrate its functionality in such detection assays.
Results and discussion
To characterize the binding affinity, the MBD proteins were displayed on the surface of S. cerevisiae using the pCTCON-2 vector.22 The binding affinity of wild-type human MBD2 for a DNA oligo with a single methyl group on one strand was evaluated using equilibrium binding titrations with flow cytometry. The sequence and methylation patterns of the test DNA used for characterization are shown in Fig. 1a. These sequences are based on a region of the MGMT gene, as described previously.17,21 In the equilibrium binding titrations, expression of the MBD protein was verified by labeling the cMyc tag of the fusion protein with Alexa Fluor® 488. The cells expressing the protein were incubated with the biotinylated DNA at a range of concentrations and labeled with streptavidin, Alexa Fluor® 647. As shown in Fig. 1b, the wild type MBD2 protein binds to symmetrically methylated DNA with high affinity but shows almost no binding to the hemi-methylated DNA sample, even at concentrations as high as 100 nM. Because binding was not observed, a dissociation constant could not be determined using this method. MBD2 was the focus of this study because it was previously demonstrated to have the greatest affinity for methylated DNA of the members of the MBD family.21 However, the results demonstrating that MBD2 binds to symmetrically methylated DNA with much higher affinity than hemi-methylated DNA agree with previously reported data for MBD1 and MeCP2, shown in Fig. 1c, that show affinity differences of an order of magnitude or more between the two methylation states.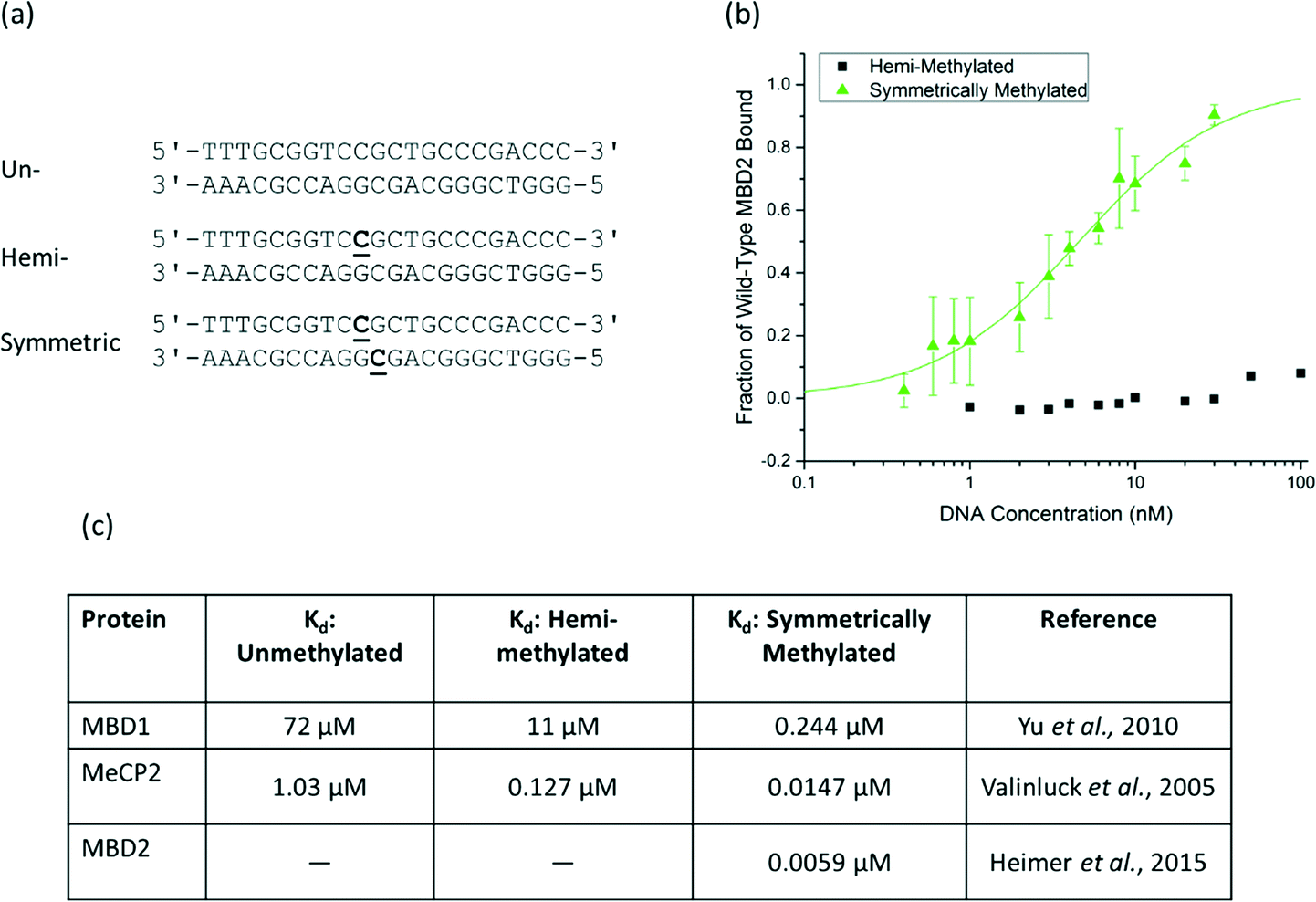 | ||
| Fig. 1 a) The DNA sequence of the probe/target pairs and methylation states used for protein assessment are shown with the methylated cytosine bases bolded and underlined. The sequence is from the MGMT gene and contains three CpG dinucleotides, one of which is methylated in the hemi-methylated and symmetrically methylated states used in