
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Evaluation of fluoropyruvate as nucleophile in reactions catalysed by N-acetyl neuraminic acid lyase variants: scope, limitations and stereoselectivity†
Jennifer
Stockwell
ab,
Adam D.
Daniels
bc,
Claire L.
Windle
bc,
Thomas A.
Harman
ab,
Thomas
Woodhall
ab,
Tomas
Lebl
d,
Chi H.
Trinh
bc,
Keith
Mulholland
e,
Arwen R.
Pearson‡
bc,
Alan
Berry
*bc and
Adam
Nelson
*ab
aSchool of Chemistry, University of Leeds, Leeds, LS2 9JT, UK. E-mail: a.s.nelson@leeds.ac.uk
bAstbury Centre for Structural Molecular Biology, University of Leeds, Leeds, LS2 9JT, UK. E-mail: a.berry@leeds.ac.uk
cSchool of Molecular and Cellular Biology, University of Leeds, Leeds, LS2 9JT, UK
dSchool of Chemistry, University of St Andrews, St Andrews, KY16 9ST, UK
eChemical Development, AstraZeneca, Silk Road Business Park, Macclesfield, Cheshire, SK10 2NA, UK
First published on 5th November 2015
Abstract
The catalysis of reactions involving fluoropyruvate as donor by N-acetyl neuraminic acid lyase (NAL) variants was investigated. Under kinetic control, the wild-type enzyme catalysed the reaction between fluoropyruvate and N-acetyl mannosamine to give a 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 ratio of the (3R,4R)- and (3S,4R)-configured products; after extended reaction times, equilibration occurred to give a 30:70 mixture of these products. The efficiency and stereoselectivity of reactions of a range of substrates catalysed by the E192N, E192N/T167V/S208V and E192N/T167G NAL variants were also studied. Using fluoropyruvate and (2R,3S)- or (2S,3R)-2,3-dihydroxy-4-oxo-N,N-dipropylbutanamide as substrates, it was possible to obtain three of the four possible diastereomeric products; for each product, the ratio of anomeric and pyranose/furanose forms was determined. The crystal structure of S. aureus NAL in complex with fluoropyruvate was determined, assisting rationalisation of the stereochemical outcome of C–C bond formation.
:10 ratio of the (3R,4R)- and (3S,4R)-configured products; after extended reaction times, equilibration occurred to give a 30:70 mixture of these products. The efficiency and stereoselectivity of reactions of a range of substrates catalysed by the E192N, E192N/T167V/S208V and E192N/T167G NAL variants were also studied. Using fluoropyruvate and (2R,3S)- or (2S,3R)-2,3-dihydroxy-4-oxo-N,N-dipropylbutanamide as substrates, it was possible to obtain three of the four possible diastereomeric products; for each product, the ratio of anomeric and pyranose/furanose forms was determined. The crystal structure of S. aureus NAL in complex with fluoropyruvate was determined, assisting rationalisation of the stereochemical outcome of C–C bond formation.
Introduction
