
In vitro evaluation of the conjugations of neonicotinoids with transport protein: photochemistry, ligand docking and molecular dynamics studies†
Wei Pengabc,
Fei Ding*ad and
Yu-Kui Penge
aCollege of Agriculture and Plant Protection, Qingdao Agricultural University, Qingdao 266109, China. E-mail: alexf.ting@outlook.com; feiding@cau.edu.cn; Fax: +86-29-87092367; Tel: +86-29-87092367
bCollege of Food Science and Engineering, Qingdao Agricultural University, Qingdao 266109, China
cDepartment of Chemistry, China Agricultural University, Beijing 100193, China
dDepartment of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
eCenter for Food Quality Supervision & Testing, Ministry of Agriculture, College of Food Science & Engineering, Northwest A&F University, Yangling 712100, China
First published on 9th December 2015
Abstract
The main objective of this study was to assess the biological effects of neonicotinoids, together with their structure–activity relationships, by employing plasma albumin as a non-target model. Fluorescence indicated clearly that static-type quenching is the effective mechanism for the reduction of Trp-214 residue emission when c(neonicotinoid) ≤ 10 μM, yet both static and dynamic properties occurred in the system if the concentration was higher than 10 μM. The stoichiometric proportion of protein to neonicotinoid is obviously 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and subdomain IIA was discovered to possess high affinity for these chemicals. This corroborates molecular docking, site-directed mutagenesis, molecular dynamics simulations and free-energy calculations, which show that neonicotinoids are present at the warfarin–azapropazone site and yield hydrogen bonds, π–π stacking and hydrophobic interactions with several pivotal amino acid residues, i.e. Phe-211, Trp-214 and Arg-222. These noncovalent bonds caused partial conformational changes in the protein, that is, α-helix content decreased from 55.9% to 48.5% along with an increase in the contents of β-sheet, turn and random coil, as derived from synchronous fluorescence and circular dichroism. This phenomenon agrees well with the outcomes of the assignment of protein secondary structure. According to analyses of structure–activity relationships, it can be observed that neonicotinoids with the ring-closed structure (part B), e.g., imidacloprid and thiacloprid, have relatively low affinity for proteins compared with some ring-open agents such as nitenpyram and acetamiprid. These disparities may be related to the fact that ring-open neonicotinoids have great flexibility and thus take part more easily in noncovalent interactions with the amino acid residues in the active cavity. In addition, the toxicological relevance of the biorecognition of neonicotinoids by a biopolymer is also investigated here. Perhaps this investigation could use a non-target biological model for the evaluation of neonicotinoid toxicity and might also provide helpful clues for the synthesis of novel neonicotinoid agents.
:1, and subdomain IIA was discovered to possess high affinity for these chemicals. This corroborates molecular docking, site-directed mutagenesis, molecular dynamics simulations and free-energy calculations, which show that neonicotinoids are present at the warfarin–azapropazone site and yield hydrogen bonds, π–π stacking and hydrophobic interactions with several pivotal amino acid residues, i.e. Phe-211, Trp-214 and Arg-222. These noncovalent bonds caused partial conformational changes in the protein, that is, α-helix content decreased from 55.9% to 48.5% along with an increase in the contents of β-sheet, turn and random coil, as derived from synchronous fluorescence and circular dichroism. This phenomenon agrees well with the outcomes of the assignment of protein secondary structure. According to analyses of structure–activity relationships, it can be observed that neonicotinoids with the ring-closed structure (part B), e.g., imidacloprid and thiacloprid, have relatively low affinity for proteins compared with some ring-open agents such as nitenpyram and acetamiprid. These disparities may be related to the fact that ring-open neonicotinoids have great flexibility and thus take part more easily in noncovalent interactions with the amino acid residues in the active cavity. In addition, the toxicological relevance of the biorecognition of neonicotinoids by a biopolymer is also investigated here. Perhaps this investigation could use a non-target biological model for the evaluation of neonicotinoid toxicity and might also provide helpful clues for the synthesis of novel neonicotinoid agents.
Introduction
Over the last few decades, the development of neonicotinoids as important novel insecticides represents a milestone in agrochemical research. They are the only major new class of insecticides that have been developed recently.1 At the moment, commercial neonicotinoids include acetamiprid, clothianidin, dinotefuran, imidacloprid, nitenpyram, sulfoxaflor, thiacloprid and thiamethoxam. Because of their low usage dose and excellent activities against diverse types of pests, neonicotinoids account for approximately 20% of the global insecticide market and generate a profit of one billion dollars worldwide per year, especially imidacloprid.2,3In fact, previous studies have clearly shown that neonicotinoids have good selectivity for nicotinic acetylcholine receptors (nAChRs) in insects.4,5 However, molecular recognition of these agrochemical compounds in mammals is very scarce. Regrettably, accumulated toxicological data indicated that exposure to neonicotinoids may be closely related to the increased production of terrible consequences in animals and perhaps humans.6–9 Furthermore, the available literature on neonicotinoids and their degradation in mammals has demonstrated that some of them can cause carcinogenesis, hepatotoxicity and probable teratogenicity.10–12 Bhardwaj et al.13 observed moderate pathological changes in female Rattus norvegicus Wistar rats that were administered 0, 5, 10, and 20 mg kg−1 day imidacloprid in their feed in corn oil for 90 days. Bal et al.14 found that the low doses of imidacloprid could lead to deterioration in sperm motility and abnormality in sperm morphology in adult male Wistar albino rats. After three months of oral feeding with imidacloprid (8.0 mg kg−1 body weight), apoptosis of germ cells increased with fragmentation of seminal DNA. Some experiments also proved that a number of widely used pesticides, including neonicotinoids, might arouse concern owing to their probable endocrine disruptor properties, which would eventually produce detrimental outcomes in the adult reproductive system in humans.15,16 Moreover, Gawade et al.17 and Devan et al.18 suggested that continuous exposure to imidacloprid and acetamiprid during development will result in negative effects on the immune system and proposed that care should be taken to protect human beings, in particular, vulnerable population such as children and pregnant women, from neonicotinoids.