this paper. b) An equilibrium binding titration of wild-type human MBD2. The fraction of MBD bound to DNA was determined based on the normalized mean fluorescence. Wild-type MBD2 binds symmetrically methylated DNA with high specificity and shows no detectable binding to hemi-methylated DNA at the concentrations of interest. c) Reported dissociation constants for MBD1 and MeCP2 also show affinity differences of one or more orders of magnitude between symmetrically methylated and hemi-methylated DNA.17,21,24 | ||
Beginning with the error-prone PCR library generated by Heimer et al.,21 variants of the protein human MBD2 were displayed on the surface of yeast cells, and those with improved affinity for hemi-methylated DNA were selected using an equilibrium binding assay. The selection process is depicted in Fig. 2. In the early rounds of selection, cells expressing the MBD library were incubated with hemi-methylated DNA attached to magnetic beads, allowing the cells that bind to the DNA to be separated from the larger library. In later rounds, the cells isolated from the magnetic bead selections were incubated with biotinylated, hemi-methylated DNA that was then labeled with a streptavidin-conjugated fluorophore. In this assay, cells expressing proteins with the highest affinities had the largest number of fluorophore-labeled DNA molecules attached, giving the brightest signal during flow cytometry. These cells were isolated using fluorescence-activated cell sorting (FACS). The amino acid sequences of the proteins isolated after the selection procedure are shown in the ESI† (Table S1). All of the variants isolated had the K161R mutation and 70% had the F208Y mutation, two mutations described previously that allow for the formation of an additional hydrogen bond to stabilize the protein structure and to bind to the DNA backbone, respectively.21 The F187I mutation, which is adjacent to the arginine residue that interacts with the methylated cytosine base,25 was also found in 50% of the isolated proteins.