The introduction of fluorine can have a profound effect on bioactive molecules including their conformation, binding, bioavailability, metabolism, pharmacokinetics and pharmacodynamics.1 As a consequence, around 20% of prescribed drugs, and 30% of leading blockbuster drugs, contain at least one fluorine atom.2 Examples of fluorinated pharmaceuticals include the cholesterol-lowering drug Atorvastatin, and Sofosbuvir which is exploited in the treatment of Hepatitis C (Fig. 1).3,4 Moreover, fluorinated sugars can serve as valuable mechanism-based probes of carbohydrate-processing enzymes.5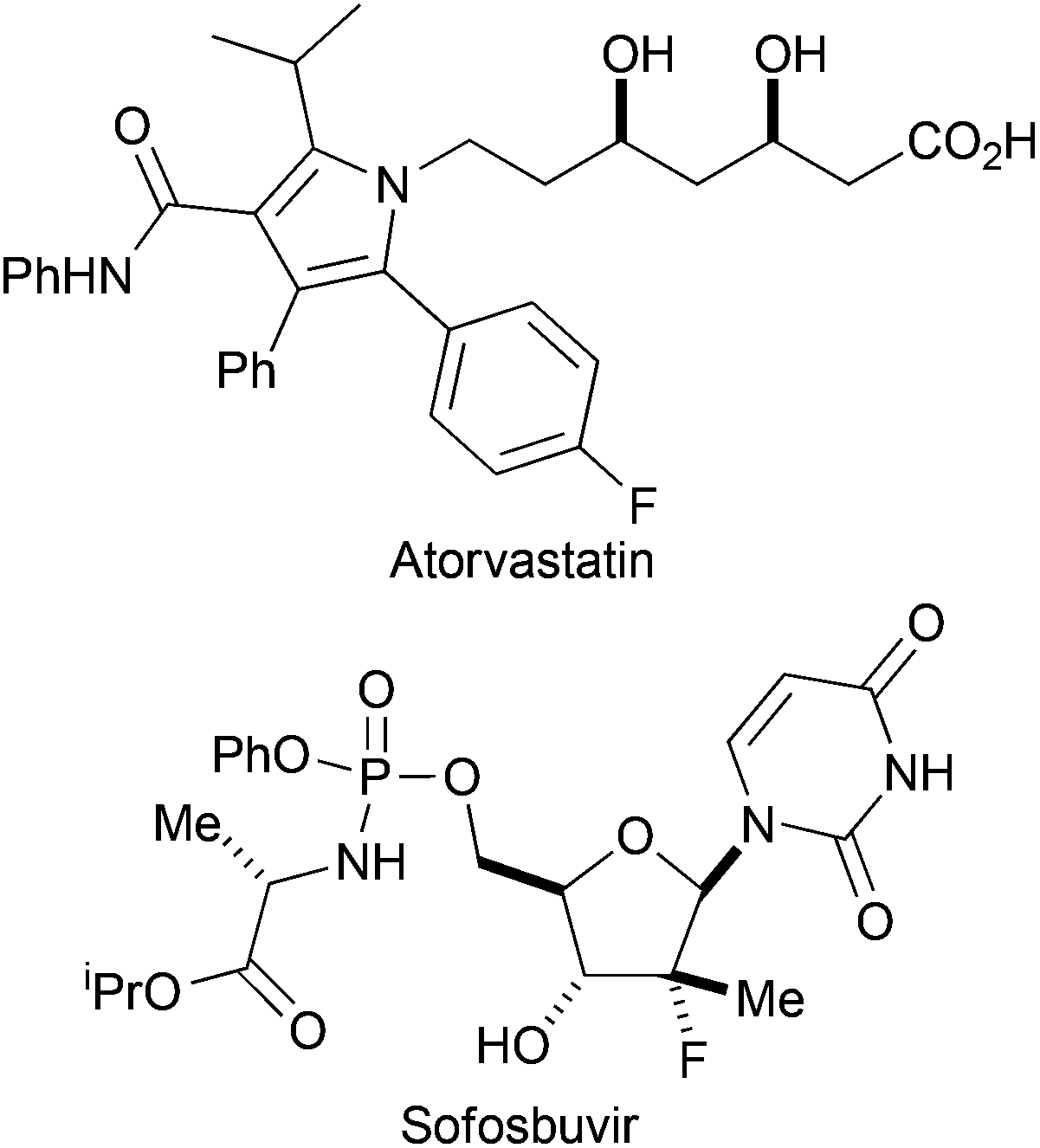 | ||
| Fig. 1 Examples of fluorinated drugs. | ||
The stereoselective synthesis of compounds with a fluorine-bearing stereocentre is a significant challenge. Most solutions to this problem rely on stereoselective C–F bond formation, for example by fluorination of allylic silanes.6 Some catalytic methods for enantioselective C–F bond formation have been developed: for example by organocatalytic α-fluorination of aldehydes7 or Pd-catalysed α-fluorination of β-keto phosphonates.8
We envisaged a complementary catalytic approach in which a F-bearing stereocentre would be controlled by formation of a neighbouring C–C bond (Scheme 1). Aldolase-catalysed reaction involving fluoropyruvate and an aldehyde 1 would yield an aldol product 2 with two new stereogenic centres. This catalytic approach would complement enantioselective aldol reactions involving fluoroacetone.9
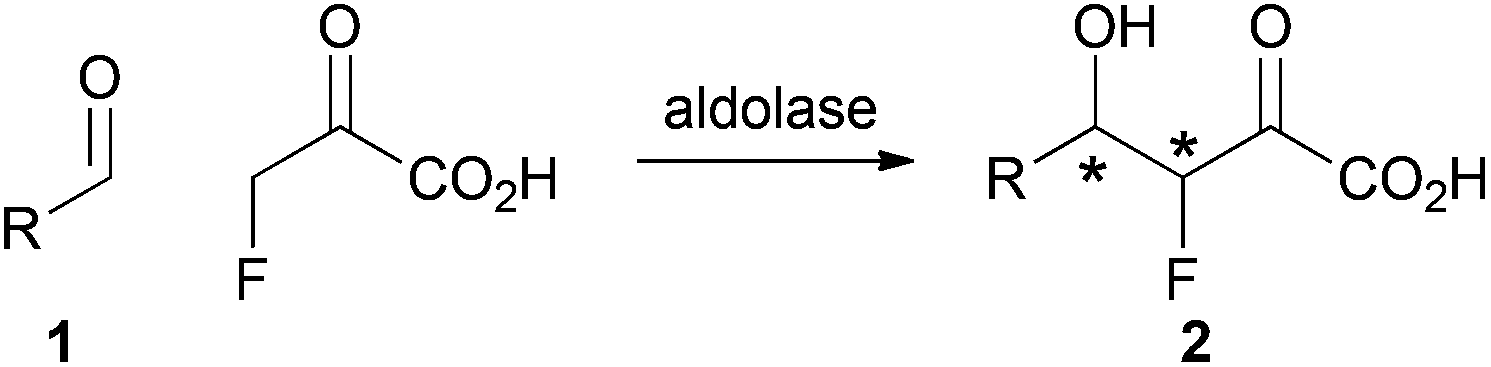 | ||
| Scheme 1 Envisaged strategy for controlling F-bearing stereocentres by C–C bond formation. | ||
N-Acetyl neuraminic acid lyase (NAL) is a Class I aldolase that catalyses the reversible aldol reaction between pyruvate and N-acetyl mannosamine (ManNAc) to give N-acetyl neuraminic acid (Neu5Ac). A combination of mutagenesis, structural biology and computational chemistry has revealed insights into its catalytic mechanism.10 Despite a report that it is not a substrate,11 fluoropyruvate is a viable donor.12 However, differing stereochemical outcomes have been reported for the NAL-catalysed reaction between fluoropyruvate and ManNAc.12 An initial aim of our study was, therefore, to clarify the stereochemical outcome of this reaction.
We also sought to investigate the catalysed reactions between fluoropyruvate and alternative aldehyde acceptors. Here, we investigated the value of synthetically-useful NAL variants that we have previously generated using directed evolution.13,14 The E192N variant of NAL is an excellent catalyst of the poorly stereoselective reaction between pyruvate and the alternative substrate (2R,3S)-2,3-dihydroxy-4-oxo-N,N-dipropylbutanamide, DHOB (Scheme 2).13 The structural basis of the modified substrate specificity of this variant has been gleaned using protein crystallography.15 In contrast, the E192N/T167G and E192N/T167V/S208V variants of NAL control the stereochemistry of C–C bond formation, and catalyse respectively the selective formation of the alternative diastereomeric products 3a and 3b (Scheme 2).14
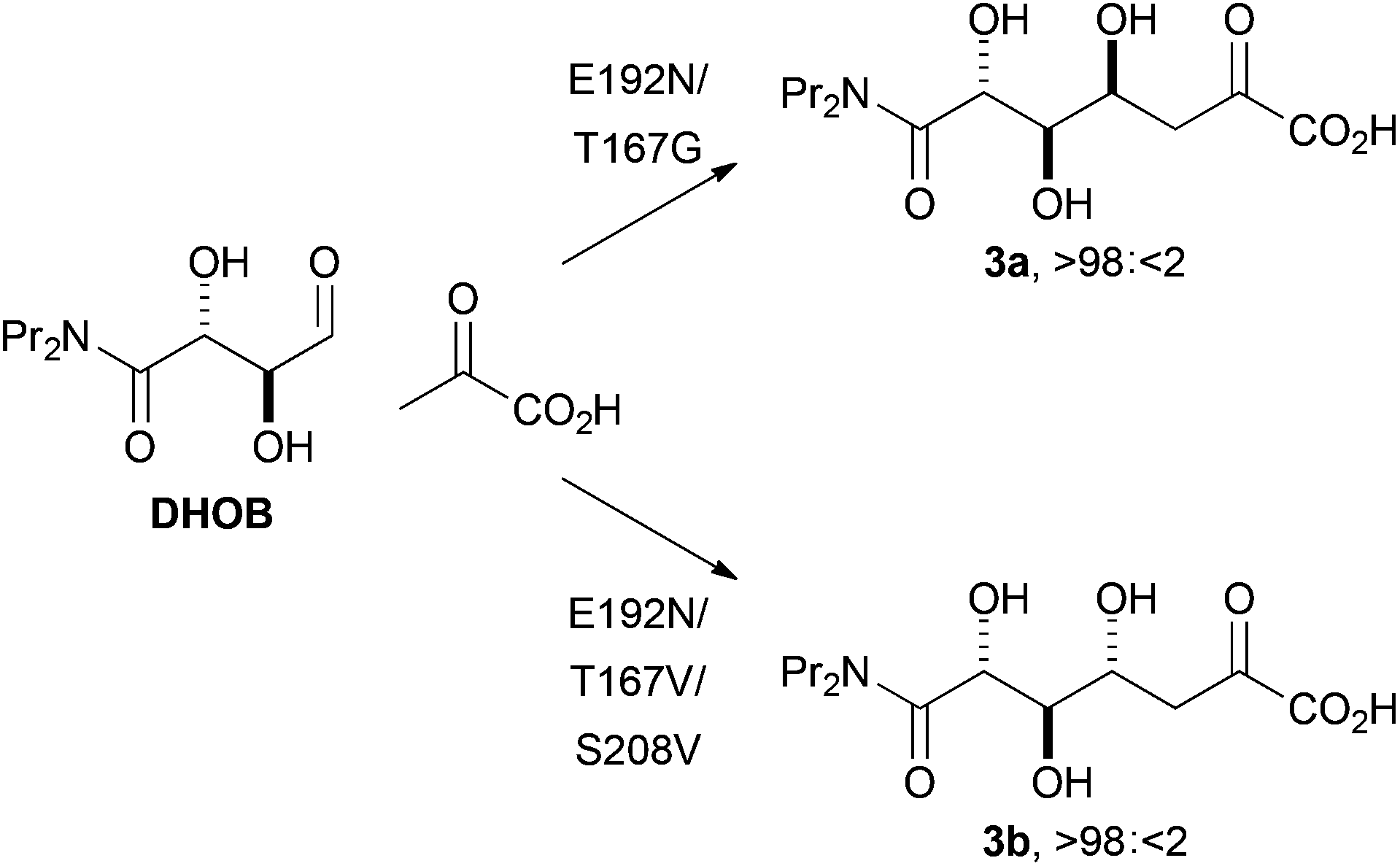 | ||