Besides the toxicological problems, nowadays the issue of pesticide residues has emerged as a great concern as well.19,20 Imidacloprid, which is currently the most extensively applied neonicotinoid insecticide in the world, has a relatively high solubility in water (0.61 g l−1) and degrades slowly in the environment.21 If in soil under aerobic conditions, it can persist with a half-life ranging from 1 to 3 years and its content has almost doubled every 5 years since the 1990s.22,23 The widespread residues of neonicotinoids in the environment may have made matters worse and could produce serious hazards to human health directly in the near future. Therefore, it is urgent that a comprehensive assessment of the toxicological action of neonicotinoids, notably by employing vital multifunctional macromolecules such as enzymes/proteins or nucleic acids as biological models, should be carried out.
In recent years, in addition to the in vivo experimental approaches, biological estimation of the molecular recognition of various ligands by biopolymers, e.g., DNA, RNA, polypeptides and proteins, is an essential part for obtaining a good comprehension of toxicological features.24,25 Albumin, which is formed in the liver, is the most abundant protein in blood plasma and contributes nearly half of blood serum proteins. One of the interesting biological functions of albumin is to transport endogenous and exogenous substances such as agrochemicals, bilirubin, colorants, fatty acids, hormones, metal ions and bioactive compounds.26,27 Moreover, albumin accounts for most of the antioxidant capacity of plasma and exhibits some types of enzymatic properties. Consequently, researchers use this protein as an excellent biomarker for evaluating many diseases, including cancer, ischemia, post-menopausal obesity and rheumatoid arthritis.28 In addition, it has the ability to treat several diseases that might need monitoring of glycemic control. It is commonly accepted today that the degree of biointeractions between biopolymers and ligands governs the absorption and dispersion of the latter into cellular tissues, affects their excretion from living organisms, and eventually influences the pharmaceutical and toxicological roles of the substance.29,30 Therefore, an investigation of the potential adverse effects of neonicotinoids through utilizing albumin as a target is completely suitable, and this kind of study could provide pivotal clues to the structural aspects that govern the overall toxic activities of neonicotinoids.
To date, many biophysical techniques have been used to study ligand recognition events, including calorimetry, chromatography, crystallography, electrophoresis, equilibrium dialysis, fluorescence, light scattering, nuclear magnetic resonance, surface-enhanced Raman spectroscopy, surface tension, ultracentrifugation, and ultrafiltration.31–33 Among these, fluorescence spectroscopy has been confirmed to be one of the most fundamental qualitative and quantitative ways to analyze non-covalent biomacromolecule–ligand reactions.34 Furthermore, molecular modeling can often be used to demonstrate binding interactions via reasonable computational calculations, and it is also usually utilized to scrutinize quantitative structure–affinity relationships.35 In two more recent qualitative investigations, Mikhailopulo et al.36 and Wang et al.37 studied the interactions between albumin and imidacloprid by steady-state fluorescence, but these studies did not determine the type of reaction, binding domain, structural changes, key non-covalent bonds and critical amino acid residues. Very recently, we preliminarily investigated the biointeractions of imidacloprid and its major metabolites with some model biopolymers such as bovine serum albumin, human hemoglobin and lysozyme from chicken egg white;38,39 however, the precise recognition features and recognition location, conformational transitions, dynamic recognition processes, binding free energies, structure–activity relationships of neonicotinoids and toxicological relevance are still unresolved. These crucial information, in particular, dynamic reaction behaviors, may benefit our understanding of the biological toxicity and biotransformation of neonicotinoids in the human body.
Given the above-mentioned background, our current contribution was to study the nature of recognition, stoichiometry, binding location, structural transitions, and dynamic interaction patterns along with the free energy in the presence of neonicotinoids (structures shown in Fig. 1) by a combination of steady-state and time-resolved fluorescence, chemical denaturation, use of an extrinsic 8-anilino-1-naphthalenesulfonic acid (ANS) probe, circular dichroism (CD), in silico docking, site-directed mutagenesis, and molecular dynamics simulations, as well as the decomposition of free energy. In particular, the structure–activity relationships and toxicological relevance of neonicotinoid agents are further discussed in this study. Possibly, this study will give a beneficial understanding for the determination of the toxicological profiles of neonicotinoids, their structure–activity relationships and the chemical nature of biorecognition between neonicotinoids and biological biomacromolecules.