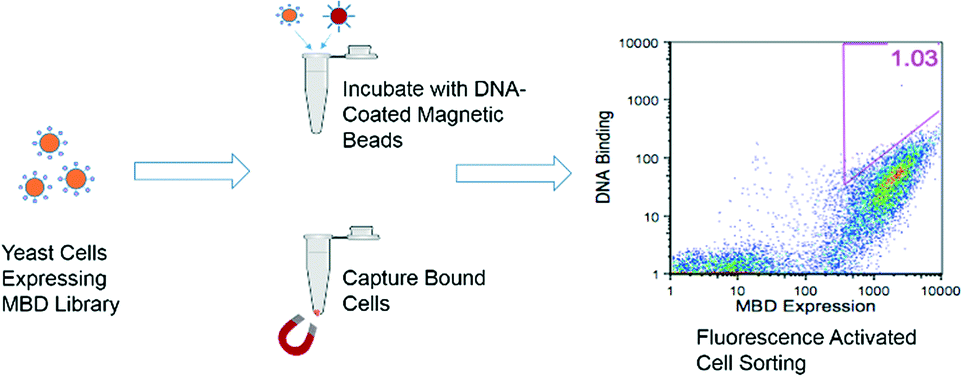 | ||
| Fig. 2 MBD variants were expressed on the surface of S. cerevisiae. Cells expressing variants with improved affinity for hemi-methylated DNA were selected first with magnetic beads coated in hemi-methylated DNA and then with fluorescence activated cell sorting. | ||
Six unique protein variants were compared (Fig. S3†), and the top-performing protein, variant h4, was characterized by equilibrium binding titration. Fig. 3a shows the improvement in the binding affinity of the engineered protein for hemi-methylated DNA over that of the wild-type MBD2 protein. Completion of equilibrium binding titrations in triplicate gives a dissociation constant of 5.6 ± 1.4 nM, a value nearly identical to the wild type protein's dissociation constant with symmetrically methylated DNA. Fig. 3b shows that the new protein binds to hemi-methylated DNA and symmetrically methylated DNA with similar affinity while retaining good specificity for these constructs over unmethylated DNA. Three of the four mutations in variant h4, K161R, F187I, and F208Y, have been described previously. The fourth mutation, T200S, is a small change from a threonine to the slightly smaller serine. Depicted in Fig. 4, which is based on the NMR structure for chicken MBD2,25 this mutation is located far from the DNA binding site. This residue is not conserved across the MBD family: it is found as alanine in MBD1, threonine in human MBD2, asparagine in MBD4, and valine in MeCP2. However, none of the wild type MBD proteins nor any of the proteins isolated from the library except for variant h4 have the S200 residue. Nevertheless, this mutation appears to play an important role in binding to hemi-methylated DNA.