| Scheme 2 Stereoselective aldol reactions catalysed by aldolases generated by directed evolution.14 The products are drawn in open chain form for clarity. | ||
Results and discussion
Evaluation of wild-type NAL in the synthesis of fluorinated analogues of N-acetyl neuraminic acid
Initially, the reaction between fluoropyruvate and ManNAc catalysed by wild-type NAL was investigated (Panel A, Scheme 3). Accordingly, the reaction was performed at 37 °C in an NMR tube (20 mM sodium fluoropyruvate and 100 mM N-acetyl mannosamine in 20 mM Tris-HCl pH 7.4 buffer) and followed by 19F NMR spectroscopy. After 500 min, the fluoropyruvate was >98% consumed, and a 90:10 mixture of the diastereomeric products 4a and 4d had been formed (which vary only in their configuration at C-3). However, after a prolonged reaction time (∼5 weeks), with regular addition of more enzyme, the ratio of products had switched to 30:70 in favour of 4d. The alternative possible diastereomeric products (4b and 4c; Panel B) were not detected. These data suggest that 4a is the kinetic product of the reaction, and that 4d is the thermodynamic product. The relative thermodynamic stability of 4d may stem from the stabilising gauche interaction between fluorine and vicinal electronegative atoms.16 As with previous studies, the reaction was found to yield selectively (4R)-configured products. The different ratios of products under kinetic and thermodynamic control may account for the contrasting selectivities reported in previous studies.12
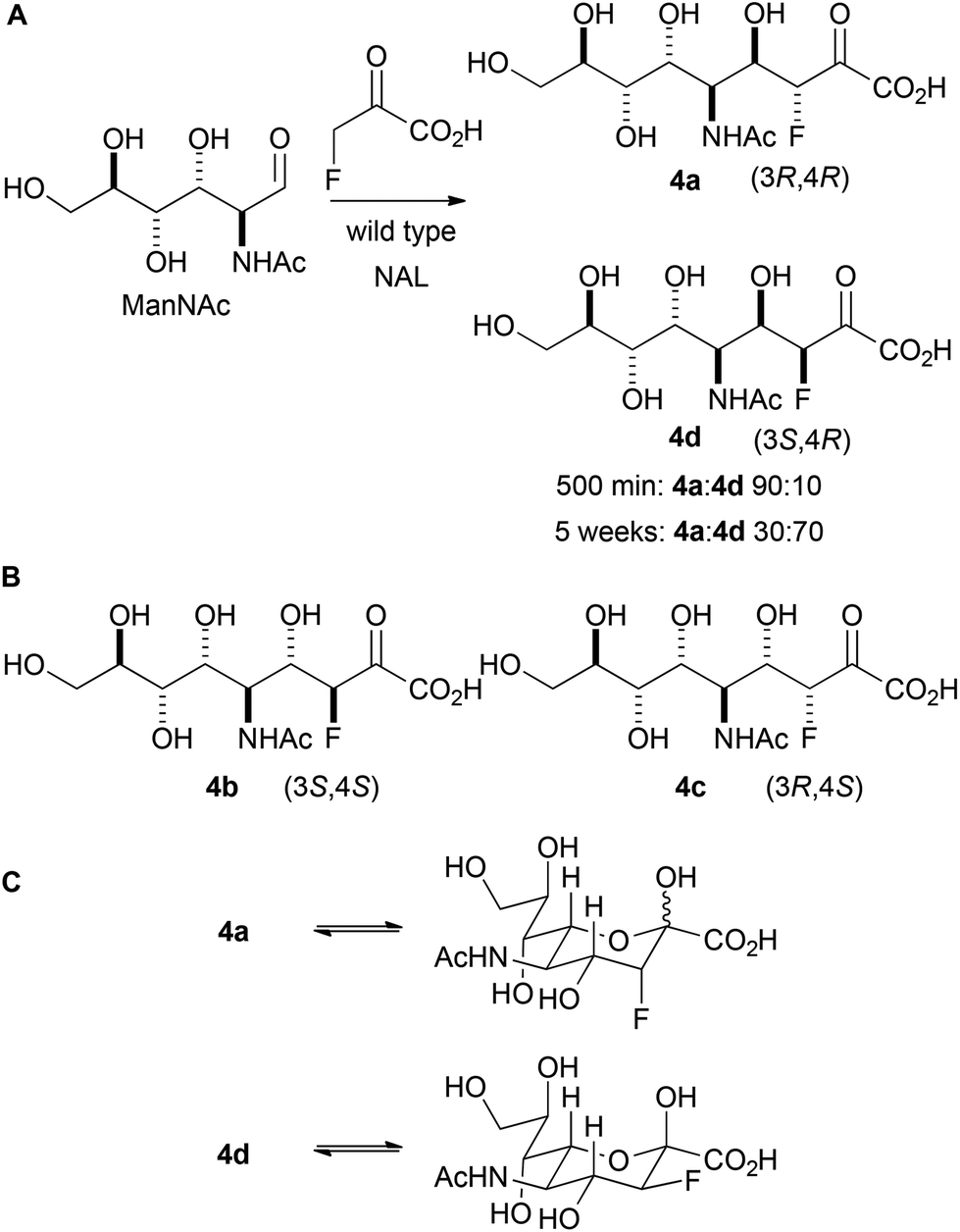 | ||
| Scheme 3 Reaction between fluoropyruvate and ManNAc catalysed by wild-type NAL. A: The stereochemical outcome is determined by the reaction time. The products are depicted in open chain form for clarity. NAL was regularly added to the 5 week reaction. B: Diastereomeric products that were not observed. C: Cyclised forms of 4a and 4d. | ||
The interpretation of the spectroscopic data was greatly assisted by the preparation of standard samples of the products 4a and 4d.12a The reaction between ManNAc and sodium fluoropyruvate, catalysed by wild-type NAL, was conducted at 37 °C in 100 mM Tris-HCl pH 7.4 buffer, and the products purified by column chromatography. After 24 h reaction, the product 4a was obtained in 34% yield; whilst after reaction for >1 week, the product 4d was obtained in 43% yield. In both cases, a single pyranose anomer predominated (Table 1; Panel C, Scheme 3). The configuration of 4a and 4d was determined by careful analysis of vicinal coupling constants.17 In both pyranose anomers of 4a, there was a large coupling constant between fluorine and H-4 (∼30 Hz) and a small coupling constant between H-3 and H-4 (2.1 Hz in the major anomer) (Table 1). In contrast, in the major anomer of 4d, there was a small coupling constant between fluorine and H-4 (∼12 Hz) and a large coupling constant between H-3 and H-4 (8.8 Hz).