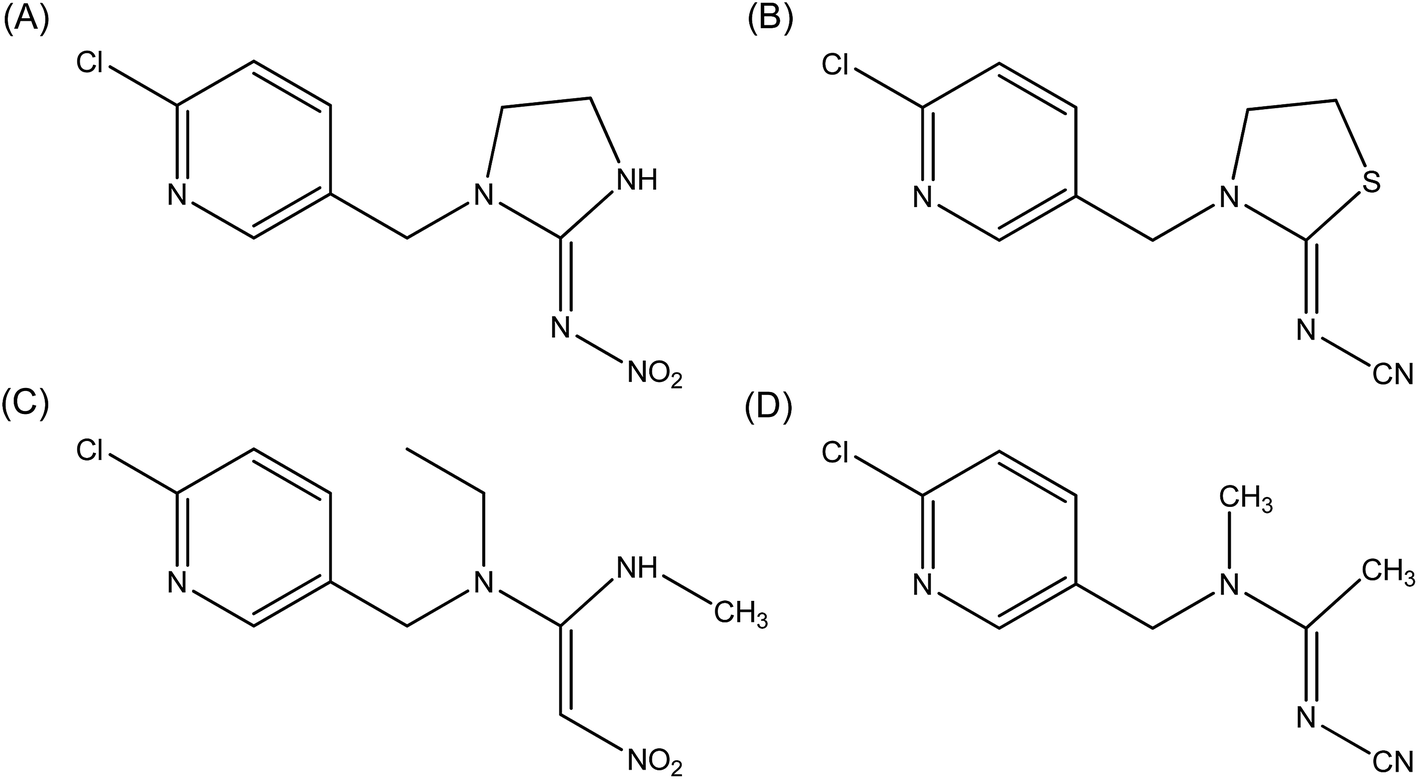 | ||
| Fig. 1 Molecular structures of imidacloprid (A), thiacloprid (B), nitenpyram (C) and acetamiprid (D). | ||
Experimental
Materials
Human serum albumin (A3782, lyophilized powder, fatty acid-free, globulin-free, ≥99%), imidacloprid (37894, analytical standard) and 8-anilino-1-naphthalenesulfonic acid (A1028, ≥97%) employed in this assay were obtained from Sigma-Aldrich (St. Louis, MO) and utilized without further purification, and ultrapure water was prepared by a Super-Q® Plus Water Purification System from EMD Millipore Corporation (Billerica, MA). All experiments were conducted in Tris (0.2 M)–HCl (0.1 M) buffer with a pH of 7.4 and an ionic strength of 0.1 M in the presence of sodium chloride, and the pH was measured with an Orion Star A211 benchtop pH meter (Thermo Scientific, Waltham, MA). Albumin was stored in a refrigerator at ∼277 K and dilutions of the albumin stock solution (10 μM) in Tris–HCl buffer solution were prepared immediately before use. The concentration of albumin was determined by the method of Lowry et al.40 All other chemicals used were of analytical reagent grade and acquired from Sigma-Aldrich.Steady-state fluorescence
Steady-state fluorescence spectra were obtained with a quartz cuvette of 1.0 cm path length using an F-7000 fluorescence spectrophotometer (Hitachi, Japan) equipped with a thermostatic bath. Both excitation and emission slits were fixed at 5.0 nm and fluorescence emission spectra were recorded by exciting continuously mixed albumin at 295 nm to generate fluorescence of tryptophan (Trp). Steady-state fluorescence spectra were recorded in the wavelength range of 300–500 nm at a scanning speed of 240 nm min−1. Spectra for a blank sample that contained a Tris–HCl buffer solution of imidacloprid in relevant quantities were deducted from all fluorescence measurements.Ligand docking
Molecular docking of albumin–neonicotinoid adducts was performed on a SGI Fuel 550L visual workstation (Silicon Graphics International Corp., Milpitas, CA). The X-ray diffraction crystallographic structure of the protein (entry code 1AO6), solved at an atomic resolution of 2.5 Å,41 was acquired from the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (http://www.rcsb.org/pdb). After being input into the commercial software Sybyl version 7.3 (http://www.certara.com), the structure of the protein was thoroughly examined for exact allocation of atom and bond types. Hydrogen atoms were theoretically added utilizing the menus of both Sybyl Biopolymer and Build/Edit. To avoid unfavorable amino acid/amino acid interactions and repellent steric hindrance, the positions of hydrogen atoms only were energy-minimized by Powell's conjugate direction method with energy gradient convergence criteria of 0.05 kcal mol−1 for 1500 iterations. This action does not alter the locations of heavy atoms, and the potential of the three-dimensional structure of the protein was determined based on the AMBER force field with Kollman charges. The two-dimensional configurations of neonicotinoids were obtained from the PubChem database (http://pubchem.ncbi.nlm.nih.gov) and the original structures of these ligands were generated by the Sybyl 7.3 program.Furthermore, both hydrogen atoms and Gasteiger–Hückel partial charges were added and assigned, respectively, to each neonicotinoid and AM1-BCC charges42 were also added to the insecticides imidacloprid and thiacloprid to ensure the rationality of the added charges and further compare the differences between the two docking results. To confirm the reasonableness of the initial docking conformations, which were generated by Sybyl 7.3, the crystal structures of imidacloprid (entry code 3WTL) and thiacloprid (entry code 3WTJ),43 respectively, were downloaded from the Protein Data Bank, and the ligand molecules derived from the two crystal structures were directly docked onto albumin. The irrationality of the original conformation that was induced by the addition of charges and force fields was excluded, and the docking results were expressed in the form of superposition pictures.