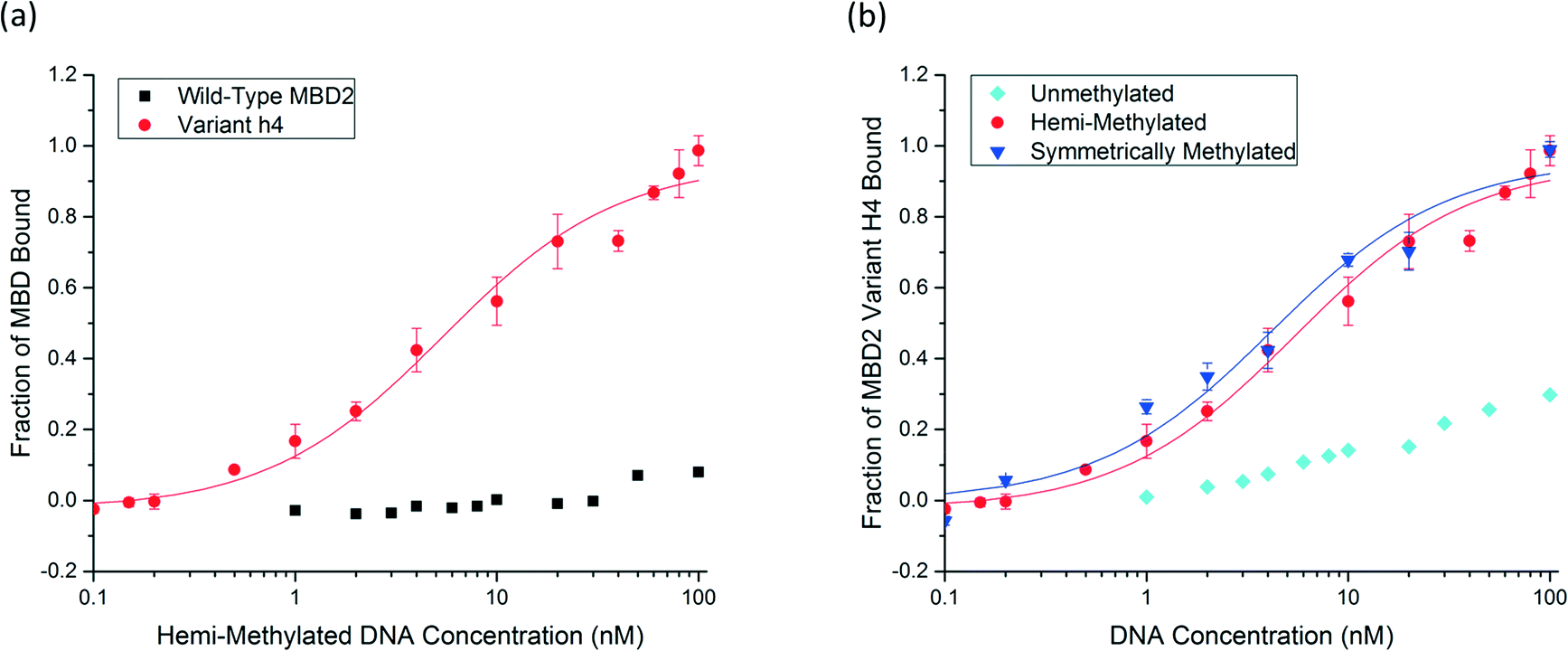 | ||
| Fig. 3 a) An equilibrium binding titration shows the binding affinity for the engineered variant H4 in comparison with the wild-type hMBD2 protein. b) The engineered hMBD2 protein H4 is characterized by equilibrium binding titrations with hemi-methylated, symmetrically methylated, and unmethylated DNA. | ||
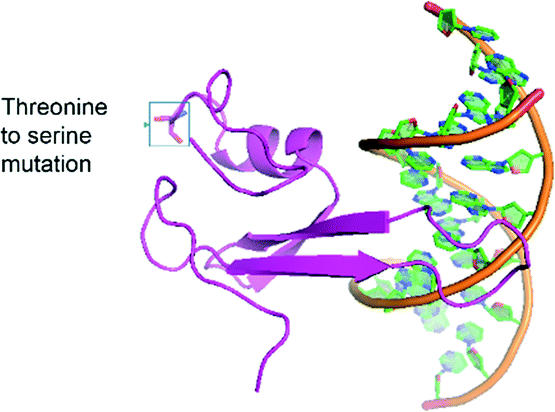 | ||
| Fig. 4 The structure of the engineered protein, variant H4, is shown in association with DNA with the T200S mutation highlighted. This structure was constructed using the SWISS-MODEL system26 based on the crystal structure of chicken MBD2.25 | ||
To determine whether the new protein can function to distinguish between hemi-methylated and unmethylated DNA in the interfacial binding assays previously developed,17,18 binding experiments were performed with soluble MBD2 variant h4 and DNA arrays printed on agarose-coated glass slides. The MBD2 variant h4 was cloned into the pET30b bacterial expression vector and expressed as a fusion protein with eGFP and a biotin acceptor sequence. The slides were printed with hemi-methylated DNA as well as unmethylated DNA. Biotinylated MBD bound to the DNA was labeled with streptavidin, Alexa Fluor® 647 and detected by fluorescence imaging. In the resulting image, found in Fig. 5a, MBD bound to the hemi-methylated DNA is easily visible while the spots printed with unmethylated DNA show little binding and are very difficult to identify by eye, a visual distinction that is confirmed by the quantitative results shown in Fig. 5b.