| Product | Form (proportion) | δ F/ppm | δ 3H/ppm | δ 4H/ppm | δ 5H/ppm | δ 6H/ppm |
2
J
HFa/Hz |
3
J
HFa/Hz |
3 J 3H–4H/Hz | 3 J 4H–5H/Hz | 3 J 5H–6H/Hz |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Determined by analysis of the 296 MHz 19F NMR spectrum. b Not measured. | |||||||||||
| 4a | Major pyranose (98%) | −208.1 | 4.65 | 3.93 | 4.07 | 3.87 | 49.3 | 30.0 | 2.1 | 10.6 | 10.6 |
| Minor pyranose (2%) | −217.9 | NMb | NMb | NMb | NMb | 51.3 | 29.9 | NMb | NMb | NMb | |
| 4d | Major pyranose (96%) | −199.3 | 4.47 | ∼3.90 | ∼3.90 | ∼3.90 | 49.7 | 12.0 | 8.8 | NMb | NMb |
| 16a | Major pyranose (92%) | −206.0 | 4.78 | 3.94 | 3.88 | 4.75 | 49.9 | 32.5 | 3.4 | 9.7 | 9.2 |
| Minor pyranose (8%) | −216.8 | NMb | NMb | NMb | NMb | 51.4 | 32.8 | NMb | NMb | NMb | |
| 16c | Major pyranose (35%) | −190.5 | 4.85 | 4.39 | 4.12 | 4.65 | 50.5 | 24.0 | 4.8 | 5.0 | 6.1 |
| Major furanose (25%) | −194.5 | 4.72 | 4.02 | 3.96 | NMb | 43.7 | 4.7 | 1.7 | NMb | NMb | |
| Minor pyranose (30%) | −201.9 | 5.03 | 4.46 | 3.95 | 4.57 | 53.1 | 18.7 | 5.5 | 5.6 | 7.2 | |
| Minor furanose (10%) | −207.4 | 4.86 | 4.30 | 4.18 | 4.75 | 48.5 | 10.1 | 7.3 | NMb | 5.4 | |
| ent-16d | Pyranose (>98%) | −199.8 | 4.60 | 3.95 | 3.78 | 4.62 | 49.3 | 13.3 | 9.3 | 9.3 | 9.7 |
| 17a | Major pyranose (98%) | −207.8 | 4.90 | 4.16 | 4.23 | 4.83 | 49.0 | 29.1 | 2.2 | 10.9 | 10.0 |
| Minor pyranose (2%) | −218.5 | NMb | NMb | NMb | NMb | 50.2 | 28.8 | NMb | NMb | NMb | |
The catalysis of the cleavage of the reaction products 4a and 4d was also studied using an established coupled enzyme assay13b (Table 2). The cleavage of the fluorinated N-acetyl neuraminic acid analogue 4a was much less efficient than that of Neu5Ac itself (kcat/KM: 0.11 min−1 mM−1 for 4a compared with 260 min−1 mM−1 for Neu5Ac). However, the catalysis of the cleavage of the diastereomeric fluorinated analogue 4d was even less efficient and was not detectable under the conditions of the assay. This observation is consistent with (3R,4R)-configured 4a being the kinetic product of the NAL-catalysed reaction between fluoropyruvate and ManNAc.
Preparation of substrate precursors
To enable evaluation of alternative potential substrates, a range of alkene precursors was prepared: ozonolysis of these alkenes (10, ent-10, 15 and ent-15) would yield the corresponding aldehydes (DHOB, ent-DHOB, AHOB§ and ent-AHOB). The alkene13aent-10 was prepared using a route that was analogous to an established18 synthesis of 10 (Scheme 4). Thus, treatment of the lactone 5 (derived from lyxose19) with concentrated hydrochloric acid in acetone gave the corresponding acetonide206. Treatment of 6 with iodine and triphenylphosphine gave the corresponding iodolactone 7, which was followed by reductive ring-opening to give the carboxylic acid ent-8 (whose enantiomer had been used to prepare1810). Finally, amide formation (→9) and deprotection gave the required alkene ent-10.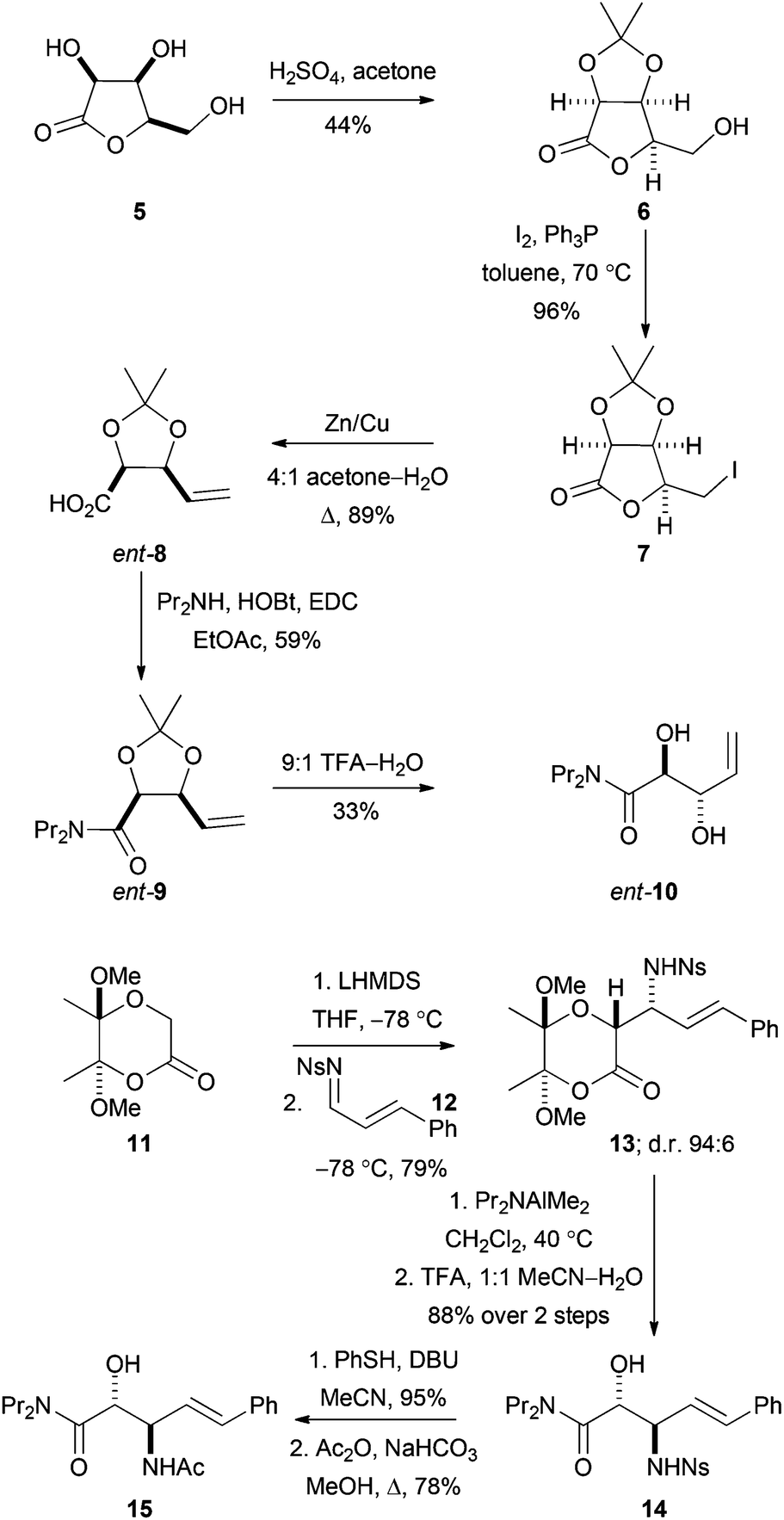 | ||
| Scheme 4 Synthesis of precursors of aldehyde substrates. In addition, the alkene ent-15 was prepared from the enantiomeric lactone starting material 11; and 10 was prepared using an established route.16 | ||
The alkenes 15 and ent-15 were prepared from the known21 enantiomerically pure lactones 11 and ent-11 (see Scheme 4 for the synthesis of 15). Treatment of the lactone 11 with LHMDS at −78 °C, and reaction with the N-sulfonyl imine 12, gave the product 13 as a 94:6 mixture of diastereomers; the relative configuration of the major diastereomer was determined by subsequent conversion into a cyclic derivative (see below). The lactone 13 was ring-opened by treatment with Pr2NAlMe2 and, following acetal hydrolysis, the β-amino amide derivative 14 was obtained in 88% yield. Finally, desulfonylation of 14, followed by acetylation, gave the required alkene 15.
