
Sulfonyl radical-enabled 6-endo-trig cyclization for regiospecific synthesis of unsymmetrical diaryl sulfones†
Rong
Fu
a,
Wen-Juan
Hao
*a,
Ya-Nan
Wu
a,
Nan-Nan
Wang
a,
Shu-Jiang
Tu
*a,
Guigen
Li
bc and
Bo
Jiang
 *ab
*ab
aSchool of Chemistry and Chemical Engineering, Jiangsu Key Laboratory of Green Synthetic Chemistry for Functional Materials, Jiangsu Normal University, Xuzhou 221116, P. R. China. E-mail: wjhao@jsnu.edu.cn; laotu@jsnu.edu.cn; jiangchem@jsnu.edu.cn; Fax: +8651683500065; Tel: +8651683500065
bDepartment of Chemistry and Biochemistry, Texas Tech University, Lubbock, Texas 79409-1061, USA
cInstitute of Chemistry & BioMedical Sciences, Collaborative Innovation Center of Chemistry for Life Sciences, Nanjing University, Nanjing 210093, P. R. China
First published on 23rd August 2016
Abstract
A new sulfonyl radical-triggered 6-endo-trig cyclization of unactivated 1,5-enynes under catalytic oxidation conditions for flexible synthesis of unprecedented unsymmetrical diaryl sulfones was established. This transformation could also proceed smoothly using 2-arylalkynyl-1,1′-biphenyls, delivering new sulfonylated phenanthrenes. The synthetic utility of these transformations results in subsequent C–S and C–C bond-forming reactions to effectively build up functional sulfones with potential significance.
Unsymmetrical diaryl sulfones constitute the core structures of various drugs and pharmacologically active compounds. These have been shown to display antifungal and antitumor activities or serve as inhibitors of the human immunodeficiency virus-type 1 (HIV-1) reverse transcriptase.1 Therefore, great efforts have been devoted to the preparation of unsymmetrical diaryl sulfones. Known procedures for unsymmetrical aryl sulfone formation include nucleophilic substitution between sulfonate esters and organometallic reagents (Scheme 1, route i),2 arylsulfonylation of arenes (route ii)3 or diaryliodonium salts (route iii),4 and coupling of aryl halides with sulfinates (route iv),5 oxidation of aryl sulfides (route v),6 and metal-catalyzed C(sp2)–H arylsulfonylation of arenes (DG = directed groups, route vi),7 as well as domino [3 + 3]cyclization (route vii).8 Despite these significant advances, to the best of our knowledge, catalytic sulfonyl-radical triggered 1,5-enyne-cyclization for the regiospecific construction of unsymmetrical diaryl sulfones has not yet been documented (Scheme 1, route viii).
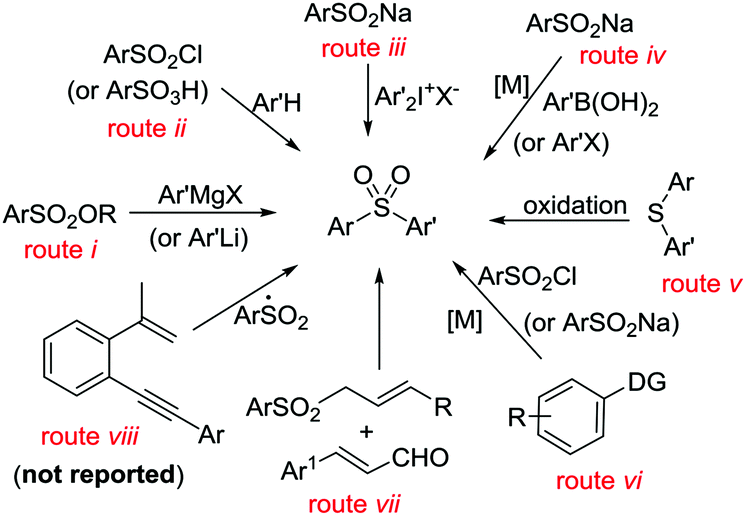 | ||
| Scheme 1 Various methods for the synthesis of diaryl sulfones. | ||
Moreover, catalytic 1,5-enyne-cyclizations have become an important platform for the collection of complicated hetero-and carbocycles.9 These molecules have been widely used for selective domino cyclizations via synergistic additions across C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C
C and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bonds.10 Thus far, two main 1,5-enyne-cyclization strategies involving metal catalysis11 or electrophilic cyclization12 have been developed. In contrast, oxidative radical 1,5-enyne-cyclization for accessing polycycles has been seldom investigated.13 Recently, our group developed an arylsulfonyl-radical triggered 5-exo-dig/6-endo-trig bicyclization for the assembly of benzo[b]fluorens using carbonyl-activated 1,5-enynes (Scheme 2a).14 High selectivity, in which the arenesulfonyl radicals add to the alkyne (Scheme 2c, route b) rather than to the alkene (Scheme 2b, route a) is of particular interest to us. We speculated that the addition of sulfonyl radicals to the CC bond might be a reversible process and the regioselective sulfonyl radical addition to alkynyl unit could give thermodynamically stable unsymmetrical diaryl sulfones. Herein, we would like to report this new catalytic sulfonyl radical-triggered unactivated 1,5-enyne-cyclization, leading to the regiospecific formation of unsymmetrical diaryl sulfones 4 (Scheme 2d). Using 2-arylalkynyl-1,1′-biphenyls as a replacement for 1,5-enynes, this transformation could also proceed smoothly, enabling sulfonyl radical-triggered 6-endo-trig cyclization and homolytic aromatic substitution (HAS) to deliver unsymmetrical diaryl sulfones 6 with different substitution patterns (Scheme 2e).