The AutoDock 4.2 program,44 which uses a fully automatic flexible molecular docking algorithm, was employed to determine the possible conformation of the ligands that binds to the protein, and then the ligands were docked onto the protein by utilizing a Lamarckian genetic algorithm (LGA).45 A grid box was defined before docking to cover the entire system including albumin and neonicotinoids with a size of 126 Å × 126 Å × 126 Å (x × y × z) and a grid spacing of 0.56 Å. A hybrid genetic algorithm (i.e. LGA) was used to ascertain the probable ligand-binding location on the protein. All the conformations that were generated after docking were clustered with a tolerance of root-mean-square deviation (RMSD) of 2.0 Å from the structural candidates (20) with the lowest energy. For each docking procedure, three conformations with the lowest energy (RMSD < 1.0 Å) were overlaid to select the most suitable docking conformation. The PyMOL computer program (http://www.schrodinger.com), which is a user-sponsored molecular visualization system, could ultimately be applied to exhibit the results of in silico docking.
Site-directed mutagenesis
Site-directed mutagenesis processes were accomplished by utilizing the module “Mutate Monomers” in Sybyl version 7.3 and the Trp-214, Phe-211 and Arg-222 residues in albumin were mutated to the alanine (Ala) residue, which has non-aromatic and non-polar properties. To guarantee the stability of the conformations, the mutated proteins were subjected to 3000 simplex minimization steps based on the AMBER force field with Kollman charges. Then, the molecular docking of mutated albumin–imidacloprid was performed, where the other parameters were in full agreement with the abovementioned native albumin–imidacloprid complex, and the results of the mutations were further validated using molecular dynamics (MD) simulations.Molecular dynamics simulations and the decomposition of free energies
MD simulations of the native and mutated albumin–neonicotinoids were carried out using the GROMACS program version 4.5.5 with the GROMOS96 ffG43a1 force field.46,47 Simulation procedures were executed under physiological conditions (pH = 7.4) and the amino acid residues that possessed acid and basic character were adjusted to the protonated states in neutral conditions. The respective initial conformations of albumin and neonicotinoids were obtained from the original X-ray diffraction crystal structure that was measured at a resolution of 2.5 Å (entry code 1AO6) and the optimal structures that originated from molecular docking. The topology of albumin was generated by the GROMACS package directly, whereas that of neonicotinoids was generated by PRODRG2.5 Server.48 The simulation systems were solvated in a periodic cubic box (with dimensions of 7.335 × 6.155 × 8.119 nm3) filled with TIP3P water molecules and an approximate number (12) of sodium counterions to neutralize the charge.49 In total, there were 51230 crystallographic solvent molecules, and the shortest distance between the complex and the edge of the box was set to 10 Å. Simulations were conducted utilizing the isothermal-isobaric (NPT) ensemble with an isotropic pressure of 1 bar, and the temperature of the neonicotinoids, albumin and solvent (i.e. water and counterion) was, respectively, coupled to an external bath held at 300 K using the Berendsen thermostat with a relaxation time of 0.2 ps.50,51 The LINCS algorithm was employed to constrain bond lengths,52–54 and long-range electrostatic interactions beyond 10 Å were modeled using the particle mesh Ewald (PME) method with a grid point density of 0.1 nm and an interpolation order of 4.55,56 Cutoffs of 10 Å and 14 Å were used for Coulomb and van der Waals interactions, respectively. The MD integration time step was 2.0 fs, covalent bonds were not constrained, and the system configurations were saved every 2.0 ps. To reduce the collisions of atoms with each other, both gradient descent and conjugate-gradient algorithms were utilized to optimize the whole system.57,58 First, the solvated starting structure was preceded by a gradient descent of 1000 steps and then by conjugate-gradient energy minimization. Subsequently, equilibration for 100 ps under position restraints was run to remove possible unfavorable interactions between the solute and the solvent and, after thorough equilibration, MD simulations of the native protein–neonicotinoid and mutated protein–neonicotinoid were run for 30 ns and 50 ns, respectively. Furthermore, pure albumin was selected to perform a MD simulation for a time period of 10 ns and the outcomes of simulations were finally displayed via Visual Molecular Dynamics 1.9.2.59 Discovery Studio Visualization 4.5 (Accelrys, San Diego, CA) software was used to display the pictures of the MD simulations. The Dictionary of Protein Secondary Structure (DSSP) program,60,61 together with the do_dssp tool embedded in GROMACS 4.5.5, was employed to standardize the assignment of secondary structures in this study.