Evaluation of variant NALs in the catalysis of reactions involving fluoropyruvate
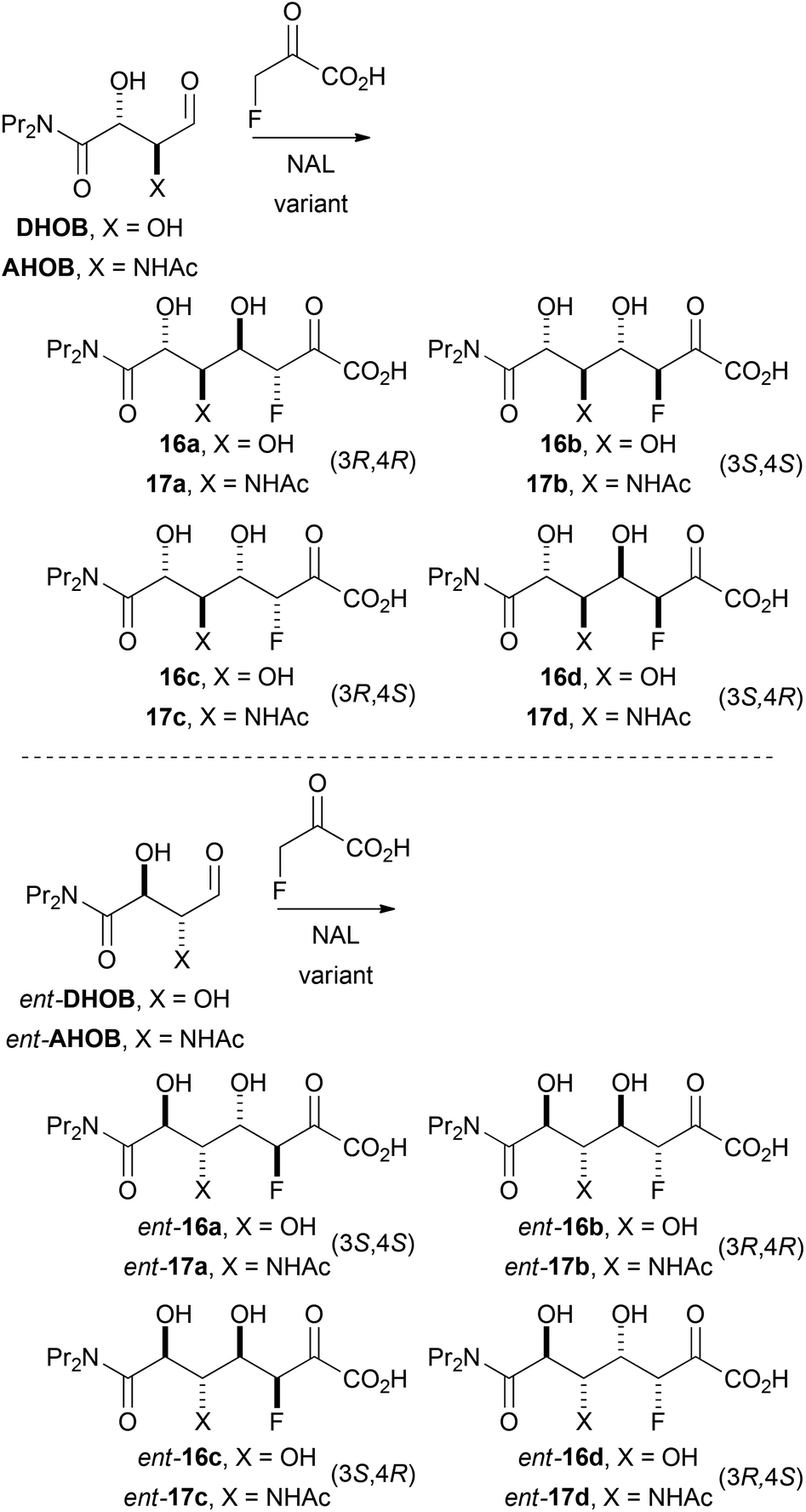 | ||
| Scheme 5 Possible diastereomeric products of aldolase-catalysed reactions with fluoropyruvate as nucleophile. | ||
| Substratea | Variant | Specific activityb/nmol min−1 nmol−1 | Product | Ratioca:b:c:d |
(3R,4R):(3S,4S):(3R,4S):(3S,4R)c |
|---|---|---|---|---|---|
| a Prepared by ozonolysis of the corresponding alkene (10, ent-10, 15 or ent-15). b Consumption of fluoropyruvate (nmol min−1 per nmol protein) determined by 296 MHz 19F NMR spectroscopy. c Kinetic ratio of diastereomeric products determined by 296 MHz 19F NMR spectroscopy. d Not measured. e Not detectable. | |||||
| DHOB | E192N | 9.1 | 16 | 40:0:50:10 |
40:0:50:10 |
| DHOB | E192N/T167V/S208V | 0.56 | 16 | 0:0:100:0 |
0:0:100:0 |
| DHOB | E192N/T167G | 0.06 | 16 | 30:0:70:0 |
30:0:70:0 |
| ent-DHOB | E192N | 0.46 | ent-16 | 10:0:0:90 |
0:10:90:0 |
| ent-DHOB | E192N/T167V/S208V | 0.03 | ent-16 | 0:0:0:100 |
0:0:100:0 |
| ent-DHOB | E192N/T167G | 0.12 | ent-16 | 20:0:0:80 |
0:20:80:0 |
| AHOB | E192N | 1.1 | 17 | 60:0:40:0 |
60:0:40:0 |
| AHOB | E192N/T167V/S208V | NDe | — | ||
| AHOB | E192N/T167G | 0.03 | 17 | NMd | |
| ent-AHOB | E192N | 0.07 | ent-17 | NMd | |
| ent-AHOB | E192N/T167V/S208V | NDe | — | ||
| ent-AHOB | E192N/T167G | 0.03 | ent-17 | NMd | |
The rate of consumption of fluoropyruvate was highest with the combination of DHOB and the E192N variant (9.1 nmol min−1 per nmol protein). This observation is, perhaps, unsurprising given that E192N was obtained via a directed evolution approach that sought to optimise catalysis of cleavage to yield DHOB.13 However, it is notable that the E192N variant – in addition to the wild-type enzyme – accepts fluoropyruvate as an alternative donor.