C bonds.10 Thus far, two main 1,5-enyne-cyclization strategies involving metal catalysis11 or electrophilic cyclization12 have been developed. In contrast, oxidative radical 1,5-enyne-cyclization for accessing polycycles has been seldom investigated.13 Recently, our group developed an arylsulfonyl-radical triggered 5-exo-dig/6-endo-trig bicyclization for the assembly of benzo[b]fluorens using carbonyl-activated 1,5-enynes (Scheme 2a).14 High selectivity, in which the arenesulfonyl radicals add to the alkyne (Scheme 2c, route b) rather than to the alkene (Scheme 2b, route a) is of particular interest to us. We speculated that the addition of sulfonyl radicals to the CC bond might be a reversible process and the regioselective sulfonyl radical addition to alkynyl unit could give thermodynamically stable unsymmetrical diaryl sulfones. Herein, we would like to report this new catalytic sulfonyl radical-triggered unactivated 1,5-enyne-cyclization, leading to the regiospecific formation of unsymmetrical diaryl sulfones 4 (Scheme 2d). Using 2-arylalkynyl-1,1′-biphenyls as a replacement for 1,5-enynes, this transformation could also proceed smoothly, enabling sulfonyl radical-triggered 6-endo-trig cyclization and homolytic aromatic substitution (HAS) to deliver unsymmetrical diaryl sulfones 6 with different substitution patterns (Scheme 2e).
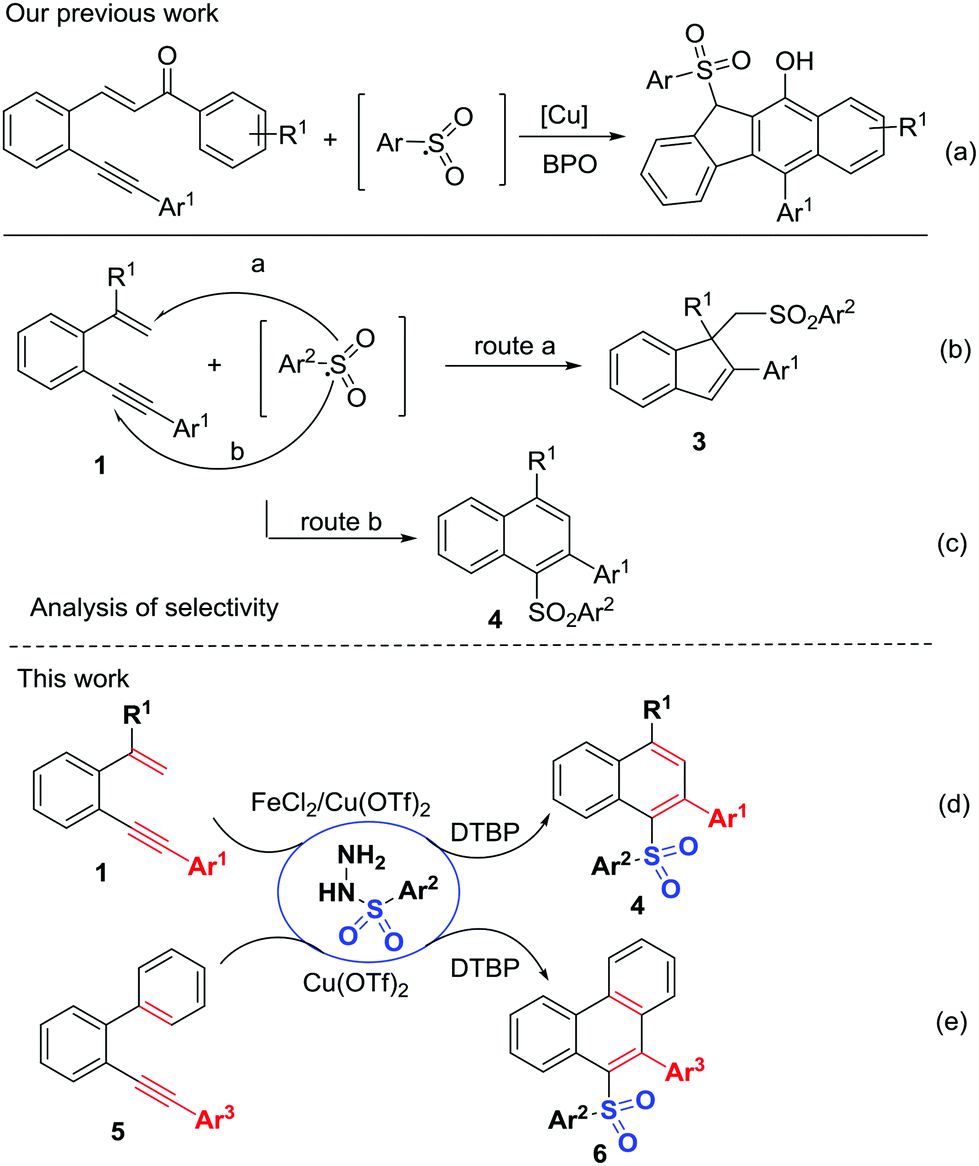 | ||
| Scheme 2 Sulfonyl radical-triggered cyclization. | ||
Our initial investigation commenced with the reaction of unactivated 1,5-enyne 1a with commercially available tosylhydrazide (2a) under air conditions using di-tert-butyl peroxide (DTBP) in 1,2-dichloroethane (DCE) at 120 °C. Theoretically, this reaction may undergo two different pathways to deliver arylsulfonylated indenes 3a and naphthalenes 4a, respectively (via pathways shown in Scheme 2b and c). In fact, only product 4a was obtained in a 29% chemical yield. Product 3a was not isolated (see the ESI, Table S1,† entry S1). Encouraged by the aforementioned result, we next optimized reaction conditions. First, the solvent effect was evaluated. The reaction in 1,4-dioxane led to a lower yield (entry S2). Exchanging 1,4-dioxane for N,N-dimethylformamide (DMF) completely suppressed the reaction process (entry S3). In the case of acetonitrile (CH3CN), slightly higher conversion was observed and 34% yield of 4a was provided (entry S4). Using 10 mol% of FeCl2 further facilitated this radical-triggered 1,5-enyne-cyclization and afforded a higher 38% yield (entry S5). The yield could be improved to 43% when the dosage of DTBP was increased to 5.0 equivalents in CH3CN at 120 °C (entry S6). Further increasing the load of DTBP is adverse to this transformation (entry S7). Using 10 mol% of Cu(OTf)2 as a catalyst gives access to the desired product 4a in