In addition, the binding free energies for these molecular interactions were computed based on the following relationships:62,63
| ΔGbind = Gcomplex − (Gprotein + Gligand) | (1) |
| ΔGbind = ΔGgas − ΔGsol | (2) |
| ΔGgas = ΔHgas − TΔS ≈ ΔEMM − TΔS | (3) |
| ΔGbind ≈ ΔEMM + ΔGsol − TΔS | (4) |
| ΔEMM = ΔEinternal + ΔEvdW + ΔEele | (5) |
| ΔGsol = ΔGGB + ΔGSA | (6) |
| GSA = γ × SASA + β | (7) |
In these equations, the binding free energy ΔGbind is calculated from the contributions of the gas phase energy ΔGgas and solvation energy ΔGsol, where ΔGgas consists of ΔEMM and TΔS. The molecular mechanics energy (ΔEMM) consists of the internal energy (ΔEinternal), van der Waals energy (ΔEvdW) and electrostatic energy (ΔEele). The polar solvation component (ΔGGB) is determined using the generalized Born method and the non-polar solvation component (ΔGSA) is estimated by utilizing the solvent-accessible area with the γ parameter set to 0.00542 kcal (mol Å)−1 and the β parameter set to 0.92 kcal mol−1. The solvent-accessible surface area (SASA) is calculated using the linear combination of pairwise overlaps (LCPO) model.64 The error bar for the standard error (SE) is calculated by
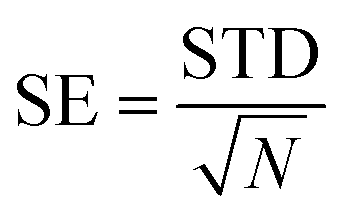 | (8) |
Results and discussion
Tryptophan fluorescence studies
Basically, there are three amino acid residues that display intrinsic fluorescence, i.e., phenylalanine (Phe), tryptophan (Trp) and tyrosine (Tyr), but only Trp and Tyr are utilized experimentally, because their quantum yield is high enough to provide a good fluorescence signal.65 Therefore, the intrinsic fluorescence of a protein is frequently used to measure the association parameter, binding mode and rate constant of a specific binding equilibrium. To monitor the reaction between a neonicotinoid and albumin, steady-state fluorescence spectra of the protein at various concentrations of the neonicotinoid were obtained and are shown in Fig. 2. The intrinsic fluorescence of albumin is normally due to the emission of Trp residues when excited at 295 nm and the contributions from Tyr residues could be ignored. Under the experimental conditions, neonicotinoids exhibit no fluorescence emission in the range of 300–500 nm and do not interfere with the fluorescence of the protein. Clearly, albumin displayed a strong fluorescence emission peak at 338 nm following excitation at 295 nm, and its intensity decreased regularly with the addition of the neonicotinoid. These phenomena indicate that conjugation occurred between the protein and neonicotinoid and the ligand was situated in the region where Trp-214 is located or close to the individual amino acid residue.66 An analogous report has been provided by Bekale et al.67 for the biorecognition of polyethylene glycols by milk β-lactoglobulin.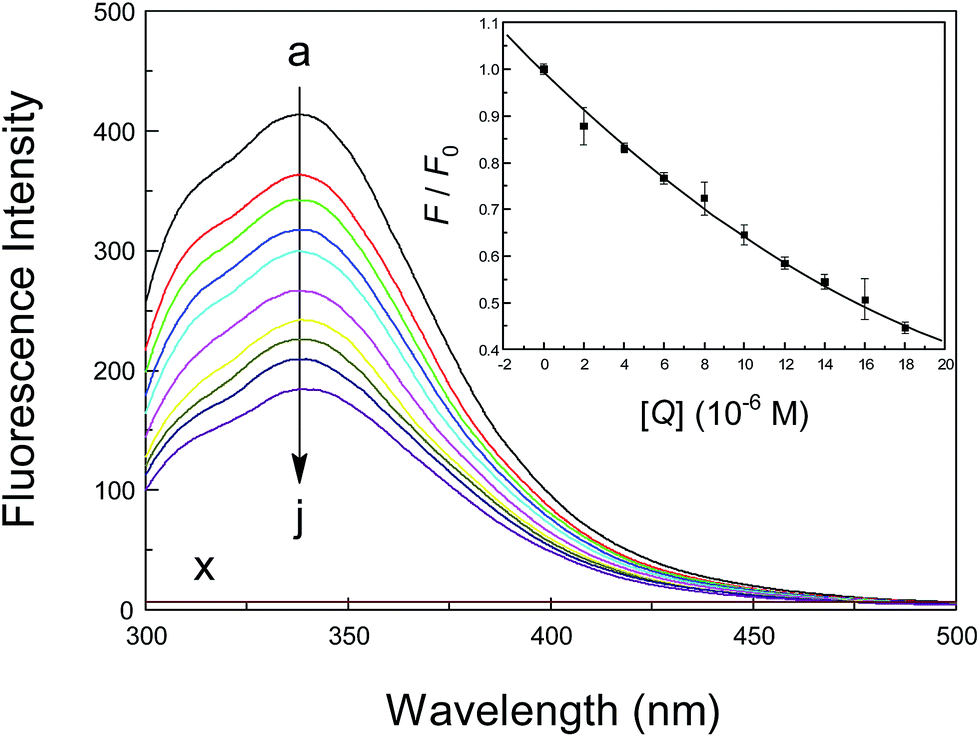 | ||
| Fig. 2 Fluorescence emission spectra of albumin (1.0 μM) at λex = 295 nm in the presence of different concentrations of imidacloprid. c(imidacloprid) = 0, 2.0, 4.0, 6.0, 8.0, 10, 12, 14, 16 and 18 μM (a → j), (x) 18 μM imidacloprid only; pH = 7.4, T = 298 K. The inset shows the extent of fluorescence quenching of albumin plotted as the extinction of intensity (F/F0) against the imidacloprid concentrations corresponding to the fluorescence emission spectra. All data were corrected for the fluorescence of imidacloprid and each point is the mean of three independent observations ±S.D. in the range of 1.07–4.39%. | ||
The sensitivity of the fluorescence of indole in proteins is the central element in the variety of fluorescence observed between different proteins and ligands, and the study of fluorescence mechanisms has been regarded as an effective method of inspecting the dynamics and conformations of proteins. Measurements of fluorescence lifetime have therefore been employed to indicate the existence of disparate and distinctive protein conformations, and can offer direct mechanistic information about the time dependence of the protein–ligand recognition processes. To illustrate the nature of albumin–neonicotinoid conjugation, representative