Catalysis by the E192N variant was significantly less efficient with the other substrates investigated. For example, with AHOB, in which the α-hydroxy group of DHOB has been replaced with an α-NHAc group, the rate of consumption of fluropyruvate was about 8-fold slower. Switching to the enantiomeric substrate series was also detrimental to catalysis: the rate of consumption of fluoropyruvate was about 20-fold slower with ent-DHOB (compared to DHOB) and about 15-fold slower with ent-AHOB (compared to AHOB).
In addition, the E192N/T167G and E192N/T167V/S208V variants are less efficient catalysts than the E192N variant. For example, with DHOB as substrate, the rate of consumption of fluoropyruvate was about 15- and 150-fold slower with the E192N/T167G and E192N/T167V/S208V variants respectively than with the E192N variant. These variants were generated to catalyse complementary stereoselective reactions between pyruvate and DHOB (Scheme 2): a reduction in the efficiency of catalysis (compared to the E192N variant) was also observed with pyruvate as the donor substrate.14
Preparation and characterisation of reaction products
The determination of the stereoselectivity of the reactions was complicated by the possibility of four diastereomeric products, each of which might exist in different anomeric and pyranose/furanose forms. To assist analysis, selected reactions were conducted preparatively, and the products purified and characterised (Table 4). In each case, the aldehyde substrate and sodium fluoropyruvate were dissolved in 50 mM Tris-HCl pH 7.4 buffer, and the relevant NAL variant added. The conversion of each reaction was determined by analysis of the crude product by 296 MHz 19F NMR spectroscopy.| Substratea (eq.) | Variant | Productb | Time/day (conversionc/%) | Yieldd/% (ratioe) |
|---|---|---|---|---|
| a Prepared by ozonolysis of the corresponding alkene (10, ent-10, 15 or ent-15). b See Table 1 for details of ratios of anomers and pyranose/furanose forms. c Determined by analysis of the crude product by 296 MHz 19F NMR spectroscopy. d Yield of purified product based on the limiting reactant. e Determined by 296 MHz 19F NMR spectroscopy. f Small samples of each diastereomer could be obtained by reverse-phase HPLC. g Not measured. h After purification by mass-directed HPLC. | ||||
| DHOB (2 eq.) | E192N | 16a and 16c | 2 (>99) | 33f (40:60) |
| DHOB (2 eq.) | E192N/T167 V/S208V | 16c | 2 (95) | 41 |
| ent-DHOB (1 eq.) | E192N/T167 V/S208V | ent-16d | 1 (NMg) | 52 |
| AHOB (5 eq.) | E192N | 17a | 5 (50) | 7h |
In two cases, the aldolase-catalysed reactions were highly diastereoselective, and >98:<2 mixtures of diastereomeric products were obtained after ion exchange chromatography. Thus, with the E192N/T167V/S208V variant, fluoropyruvate and DHOB reacted to give 16c which was isolated in 41% yield. Similarly, with the same NAL variant, fluoropyruvate and ent-DHOB reacted to give ent-16d which was isolated in 52% yield. However, with the E192N variant, fluoropyruvate and DHOB were converted into a 40:60 mixture of 16a and 16c from which it was possible to obtain small samples of both products after reverse-phase HPLC purification. Similarly, using E192N, fluoropyruvate and AHOB reacted to give a diastereomeric mixture of products, from which a small sample of 17a could be obtained by mass-directed HPLC.
The fluorinated products 16a, ent-16d and 17a existed in pyranose forms (Panel A, Fig. 2). In both pyranose anomers of 16a and 17a, there was a large coupling constant between fluorine and H-4 (∼30 Hz); in addition, in the major anomer of each compound, there was a small coupling constant between H-3 and H4 (16a: 3.4 Hz; 17a: 2.2 Hz) (Table 3). These data imply that 16a and 17a are (3R,4R)-configured (and indirectly enabled determination of the relative configuration of 13). In contrast, ent-16d had a small coupling constant between the equatorially-positioned fluorine and H-4 (13.3 Hz) and a large coupling constant between the axial protons H-3 and H-4 (9.3 Hz). The configuration of 16a and ent-16d was corroborated by the observation of nOe interactions between the axial protons at H-4 and H-6. The analysis of 16c was hugely complicated by the existence of both pyranose and furanose anomers (Panel B, Fig. 2); however, 1H/19F HSQC-TOCSY spectroscopy enabled extraction of the 1H NMR spectra of each of the four species that were present (Table 1 and ESI†). The pyranose anomers of 16c have axially-oriented fluorine and 4-OH groups which cannot enjoy a stabilising gauche interaction.16
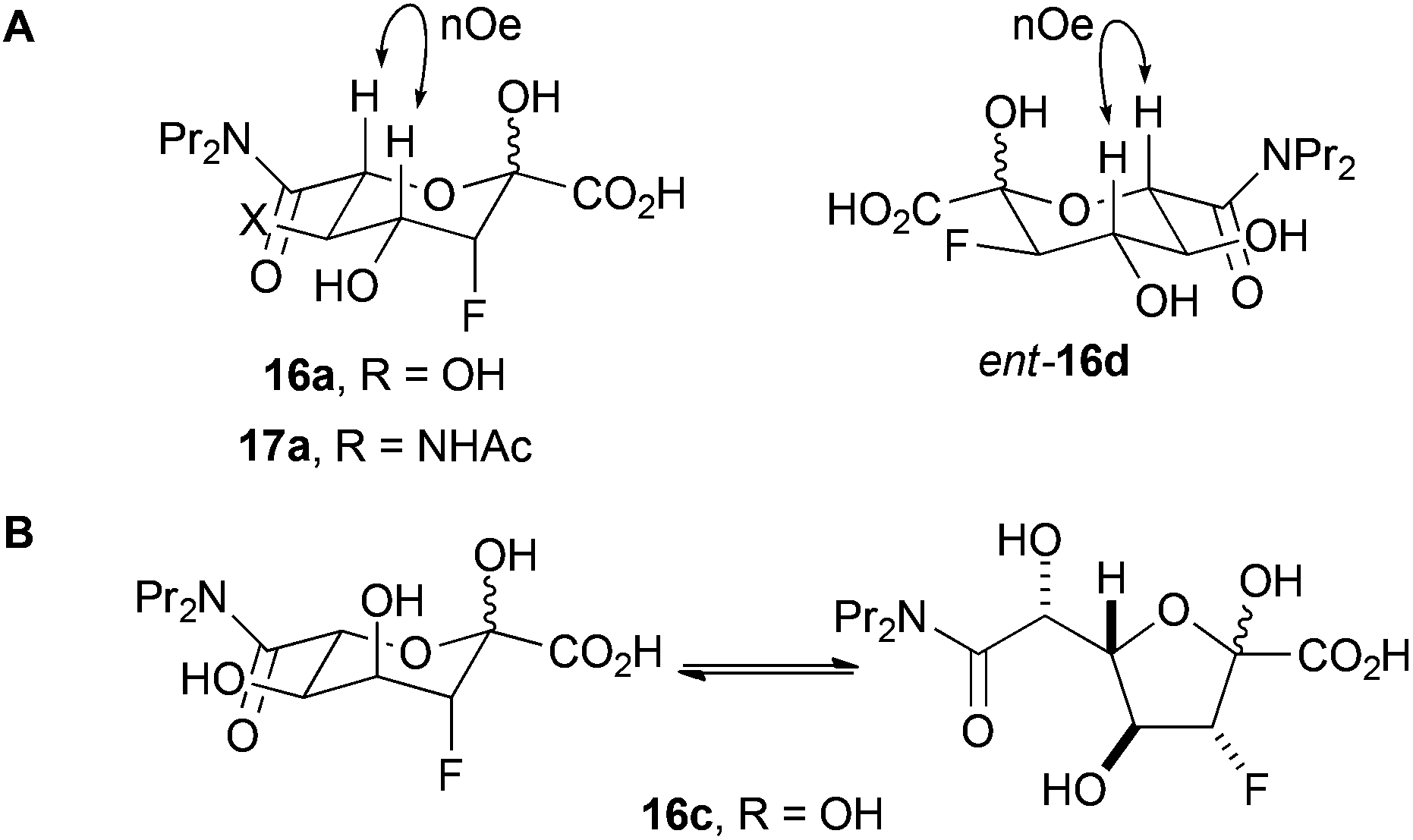 | ||
| Fig. 2 Forms of reaction products. Panel A: The products 16a, ent-16d and 17a exist predominantly in pyranose forms. Panel B: the product 16c exists as a mixture of pyranose and furanose anomers. | ||
:0:50:10 mixture of 16a, 16b, 16c and 16d; this poor stereoselectivity parallels that observed with this enzyme in the reaction between pyruvate and DHOB.13a,14 In contrast, the E192N/T167V/S208V variant yielded selectively the (3R,4S)-configured product 16c. This variant was generated14 by directed evolution to yield selectively the 4S-configured product (3b) with pyruvate as nucleophile (Scheme 2): it is remarkable that the 4S selectivity is retained with an alternative nucleophile (fluoropyruvate). However, in contrast, selectivity for 4R-configured products was not observed with the E192N/T167G variant: with this variant, the reaction between DHOB and fluoropyruvate was very inefficient, and a 30:70 mixture of 16a and 16c was obtained.