a 30% yield (entry S8). Next, we found that the combination of FeCl2 with Cu(OTf)2 is beneficial to the reaction efficiency (entry S9). Delightfully, this transformation worked even more efficiently in the presence of 0.5 equivalent of DBU, affording a 77% yield of 4a (entry S11). Other bases such as tBuOLi, Cs2CO3 and Et3N met little success with respect to the reaction yields (entries S12–S14). The use of molecular sieves resulted in a low conversion (entry S15). Subsequent screening of other oxidants like tert-butyl peroxy benzoate (TBPB), dicumyl peroxide (DCP) and tert-butyl hydroperoxide (TBHP, 70% in water) did not improve this transformation (entries 16–19). Fine-tuning of the stoichiometric ratio of 1a and 2a and the reaction temperature did not ameliorate the reaction efficiency (entries 20–22).
Under the optimized reaction conditions, we then explored the scope of this bimetallic Cu/Fe-co-catalyzed radical cyclization using a variety of unactivated 1,5-enynes 1 and sulfonyl hydrazides 2 (Scheme 3). The reaction was first repeated using unactivated 1,5-enyne 1a and substrates 2a–2h, which possessed electron-donating and electron-withdrawing groups at different positions of the phenyl ring. All of the sulfonyl hydrazides participate in the reaction, leading to the corresponding unsymmetrical diaryl sulfones 4a–4h with yields ranging from 31% to 77%. A variety of functional groups including methyl, t-butyl, methoxy, chloro and bromo can tolerate the oxidative conditions well. The sterically more demanding o-chlorophenyl (2e), 1-naphthyl (1-Np) (2g) and 2-naphthyl (2-Np) (2h) counterparts were all adaptable sulfonyl components, enabling their radical-initiated 6-endo-trig cyclizations to access the desired products 4e and 4g–4h, under the standard conditions, albeit with relatively lower yields (31%–52%). Unfortunately, benzylsulfonyl hydrazide (2i) was proven to be an ineffective substrate, perhaps due to instability of the sulfonyl radicals generated in situ under oxidative conditions. Next, the scope with respect to the 1,5-enyne partner was systematically explored. As we expected, the reaction could be extended to different functionalities including methyl (1b), methoxy (1c, p-methoxyphenyl = PMP) and fluoro (1d) on the arylalkynyl moiety, with the corresponding products 4j–4q afforded in 42%–68% yields through domino radical-initiated 6-endo-trig cyclization of unactivated 1,5-enynes. Similarly, methyl group was replaced with a phenyl group on the alkenyl unit (1,5-enyne 1e), confirming the reaction efficiency, and 4r was afforded in 64% yield. Note that this is the first reported procedure for the oxidative synthesis of these new unsymmetrical diaryl sulfones through a bimetallic Cu/Fe-co-catalyzed radical 6-endo-trig cyclization of unactivated 1,5-enynes.