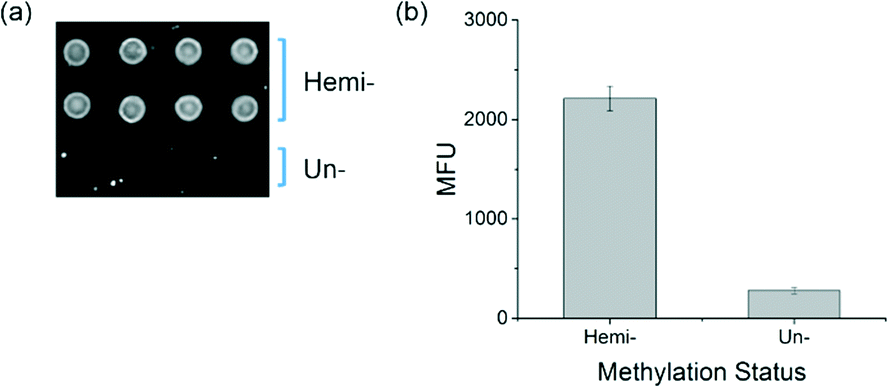 | ||
| Fig. 5 a) An image of the DNA array after labeling of MBD2 variant h4 shows that the hemi-methylated and unmethylated spots are easily distinguishable. b) Quantitative analysis shows a 7.8-fold higher signal from binding to hemi-methylated DNA as compared to unmethylated DNA in the arrays. | ||
Conclusions
The chemical conversion-based methods currently used for clinical methylation analyses have many disadvantages, including DNA degradation during sample treatment, and affinity agent-based methylation assays are a promising alternative. There are also limitations that must be overcome before these assays will be useful for clinical applications, however. Protein–DNA binding assays are highly dependent on the concentration of probe/target duplexes on the surface of the slide. Therefore, one key limitation is that the assay is not currently sensitive enough to detect the pM DNA concentrations that could be obtained from many patient samples. Further work to improve both the probe density and the efficiency of DNA hybridization could lead to higher concentrations of probe/target duplexes, effectively addressing this problem. Another disadvantage of the assay as described in this paper is that equipment for fluorescence detection was also required for assay readout; however, an alternative detection mechanism that allows detection by eye has already been developed for this system.18Compared to other protein-based assays, a method that doesn't require methylated capture probes can be developed more quickly and easily into an affordable test suitable for clinical use. Here, an MBD protein was developed to bind hemi-methylated DNA by introducing four mutations into human MBD2 through directed evolution with yeast surface display. The resulting protein variant binds to singly, hemi-methylated DNA with a dissociation constant of 5.6 ± 1.4 nM while the wild-type protein has an affinity too low to be quantified using the yeast surface display method. Specificity over unmethylated DNA was retained with the engineered protein, and the protein was proven to distinguish between hemi-methylated and unmethylated DNA in an interfacial binding assay performed with DNA affixed to a slide. These results demonstrate that variant h4 with unmethylated DNA probes can be used in place of the wild-type MBD proteins with methylated probes used in previously developed epigenotyping assays.
Acknowledgements
This work was supported by an NSF graduate research fellowship (1122374 to KS), the Massachusetts Institute of Technology Center for Environmental Health Sciences (through support from the National Institutes of Health-National Institute of Environmental Health Sciences) [P30-ES002109], and partially by the National Institutes of Health-National Cancer Institute [P30CCA14051 to the Massachusetts Institute of Technology Flow Cytometry Core Facility]. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation. We thank Dr. Brandon Heimer for sharing his expertise and providing advice related to this project.Notes and references
- M. E. Hegi, A. C. Diserens, S. Godard, P. Y. Dietrich, L. Regli, S. Ostermann, P. Otten, G. Van Melle, N. De Tribolet and R. Stupp, Clin. Cancer Res., 2004, 10, 1871–1874 CrossRef CAS PubMed.
- J. M. Cunningham, E. R. Christensen, D. J. Tester, C. Y. Kim, P. C. Roche, L. J. Burgart and S. N. Thibodeau, Cancer Res., 1998, 58, 3455–3460 CAS.
- J. G. Herman, A. Umar, K. Polyak, J. R. Graff, N. Ahuja, J.-P. J. Issa, S. Markowitz, J. K. V. Willson, S. R. Hamilton, K. W. Kinzler, M. F. Kane, R. D. Kolodner, B. Vogelstein, T. A. Kunkel and S. B. Baylin, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6870–6875 CrossRef CAS.
- M. L. Veigl, L. Kasturi, J. Olechnowicz, A. Ma, J. D. Lutterbaugh, S. Periyasamy, G.-M. Li, J. Drummond, P. L. Modrich, W. D. Sedwick and S. D. Markowitz, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 8698–8702 CrossRef CAS.
- T. F. Imperiale, D. F. Ransohoff, S. H. Itzkowitz, T. R. Levin, P. Lavin, G. P. Lidgard, D. A. Ahlquist and B. M. Berger, N. Engl. J. Med., 2014, 370, 1287–1297 CrossRef CAS PubMed.