The effect of the structure of the aldehyde substrate on stereoselectivity was also investigated. AHOB has an α-NHAc group in place of the α-hydroxy group of DHOB; with AHOB and the E192N variant, similarly poor stereoselectivity was also observed with this substrate (17a:17b:17c:17d 60:0:40:0). In the enantiomeric series, ent-DHOB gave predominantly the (3R,4S)-configured product ent-16d with all three NAL variants; here, the NAL variant had only a small effect on the stereoselectivity of the aldol reaction.
To gain an insight into the structural basis of stereoselectivity, the crystal structure of S. aureus NAL was determined in complex with fluoropyruvate (PDB: 5A8G) (Panel A, Fig. 3); the structure and kinetic properties of S. aureus NAL have been previously shown to be extremely similar to those of E. coli NAL.22 The formation of a Z-configured enamine was observed, which presents only one face to aldehyde substrates. Reaction of this face of the (Z)-enamine intermediate would necessarily lead to the formation of 3R-configured products. The structure10 of an aldol product (4-epi-Neu5Ac) in complex with NAL (the Y137A variant of the E. coli enzyme) (PDB: 4BWL) is provided for comparison (Panel B, Fig. 3). Previous studies have shown that an analogue of DHOB – (2R,3S)-2,3-trihydroxy-4-oxo-N,N-dipropyl butanamide – can bind to the E192N variant of E. coli NAL in two distinct conformations (Panel B, Fig. 3).15 Aldehyde substrates may react via conformations that allow protonation by the general acid Y137.10 The facial selectivity of the reaction of the aldehyde determines the configuration of the product – (3R,4R) or (3R,4S) – obtained (Panel C, Fig. 3). In some cases, the ratio of C-4 epimers could be changed by altering the specific NAL variant used.
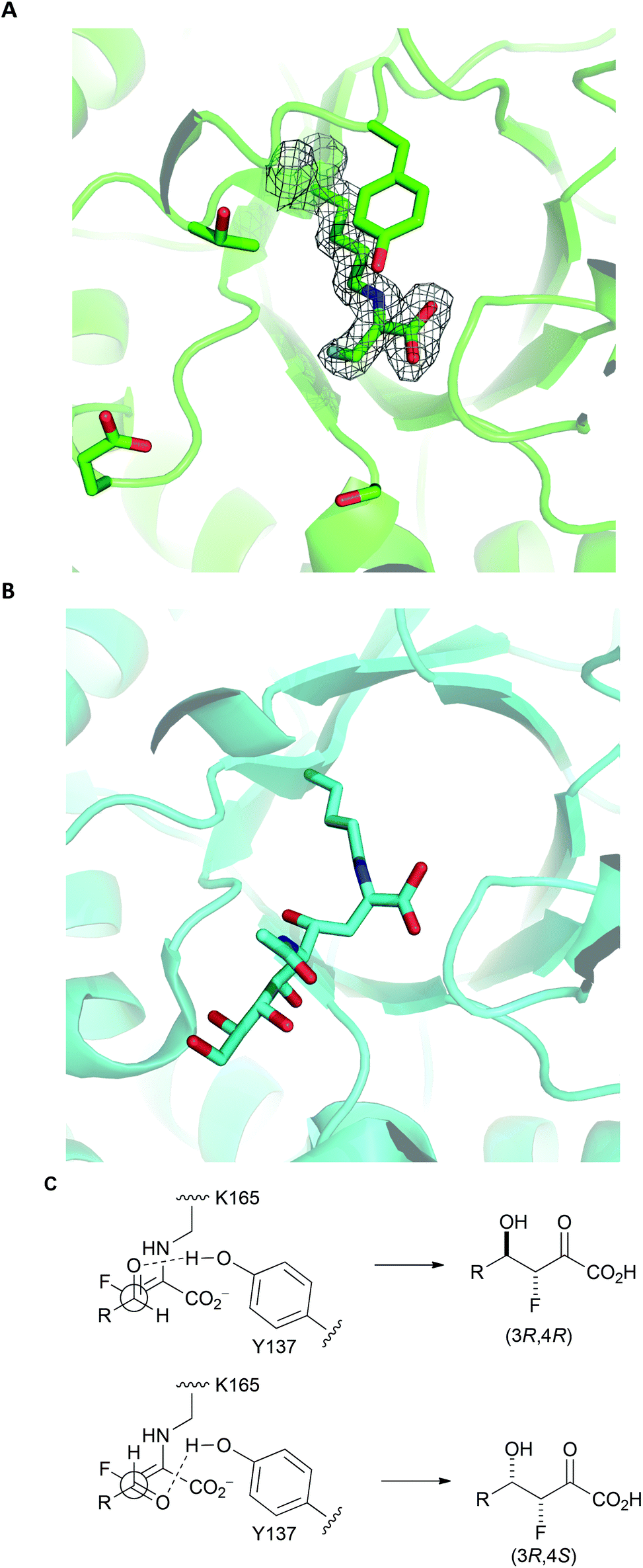 | ||
| Fig. 3 Rationale for stereoselectivity of NAL-catalysed reactions between fluoropyruvate and aldehyde substrates. Panel A: S. aureus NAL in complex with fluoropyruvate with general acid Y137 and residues that have key roles in recognition and stereocontrol (T167, E192 and S208) shown (PDB: 5A8G). The top face (as depicted) of the Z-configured enamine is poised to react with an aldehyde substrate. Panel B: Y137A variant of E. coli NAL in compex with 4-epi-Neu5Ac (PDB: 4BWL). Panel C: Possible stereochemical outcomes of the reaction of the top face of the Z-configured enamine with an aldehyde substrate. | ||
Conclusions
NAL variants can be useful catalysts of reactions between fluoropyruvate and aldehyde substrates. Wild-type NAL catalysed the reaction between fluoropyruvate and ManNAc, albeit much less efficiently than with pyruvate as donor. It was shown that a 90:10 ratio of (3R,4R)- and (3S,4R)-configured products was obtained under kinetic control; whilst a 30:70 mixture of these products was obtained at equilibrium. The switch between kinetic and thermodynamic control may account for previous apparently conflicting reports of the outcome of this reaction.12
It was also shown that NAL variants are useful catalysts of reactions between fluoropyruvate and unnatural aldehyde substrates. The efficiency of catalysis varied widely, depending on the specific combination of NAL variant and aldehyde used. However, using the aldehyde DHOB or its enantiomer as substrate, three of the four possible diastereomeric products could be isolated.