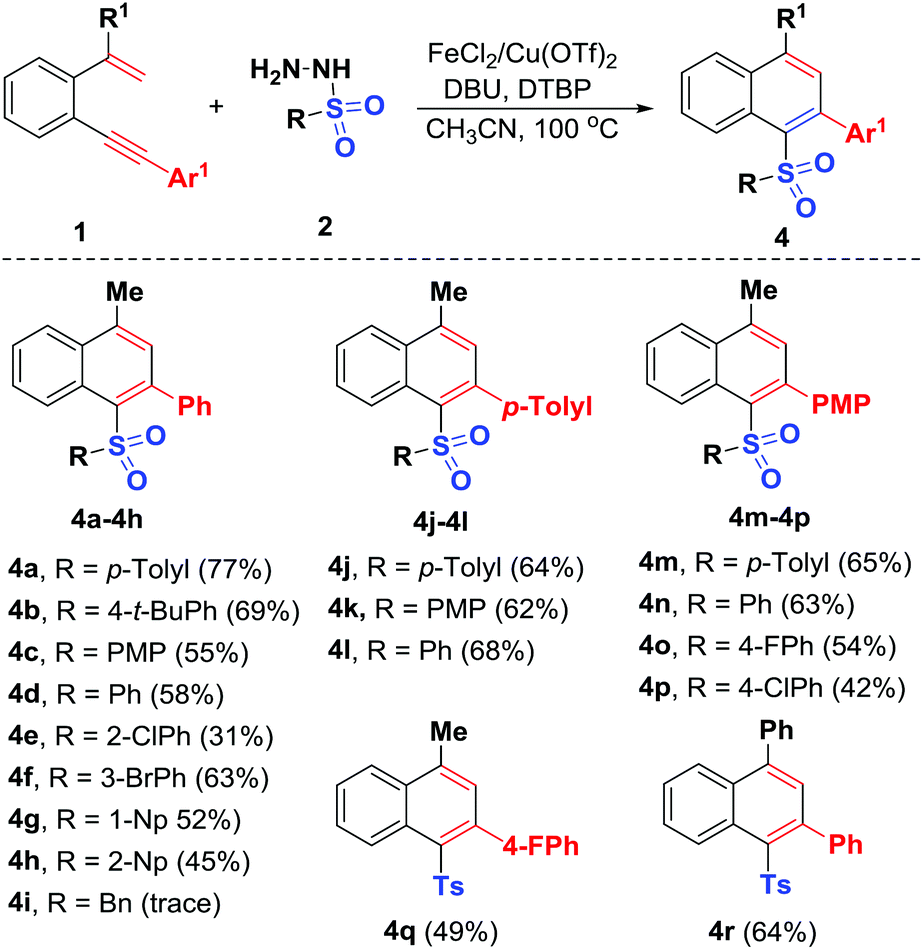 | ||
| Scheme 3 Substrate scope for synthesis of 4. (i) Reaction conditions: 1,5-enynes (1, 0.2 mmol, 1.0 equiv.), sulfonyl hydrazide (2, 0.4 mmol, 2.0 equiv.), DBU (0.1 mmol, 0.5 equiv.), FeCl2 (0.02 mmol), Cu(OTf)2 (0.02 mmol), DTBP (1.0 mmol, 5.0 equiv.), CH3CN (1.0 mL), 100 °C under air conditions. (ii) Isolated yield in brackets was based on 1. | ||
After the successful synthesis of diaryl sulfones 4, we focused our efforts on the evaluation of another new radical 6-endo-trig cyclization by replacement of 1,5-enynes with 2-arylalkynyl-1,1′-biphenyls as the sulfonyl radical acceptors. The reaction of 5a with 2a was performed under the above-described conditions, but only 15% yield of sulfonylated phenanthrene 6a was isolated (see the ESI, Table S2,† entry S1). Without FeCl2 and DBU (entries S2 and S3), the reaction gave access to the desired phenanthrene product 6a in 25% yield. Increasing the reaction temperature led to a slightly higher yield (34%, entry S5). Switching the solvent from acetonitrile to 1,2-dichloroethane (DCE) furnished 6a in a 43% isolated yield (entry S6). After careful optimizations, including solvents, catalysts and oxidants, we found that using Cu(OTf)2 (10 mol%) and DTBP (6.0 equiv.) in the presence of 4 Å molecular sieves (MS) in DCE gave the best outcome (50%, entry S16).
With the optimal conditions established, the generality of this Cu-catalyzed radical 6-endo-trig cyclization of 2-arylalkynyl-1,1′-biphenyls with various sulfonyl hydrazides was investigated carefully. As shown in Scheme 4, 2-arylalkynyl-1,1′-biphenyl 5a was first subjected to reaction with arylsulfonyl hydrazides of different electronic properties, and the expected sulfonylated phenanthrenes 6a–6j were afforded in 25%–60% yields, which may be ascribed to their low reactivity. Various arylsulfonyl hydrazides having substituents at different positions with either electronically rich (methyl, t-butyl, methoxy), neutral (H), or poor (fluoro, chloro, bromo, trifluoromethyl) groups were compatible. Similar to the above 1,5-enyne-cyclization, benzylsulfonyl hydrazide did not work in this reaction (6k). Next, the scope of the 2-arylalkynyl-1,1′-biphenyls 5 was investigated by the adoption of four examples of representative sulfonyl radical acceptors. These reactions all proceeded smoothly, delivering a series of new unsymmetrical diaryl sulfone products 6l–6s in moderate yields. Functional groups like fluoro (5b), methyl (5c), and methoxy (5e) at the para-position of the arylalkynyl moiety can tolerate these catalytic oxidation conditions well. The structures of the resulting unsymmetrical diaryl sulfone 4 and 6 were confirmed by NMR spectroscopy and HRMS. In the case of products 4a and 6n, their structures were further determined by X-ray diffraction (see the ESI†).