patterns of the fluorescence decay of the protein at various molar ratios of the neonicotinoid in Tris–HCl buffer, pH = 7.4, are shown in Fig. S1 (ESI†), and the time-resolved fluorescence lifetimes and their oscillations are also listed in Table 1. Evidently, the fluorescence decay curves agree closely with biexponential decay kinetics, which may suggest the presence of conformers in equilibrium in the folded structure of albumin. As can be seen in Table 1, the short and long lifetime are observed to be τ1 = 3.14 ns and τ2 = 7.18 ns (χ2 = 1.09), respectively, for the protein during time-resolved fluorescence decay, whereas at the maximum concentration of the neonicotinoid, the lifetime components are τ1 = 2.41 ns and τ2 = 6.31 ns (χ2 = 1.03). The biexponential decay in the present case might be ascribed to a single electronic transition of the Trp residue, which could be present as diverse conformational isomers in the protein. Actually, owing to steric effects between the side chain of the Trp residue and the polypeptide backbone, all rotamers are not equally possible.68,69 The quenching group that is closest to the indole moiety is the amino group after the protein–neonicotinoid conjugation occurs; as a result, this rotamer has the maximum population and a fluorescence lifetime of 7.18 ns. However, if amino and carbonyl groups are close to the indole ring, this rotamer might have the short lifetime of 3.14 ns of 3.14 ns. The assignments of conformers of the protein are restricted to the solution, and the presence of different rotamers of the Trp residue has been accurately confirmed by nuclear magnetic resonance.70,71 Thus, we have not tried to assign the separate constituents, but in contrast the average fluorescence lifetime has been used to provide a qualitative analysis. The average fluorescence lifetime of the protein is reduced from 5.93 ns to 5.49 ns at different neonicotinoid concentrations, which illustrates evidently that the quenching of the fluorescence of albumin Trp residues by the neonicotinoid is a combination of dynamic and static in nature, not static or dynamic quenching alone. These results are in reasonable agreement with the following analyses based on steady-state fluorescence data using the Stern–Volmer equation, and a comparable examination has been indicated by Abou-Zied et al.72 for the interpretation of fluorescence quenching of proteins in the presence of medicinal hydroxyquinoline chemicals, namely, 6-hydroxyquinoline, 7-hydroxyquinoline and 8-hydroxyquinoline.
| Samples | τ1 (ns) | τ2 (ns) | A1 | A2 | τ (ns) | χ2 |
|---|---|---|---|---|---|---|
| Free albumin | 3.14 | 7.18 | 0.31 | 0.69 | 5.93 | 1.09 |
| Albumin + imidacloprid (1:1) |
3.02 | 7.02 | 0.29 | 0.71 | 5.86 | 1.01 |
| Albumin + imidacloprid (1:2) |
2.75 | 6.73 | 0.26 | 0.74 | 5.70 | 1.15 |
| Albumin + imidacloprid (1:4) |
2.41 | 6.31 | 0.21 | 0.79 | 5.49 | 1.03 |
To determine the type of fluorescence quenching, the well-known Stern–Volmer equation was used for the analysis of emission data, and the corresponding results that were fitted from Fig. 3 are summarized in Table 2. Usually, a linear Stern–Volmer plot is frequently suggestive of fluorophores of a single type, which are all equally accessible to the ligand. Intuitively, the Stern–Volmer plot in Fig. 3 in such circumstances has an upward curvature, which is concave towards the y-axis. This outcome implies plainly that the fluorophore (Trp-214 residue) may have declined both by collision and by the formation of a complex with the same compound (neonicotinoid). The Stern–Volmer plot appears to be divided into two sections, that is, whether the concentration of the neonicotinoid is less than or greater than 10 μM. The Stern–Volmer quenching constant KSV in both cases has an opposite correlation with temperature, which clearly indicates that the biointeraction between the protein and neonicotinoid is controlled by a static reaction at low concentrations of the ligand (≤10 μM), whereas a combination of static and dynamic reactions is likely to predominate when the concentration exceeds 10 μM.