It was noted that, under kinetic control, all productive NAL variant-catalysed reactions involving fluoropyruvate yielded (3R)-configured products selectively. The crystal structure of S. aureus NAL in complex with fluoropyruvate reveals the presence of a (Z)-configured enamine. The (3R)-selectivity of NAL catalysed reactions may be rationalised in terms of selective reaction of this (Z)-configured enamine via the face that is presented to aldehyde substrates.
Acknowledgements
We thank BBSRC, EPSRC and AstraZeneca for funding studentships and EPSRC for funding core chemistry equipment (EP/K039202/1).Notes and references
- (a) F. M. D. Ismail, J. Fluorine Chem., 2002, 118, 27 CrossRef CAS; (b) K. Muller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (c) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed; (d) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015 DOI:10.1021/acs.jmedchem.5b00258.
- D. O'Hagan, J. Fluorine Chem., 2010, 131, 1071 CrossRef.
- B. D. Roth, Prog. Med. Chem., 2002, 40, 1 CrossRef CAS PubMed.
- E. Cholongitas and G. V. Papatheodoridis, Ann. Gastroenterol., 2014, 27, 331 Search PubMed.
- For an example, see: J.-H. Kim, R. Resende, T. Wennekes, H.-M. Chen, N. Bance, S. Buchini, A. G. Watts, P. Pilling, V. A. Streltsov, M. Petric, R. Liggins, S. Barrett, J. L. McKimm-Breschkin, M. Nikura and S. G. Withers, Science, 2013, 340, 71 CrossRef CAS PubMed.
- (a) B. Greedy, J.-M. Paris, T. Vidal and V. Gouverneur, Angew. Chem., Int. Ed., 2003, 115, 3413 CrossRef; (b) G. T. Giuffredi, S. Purser, M. Sawicki, A. L. Thompson and V. Gouverneur, Tetrahedron: Asymmetry., 2009, 20, 910 CrossRef CAS.
- (a) T. D. Beeson and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 8826 CrossRef CAS PubMed; (b) M. Marigo, D. Fielenbach, A. Braunton, A. Kjærsgaard and K. A. Jørgensen, Angew. Chem., Int. Ed., 2005, 44, 3703 CrossRef CAS PubMed.
- S. M. Kim, H. R. Kim and D. Y. Kim, Org. Lett., 2005, 7, 2309 CrossRef CAS PubMed.
- (a) G. Zhong, J. Fan and C. F. Barbas III, Tetrahedron Lett., 2004, 45, 5681 CrossRef CAS; (b) X.-Y. Xu, Y.-Z. Wang and L.-Z. Gong, Org. Lett., 2007, 9, 4247 CrossRef CAS PubMed; (c) X.-Y. Xu, Y.-Z. Wang, L.-F. Cun and L.-Z. Gong, Tetrahedon: Asymmetry., 2007, 18, 237 CrossRef CAS.
- A. D. Daniels, I. Campeotto, M. W. Van der Kamp, A. H. Bolt, C. H. Trinh, S. E. V. Phillips, A. R. Pearson, A. Nelson, A. J. Mulholland and A. Berry, ACS Chem. Biol., 2014, 9, 1025 CrossRef CAS PubMed.
- R. Gantt, S. Millner and S. B. Binkley, Biochemistry, 1964, 3, 1952 CrossRef CAS PubMed.
- (a) H. A. Chokhawala, H. Cao, H. Yu and X. Chen, J. Am. Chem. Soc., 2007, 129, 10630 CrossRef CAS PubMed; (b) J. Beliczey, U. Kragl, A. Liese, C. Wandrey, K. Hamacher, H. H. Coenen and T. Tierling, US Patent, 635543, 2002 Search PubMed; (c) A. G. Watts and S. G. Withers, Can. J. Chem., 2004, 82, 1581 CrossRef CAS.
- (a) T. Woodhall, G. J. Williams, A. Berry and A. Nelson, Angew. Chem., Int. Ed., 2005, 44, 2109 CrossRef CAS PubMed; (b) G. J. Williams, T. Woodhall, A. Nelson and A. Berry, Protein Eng. Des. Sel., 2005, 18, 239 CrossRef CAS PubMed.
- G. J. Williams, T. Woodhall, L. M. Farnsworth, A. Nelson and A. Berry, J. Am. Chem. Soc., 2006, 128, 16238 CrossRef CAS PubMed.
- I. Campeotto, A. H. Bolt, T. A. Harman, C. Dennis, C. H. Trinh, S. E. V. Phillips, A. Nelson, A. R. Pearson and A. Berry, J. Mol. Biol., 2010, 404, 56 CrossRef CAS PubMed.
- D. O'Hagan, J. Org. Chem., 2012, 77, 3689 CrossRef PubMed.
- (a) M. Karplus, J. Am. Chem. Soc., 1963, 85, 2870 CrossRef CAS; (b) W. R. Dolbier, Guide to Fluorine NMR for Organic Chemists, Wiley, New Jersey, 2009 Search PubMed.
- T. Woodhall, G. J. Williams, A. Berry and A. Nelson, Org. Biomol. Chem., 2005, 3, 1795 CAS.
- S.-Y. Han, M. M. Joullie, V. V. Fokin and N. A. Petasis, Tetrahedron: Asymmetry., 1994, 5, 2535 CrossRef CAS.
- M. Godskesen, I. Lundt and I. Sotofte, Tetrahedron: Asymmetry., 2000, 11, 567 CrossRef CAS.
- S. V. Ley, E. Diez, D. Dixon, R. T. Guy, P. Michel, G. L. Nattrass and T. D. Sheppard, Org. Biomol. Chem., 2004, 2, 3608 CAS.
- N. Timms, C. L. Windle, A. Polyakova, J. R. Ault, C. H. Trinh, A. R. Pearson and A. Berry, ChemBioChem, 2013, 14, 474 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ob02037a |
| ‡ Present address: Centre for Ultrafast Imaging, Institute of Nanostructure and Solid State Physics, University of Hamburg, Luruper Chausee 149, Hamburg 22761, Germany. |
| § (2R,3S)-3-acetyl-2-hydroxy-4-oxo-N,N-dipropylbutanamide. |
| This journal is © The Royal Society of Chemistry 2016 |