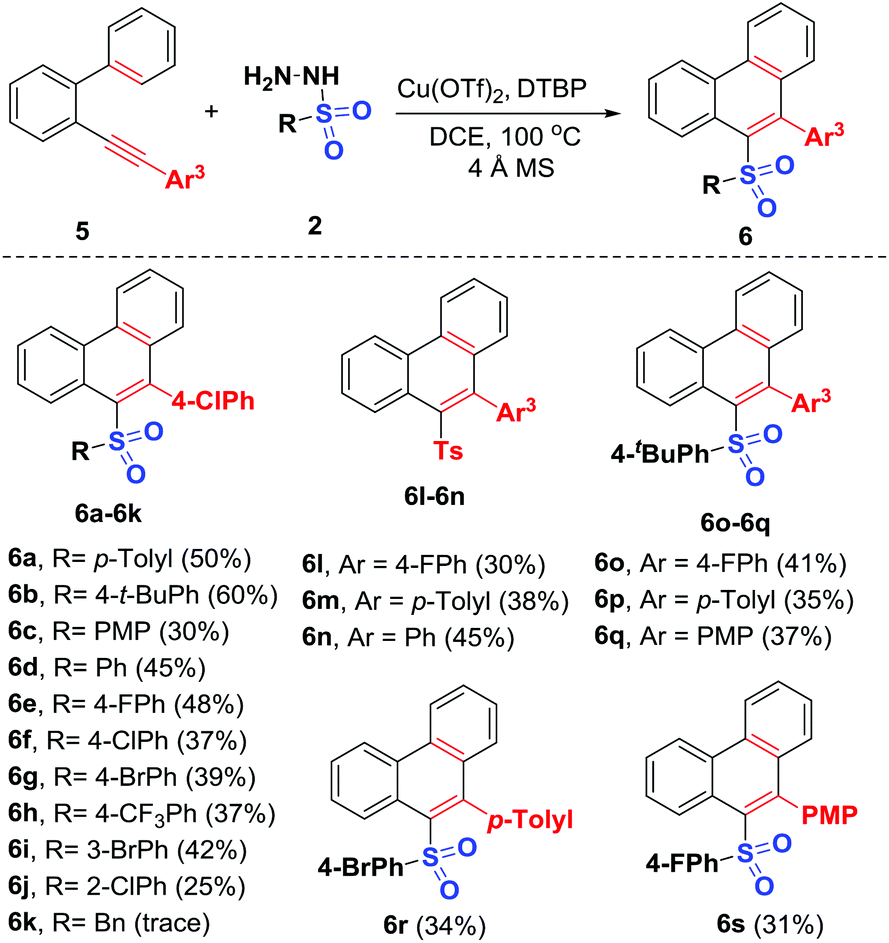 | ||
| Scheme 4 Substrate scope for synthesis of 6. (i) Reaction conditions: 5 (0.2 mmol, 1.0 equiv.), 2 (0.8 mmol, 4 equiv.), DTBP (1.2 mmol, 6.0 equiv.), Cu(OTf)2 (0.01 mmol), 4 Å MS (100 mg), DCE (1.0 mL) at 120 °C. (ii) Isolated yield in brackets was based on 5. | ||
To understand the mechanism, the following control experiments were conducted: 1,5-enyne 1a was subjected to the reaction with 5.0 equiv. of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) or butylhydroxytoluene (BHT) (Scheme 5a) without observation of expected sulfone product 4a, suggesting the existence of a radical mechanism. In the absence of DTBP, the reaction did not proceed and the starting materials were recovered (Scheme 5b). Without arylsulfonyl hydrazides, 1,5-enyne 1a failed to convert into 3-phenylnaphthalene 7 under standard conditions (Scheme 5c). These control experiments indicate that DTBP is essential for the catalytic cycles and in situ generated arylsulfonyl radicals trigger addition/6-endo-trig cyclization. Therefore, sulfonylation occurs prior to the radical cyclization step.16
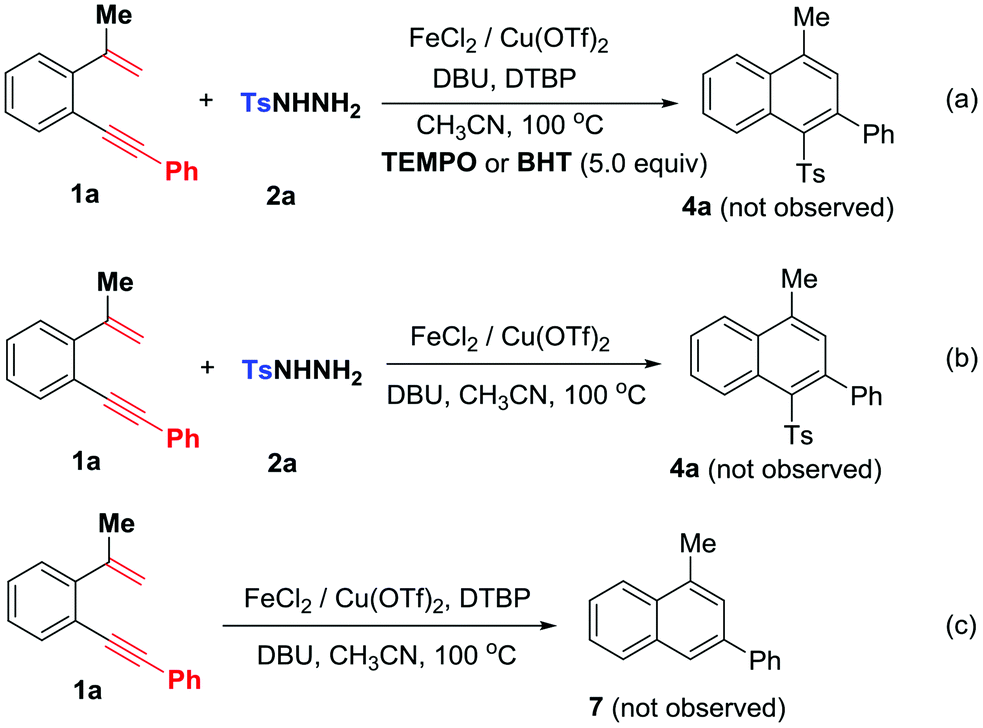 | ||
| Scheme 5 Control experiments. | ||
A plausible mechanism is proposed in Scheme 6 on the basis of the above observations and those reported in literature.15 The first step is to form the sulfonyl radical by reaction of sulfonyl hydrazides with tert-butoxy radicals generated in situ by Fe-assisted decomposition of DTBP.17 The intermolecular addition of the resulting