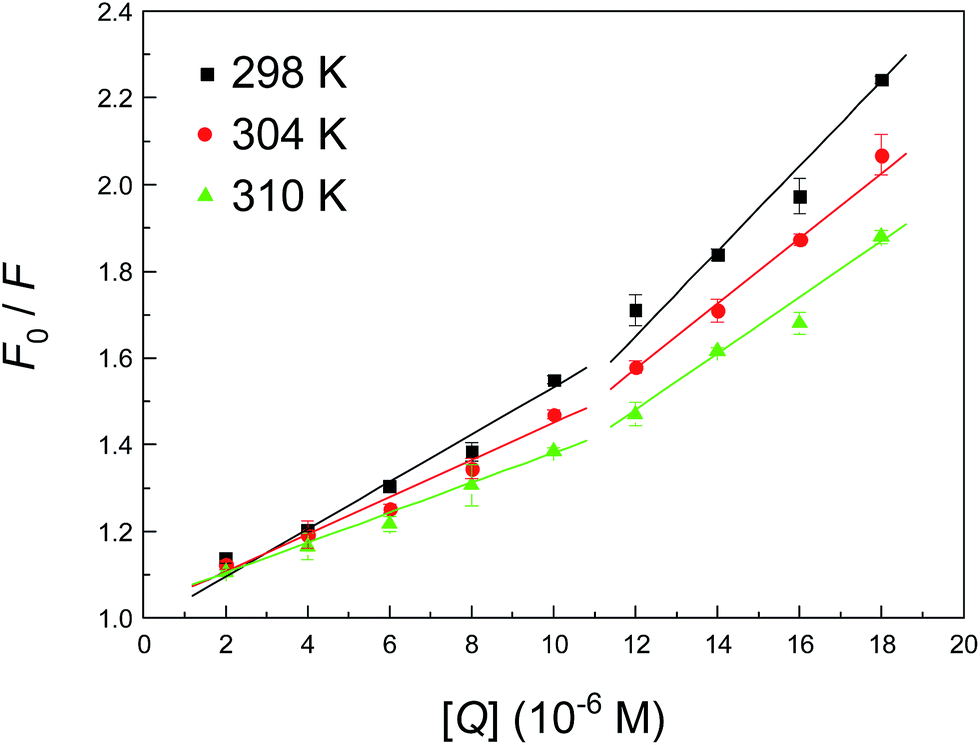 | ||
| Fig. 3 Stern–Volmer plot describing the fluorescence quenching of albumin (1.0 μM) at pH = 7.4 in the presence of different concentrations of imidacloprid. The fluorescence intensity was recorded at λex = 295 nm and the emission maximum occurred at 338 nm. Each data point is the average of three separate measurements ±S.D. in the range of 0.25–4.69%. | ||
| T (K) | c(imidacloprid) ≤ 10 μM | c(imidacloprid) > 10 μM | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| KSV (×104 M−1) | Ra | K (×104 M−1) | n | Ra | KSV (×104 M−1) | Ra | K (×104 M−1) | n | Ra | |
| a R is the correlation coefficient. | ||||||||||
| 298 | 5.005 | 0.9853 | 1.442 | 0.90 | 0.9995 | 8.647 | 0.9813 | 116.9 | 1.27 | 0.9975 |
| 304 | 4.227 | 0.9887 | 0.5559 | 0.83 | 0.9996 | 8.146 | 0.9962 | 19.50 | 1.12 | 0.9956 |
| 310 | 3.491 | 0.9946 | 0.4989 | 0.83 | 0.9961 | 6.457 | 0.9821 | 9.977 | 1.08 | 0.9973 |
In pharmacology, as well as in toxicology, the association capacity is one of the most prominent indicators when we estimate the potential pharmacological or toxicological activities of a ligand, such as a specific drug or agrochemical, with a biomacromolecule.31 Knowledge of the recognition affinity is of great significance in determining the absorption, distribution and bioavailability and even in the quantitative depiction of the dose–response relationship of a ligand.73 The equation numbered (3) in the ESI† has been employed to process raw steady-state fluorescence data, and Fig. S2† shows plots of log(F0–F)/F versus log[Q] for the protein–neonicotinoid mixture at different temperatures and the corresponding results for values of K and n are also shown in Table 2. Visibly, the association constant K at both low and high concentrations of the neonicotinoid is reduced with an increase in temperature, which suggests the emergence of a weak adduct during the association process, and the non-covalent conjugate might be decomposed in part when the temperature is increased. A primary cause of this phenomenon is that a higher temperature will typically result in the dissociation of weakly bound protein–ligand adducts and consequently a smaller amount of static quenching.
According to an idea from Dufour and Dangles,74 and also a group of several recent publications on the topic of biopolymer–ligand complexes, e.g., various drugs, emodin, flavonoids and long-chain perfluoroalkyl acids,75–78 it is quite clear that the complexation of the neonicotinoid with albumin exhibits moderate affinity with respect to other strong protein–ligand complexes, with association constants ranging from 106 to 108 M−1. Using the thermodynamic equation ΔG° = −RTlnK, we may calculate the Gibbs free energy ΔG° to be −5.67 kcal mol−1 (298 K), which shows that the formation of the protein–neonicotinoid complex is an exothermic reaction. Moreover, the value of n is approximately equal to 1, which suggests the presence of one single binding site in the protein for the neonicotinoid. As noted earlier, a unique quality of the intrinsic fluorescence of albumin is due to the Trp-214 residue in the subdomain IIA; from the value of n, the neonicotinoid-binding region is most likely close to this aromatic amino acid residue and gives rise to fluorescence quenching via conjugation.