- N. T. Potter, P. Hurban, M. N. White, K. D. Whitlock, C. E. Lofton-Day, R. Tetzner, T. Koenig, N. B. Quigley and G. Weiss, Clin. Chem., 2014, 000, 1–9 Search PubMed.
- V. M. Pratt, Clin. Chem., 2014, 60, 1141–1142 CAS.
- C. Noehammer, W. Pulverer, M. R. Hassler, M. Hofner, M. Wielscher, K. Vierlinger, T. Liloglou, D. McCarthy, T. J. Jensen, A. Nygren, H. Gohlke, G. Trooskens, M. Braspenning, W. Van Criekinge, G. Egger and A. Weinhaeusel, Epigenomics, 2014, 6, 603–622 CrossRef CAS PubMed.
- B. W. Heimer, B. E. Tam, A. Minkovsky and H. D. Sikes, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2016 DOI:10.1002/wnan.1407.
- H. Heyn and M. Esteller, Nat. Rev. Genet., 2012, 13, 679–692 CrossRef CAS PubMed.
- J. G. Herman, J. R. Graff, S. Myöhänen, B. D. Nelkin and S. B. Baylin, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 9821–9826 CrossRef CAS.
- C. Grunau, S. J. Clark and A. Rosenthal, Nucleic Acids Res., 2001, 29, E65 CrossRef CAS PubMed.
- A. Okamoto, Org. Biomol. Chem., 2009, 7, 21–26 CAS.
- A. B. Brinkman, F. Simmer, K. Ma, A. Kaan, J. Zhu and H. G. Stunnenberg, Methods, 2010, 52, 232–236 CrossRef CAS PubMed.
- R. S. Gitan, H. Shi, C.-M. Chen, P. S. Yan and T. H.-M. Huang, Genome Res., 2002, 12, 158–164 CrossRef CAS PubMed.
- C. Gebhard, L. Schwarzfischer, T.-H. Pham, E. Schilling, M. Klug, R. Andreesen and M. Rehli, Cancer Res., 2006, 66, 6118–6128 CrossRef CAS PubMed.
- Y. Yu, S. Blair, D. Gillespie, R. Jensen, D. Myszka, A. H. Badran, I. Ghosh and A. Chagovetz, Anal. Chem., 2010, 82, 5012–5019 CrossRef CAS PubMed.
- B. W. Heimer, T. A. Shatova, J. K. Lee, K. Kaastrup and H. D. Sikes, Analyst, 2014, 139, 3695–3701 RSC.
- S. Waldmüller, P. Freund, S. Mauch, R. Toder and H.-P. Vosberg, Hum. Mutat., 2002, 19, 560–569 CrossRef PubMed.
- A. Gall, B. Hoffmann, T. Harder, C. Grund, D. Höper and M. Beer, J. Clin. Microbiol., 2009, 47, 327–334 CrossRef CAS PubMed.
- B. W. Heimer, B. E. Tam and H. D. Sikes, Protein Eng., Des. Sel., 2015, 28, 543–551 CrossRef PubMed.
- G. Chao, W. L. Lau, B. J. Hackel, S. L. Sazinsky, S. M. Lippow and K. D. Wittrup, Nat. Protoc., 2006, 1, 755–768 CrossRef CAS PubMed.
- M. E. Boyd, B. W. Heimer and H. D. Sikes, Protein Expression Purif., 2012, 82, 332–338 CrossRef CAS PubMed.
- V. Valinluck, P. Liu, J. I. Kang, A. Burdzy and L. C. Sowers, Nucleic Acids Res., 2005, 33, 3057–3064 CrossRef CAS PubMed.
- J. N. Scarsdale, H. D. Webb, G. D. Ginder and D. C. Williams, Nucleic Acids Res., 2011, 39, 6741–6752 CrossRef CAS PubMed.
- M. Biasini, S. Bienert, A. Waterhouse, K. Arnold, G. Studer, T. Schmidt, F. Kiefer, T. Gallo Cassarino, M. Bertoni, L. Bordoli and T. Schwede, Nucleic Acids Res., 2014, 42, W252–W258 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6me00073h |
| This journal is © The Royal Society of Chemistry 2016 |