sulfonyl radical onto alkynyl unit of 1,5-conjugated enynes 1, followed by intramolecular 5-endo-trig cyclization gives intermediate C, which undergoes SET (single electron transfer) oxidation and subsequent deprotonation to provide sulfonylated naphthalenes 4. The mechanism for the formation of sulfonylated phenanthrene 6 is similar to the above proposed one. Although the synthesis of unsymmetrical diaryl sulfones has been achieved previously,2–8 sulfonyl radical-initiated 6-endo-trig cyclization methodology for their synthesis is very rare.18
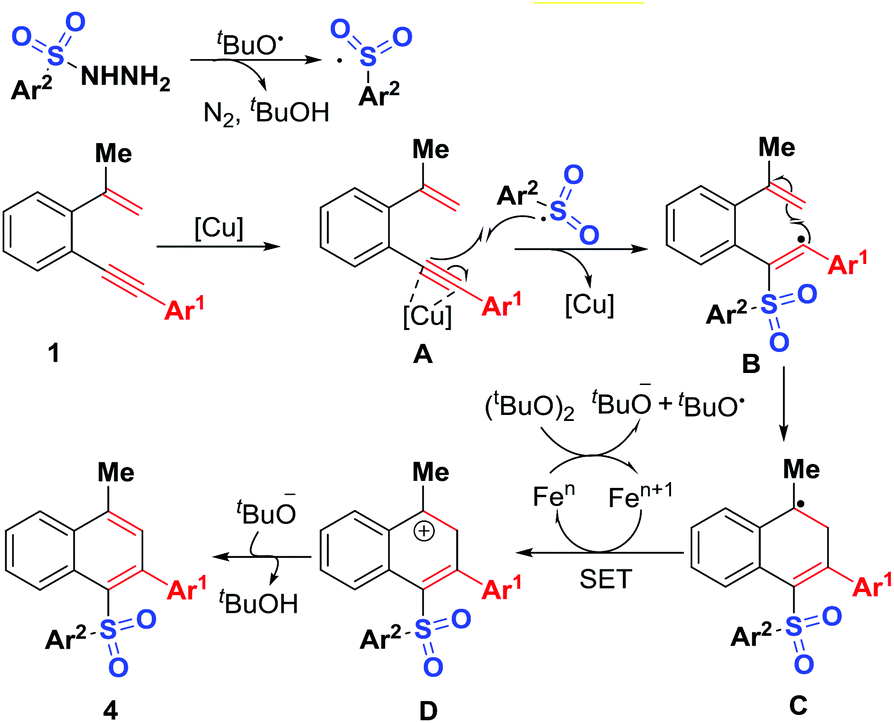 | ||
| Scheme 6 Proposed mechanism for forming products 4. | ||
To summarize, we have discovered new sulfonyl radical-triggered 6-endo-trig cyclizations of unactivated 1,5-enynes and 2-arylalkynyl-1,1′-biphenyls under convenient catalytic conditions. The regiospecific addition of in situ generated sulfonyl radicals onto unactivated triple bonds is able to trigger a 6-endo-trig cyclization and SET sequence, delivering unsymmetrical diaryl sulfones in a successive C–S and C–C bond-forming process. Both methods provide direct and practical access to a wide range of unreported aryl sulfones with potential usefulness.
We are grateful for financial support from the NSFC (No. 21232004, 21472071, 21602087), PAPD of Jiangsu Higher Education Institutions, the Outstanding Youth Fund of JSNU (YQ2015003), NSF of Jiangsu Province (BK20151163, BK 20160212), NSF of JSNU (14XLR005), the Qing Lan and NSF of Jiangsu Education Committee (15KJB150006).
Notes and references
- (a) M. Artico, R. Silvestri, E. Pagnozzi, B. Bruno, E. Novellino, G. Greco, S. Massa, A. Ettorre, A. G. Loi, F. Scintu and P. J. La Colla, Med. Chem., 2000, 43, 1886 CrossRef CAS; (b) M. Artico, R. Silvestri, S. Massa, A. G. Loi, S. Corrias, G. Piras and P. J. La Colla, Med. Chem., 1996, 39, 522–530 CrossRef CAS PubMed; (c) T. M. Williams, T. M. Ciccarone, S. C. MacTough, C. S. Rooney, S. K. Balani, J. H. Condra, E. A. Emini, M. E. Goldman and W. J. Greenlee, J. Med. Chem., 1993, 36, 1291 CrossRef CAS PubMed; (d) J. B. McMahon, R. J. Gulakowski, O. S. Weislow, R. J. Schultz, V. L. Narayanan, D. J. Clanton, R. Pedemonte, F. W. Wassmundt, R. W. Buckheit and W. D. Decker, Antimicrob. Agents Chemother., 1993, 37, 754 CrossRef CAS PubMed.
- (a) H. Gilman, N. J. Beaber and C. H. Myers, J. Am. Chem. Soc., 1925, 47, 2047 CrossRef CAS; (b) W. H. Baarschers, Can. J. Chem., 1976, 54, 3056 CrossRef CAS.
- (a) B. M. Graybill, J. Org. Chem., 1967, 32, 2931 CrossRef CAS; (b) M. Ueda, K. Uchiyama and T. Kano, Synthesis, 1984, 323 CrossRef CAS; (c) W. E. Truce, T. C. Klinger and W. W. Brand, in Organic Chemistry of Sulfur, ed. S. Oae, Plenum Press, New York, 1977 Search PubMed; (d) S. J. Nara, J. R. Harjani and M. M. Salunkhe, J. Org. Chem., 2001, 66, 8616 CrossRef CAS PubMed; (e) C. G. Frost, J. P. Hartley and A. J. Whittle, Synlett, 2001, 830 CrossRef CAS.
- (a) N. Umierski and G. Manolikakes, Org. Lett., 2013, 15, 188 CrossRef CAS PubMed; (b) X. Liu, W. Li, D. Zheng, X. Fan and J. Wu, Tetrahedron, 2015, 71, 3359 CrossRef CAS; (c) D. Kumar, V. Arun, M. Pilania and K. P. C. Shekar, Synlett, 2013, 831 CrossRef CAS; (d) N. Margraf and G. Manolikakes, J. Org. Chem., 2015, 80, 2582 CrossRef CAS PubMed; (e) N. Umierski and G. Manolikakes, Org. Lett., 2013, 15, 4972 CrossRef CAS PubMed.
- For selected examples, see: (a) S. Cacchi, G. Fabrizi, A. Goggiamani and L. M. Parisi, Org. Lett., 2002, 4, 4719 CrossRef CAS PubMed; (b) S. Cacchi, G. Fabrizi, A. Goggiamani, L. M. Parisi and R. Bernini, J. Org. Chem., 2004, 69, 5608 CrossRef CAS PubMed; (c) A. Kar, I. A. Sayyed, W. F. Lo, H. M. Kaiser, M. Beller and M. K. Tse, Org. Lett., 2007, 9, 3405 CrossRef CAS PubMed; (d) E. J. Emmett, B. R. Hayter and M. C. Willis, Angew. Chem., Int. Ed., 2013, 52, 12679 CrossRef CAS PubMed; (e) F. Hu and X. Lei, ChemCatChem, 2015, 7, 1539 CrossRef CAS. For reviews, see: (f) I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2011, 111, 1596 CrossRef CAS PubMed; (g) C. Shen, P. Zhang, Q. Sun, S. Bai, T. S. A. Hor and X. Liu, Chem. Soc. Rev., 2015, 44, 291 RSC.
- (a) R. S. Varma, R. K. Saini and H. M. Meshram, Tetrahedron Lett., 1997, 38, 6525 CrossRef CAS; (b) R. S. Ward and R. L. Diaper, Sulfur Rep., 2001, 22, 251 CrossRef CAS; (c) A. Ivachtchenko, E. Golovina, M. Kadieva, O. Mitkin, S. Tkachenko and I. Okun, Bioorg. Med. Chem., 2013, 21, 4614 CrossRef CAS PubMed.
- (a) X. Zhao, E. Dimitrijevice and V. M. Dong, J. Am. Chem. Soc., 2009, 131, 3466 CrossRef CAS PubMed; (b) Z. Wu, H. Song, X. Cui, C. Pi, W. Du and Y. Wu, Org. Lett., 2013, 15, 1270 CrossRef CAS PubMed; (c) O. Saidi, J. Marae, A. E. W. Ledger, P. Liu, M. F. Mahon, G. Kociok-Kohn, M. K. Whittlesey and C. G. Frost, J. Am. Chem. Soc., 2011, 133, 19298 CrossRef CAS PubMed; (d) D. Zhang, X. Cui, Q. Zhang and Y. Wu, J. Org. Chem., 2015, 80, 1517 CrossRef CAS PubMed; (e) W.-H. Rao and B.-F. Shi, Org. Lett., 2015, 17, 2784 CrossRef CAS PubMed.
- P. R. Joshi, S. Undeela, D. D. Reddy, K. K. Singarapu and R. S. Menon, Org. Lett., 2015, 17, 1449 CrossRef CAS PubMed.
- For review, see: (a) L. Zhang, J. Sun and S. A. Kozmin, Adv. Synth. Catal., 2006, 348, 2271 CrossRef CAS. For selected examples, see: (b) M. R. Luzung, J. P. Markham and F. D. Toste, J. Am. Chem. Soc., 2004, 126, 10858 CrossRef CAS PubMed; (c) L. Zhang and S. A. Kozmin, J. Am. Chem. Soc., 2005, 127, 6962 CrossRef CAS PubMed; (d) Y. Horino, T. Yamamoto, K. Ueda, S. Kuroda and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 2809 CrossRef CAS PubMed.
- For selected examples, see: (a) Q. Zhou and Y. Li, J. Am. Chem. Soc., 2014, 136, 1505 CrossRef CAS PubMed; (b) F. Gagosz, Org. Lett., 2005, 7, 4129 CrossRef CAS PubMed; (c) A. S. K. Hashmi, W. Yang and F. Rominger, Angew. Chem., Int. Ed., 2011, 50, 5762 CrossRef CAS PubMed.
- For selected examples, see: (a) K. Speck, K. Karaghiosoff and T. Magauer, Org. Lett., 2015, 17, 1982 CrossRef CAS PubMed; (b) C.-H. Chen, Y.-C. Tsai and R.-S. Liu, Angew. Chem., Int. Ed., 2013, 52, 4599 CrossRef CAS PubMed; (c) E. Tudela, J. Gonzalez, R. Vicente, J. Santamaria, M. A. Rodriguez and A. Ballesteros, Angew. Chem., Int. Ed., 2014, 53, 12097 CrossRef CAS PubMed; (d) J. Zhang, D. Wu, X. Chen, Y. Liu and Z. Xu, J. Org. Chem., 2014, 79, 4799 CrossRef CAS PubMed; (e) H. Zheng, R. J. Felix and M. R. Gagne, Org. Lett., 2014, 16, 2272 CrossRef CAS PubMed.
- (a) F. Huber and S. F. Kirsch, J. Org. Chem., 2013, 78, 2780 CrossRef CAS PubMed; (b) A. Pradal, A. Nasr, P. Y. Toullec and V. Michelet, Org. Lett., 2010, 12, 5222 CrossRef CAS PubMed; (c) B. Crone, S. F. Kirsch and K.-D. Umland, Angew. Chem., Int. Ed., 2010, 49, 4661 CrossRef CAS PubMed.
- R. K. Mohamed, S. Mondal, K. Jorner, T. F. Delgado, V. V. Lobodin, H. Ottosson and I. V. Alabugin, J. Am. Chem. Soc., 2015, 137, 15441 CrossRef CAS PubMed.
- Z.-Z. Chen, S. Liu, W.-J. Hao, G. Xu, S. Wu, J.-N. Miao, B. Jiang, S.-L. Wang, S.-J. Tu and G. Li, Chem. Sci., 2015, 6, 6654 RSC.
- For review, see: (a) X.-F. Wu, J.-L. Gong and X. Qi, Org. Biomol. Chem., 2014, 12, 5807 RSC. For selected examples, see: (b) J. Zhang, J. Jiang, Y. Li, Y. Zhao and X. Wan, Org. Lett., 2013, 15, 3222 CrossRef CAS PubMed; (c) Y.-L. Zhu, B. Jiang, W.-J. Hao, J.-K. Qiu, J. Sun, D.-C. Wang, P. Wei, A.-F. Wang, G. Li and S.-J. Tu, Org. Lett., 2015, 17, 6078 CrossRef CAS PubMed; (d) Z. Yang, W.-J. Hao, S.-L. Wang, J.-P. Zhang, B. Jiang, G. Li and S.-J. Tu, J. Org. Chem., 2015, 80, 9224 CrossRef CAS PubMed.
- W.-J. Hao, Y. Du, D. Wang, B. Jiang, Q. Gao, S.-J. Tu and G. Li, Org. Lett., 2016, 18, 1884 CrossRef CAS PubMed.
- For selected sulfonyl radicals generated in situ from sulfonyl hydrazides, see: (a) X. Li, X. Xu and C. Zhou, Chem. Commun., 2012, 48, 12240 RSC; (b) J. Zhang, Y. Shao, H. Wang, Q. Luo, J. Chen, D. Xu and X. Wan, Org. Lett., 2014, 16, 3312 CrossRef CAS PubMed; (c) X. Li, Y. Xu, W. Wu, C. Jiang, C. Qi and H. Jiang, Chem. – Eur. J., 2014, 20, 7911 CrossRef CAS PubMed; (d) J.-K. Qiu, W.-J. Hao, D.-C. Wang, P. Wei, J. Sun, B. Jiang and S.-J. Tu, Chem. Commun., 2014, 50, 14782 RSC; (e) Y.-L. Zhu, B. Jiang, W.-J. Hao, A.-F. Wang, J.-K. Qiu, P. Wei, D. Wang, G. Li and S.-J. Tu, Chem. Commun., 2016, 52, 1907 RSC.
- W. Wei, J. Wen, D. Yang, M. Guo, Y. Wang, J. You and H. Wang, Chem. Commun., 2015, 51, 768 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1474252 (4a) and 1474253 (6n). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00422a |
| This journal is © the Partner Organisations 2016 |