To confirm the stoichiometry between the neonicotinoid and protein that was estimated from the above discussion, the method of continuous variation (Job's plot) is adopted here. In this approach, the total molar concentrations of the neonicotinoid and protein are fixed, but their molar fractions are varied.79 Fluorescence emission spectra that correspond to complex formation are plotted against the molar fractions of the two components, and the maximum on the plot corresponds to the stoichiometry of the two species. A Job's plot for protein–neonicotinoid fluorescence at 338 nm upon excitation at 295 nm is shown in Fig. 4 and, apparently, the x-coordinate at the maximum in the curve is 0.509. This supports the formation of a 1:1 protein–ligand complex and is perfectly in agreement with the abovementioned result derived from double-logarithmic plots.
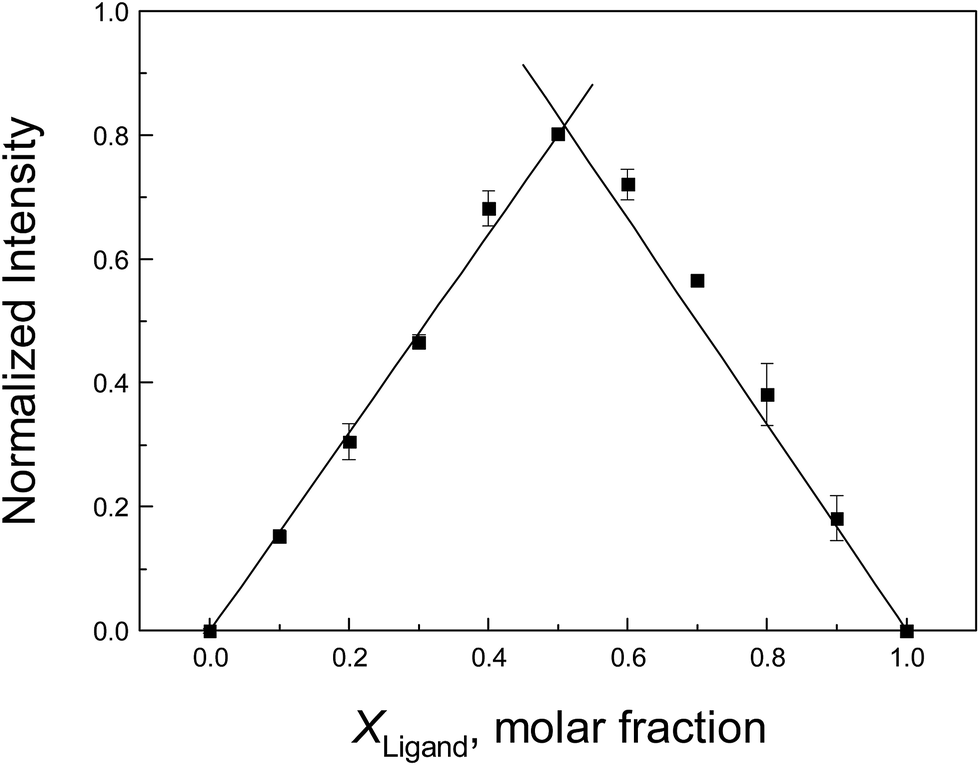 | ||
| Fig. 4 Job's plot for albumin–imidacloprid fluorescence based on the method of continuous variation (pH = 7.4, T = 298 K). All data were corrected for the fluorescence of imidacloprid and each point is the mean of three respective measurements ±S.D. in the range from 0.13–4.99%. | ||
Structural alterations
As has been discussed, time-resolved fluorescence indicates that the conformation of albumin is likely to change to accommodate neonicotinoid suitably through non-covalent bonds. The following section will primarily be centered on the identification of structural changes in the protein in the presence of neonicotinoid. To begin with, a synchronous fluorescence method was used to characterize the conformation of albumin when altered by the neonicotinoid. This comprises parallel scanning with both excitation and emission monochromators while maintaining a fixed gap in wavelengths (Δλ) or constant increase in energy (Δv) between them.80 In the pioneering study of Miller81 and Burstein et al.,82 when Δλ = 15 nm or 60 nm, the distinctive characteristics of Tyr and Trp residues are revealed. Fig. 5 shows the spectral intensity of the synchronous fluorescence of albumin in Tris–HCl buffer solution in the presence of various concentrations of neonicotinoid. It is evident that the wavelength of maximum fluorescence emission undergoes a slight red shift in the investigated range of concentrations when Δλ = 60 nm; however, almost no shift is observed when Δλ = 15 nm. The bathochromic effect indicates that the polarity around the Trp residue was increased and the hydrophobicity was decreased; thus, neonicotinoid is within easy reach of the Trp-214 residue, and then has a remarkable impact on the microenvironment of the nearby Trp-214 residue. However, the intensity of synchronous fluorescence decreased regularly with the addition of the neonicotinoid, which further demonstrates the occurrence of fluorescence quenching in the association reaction.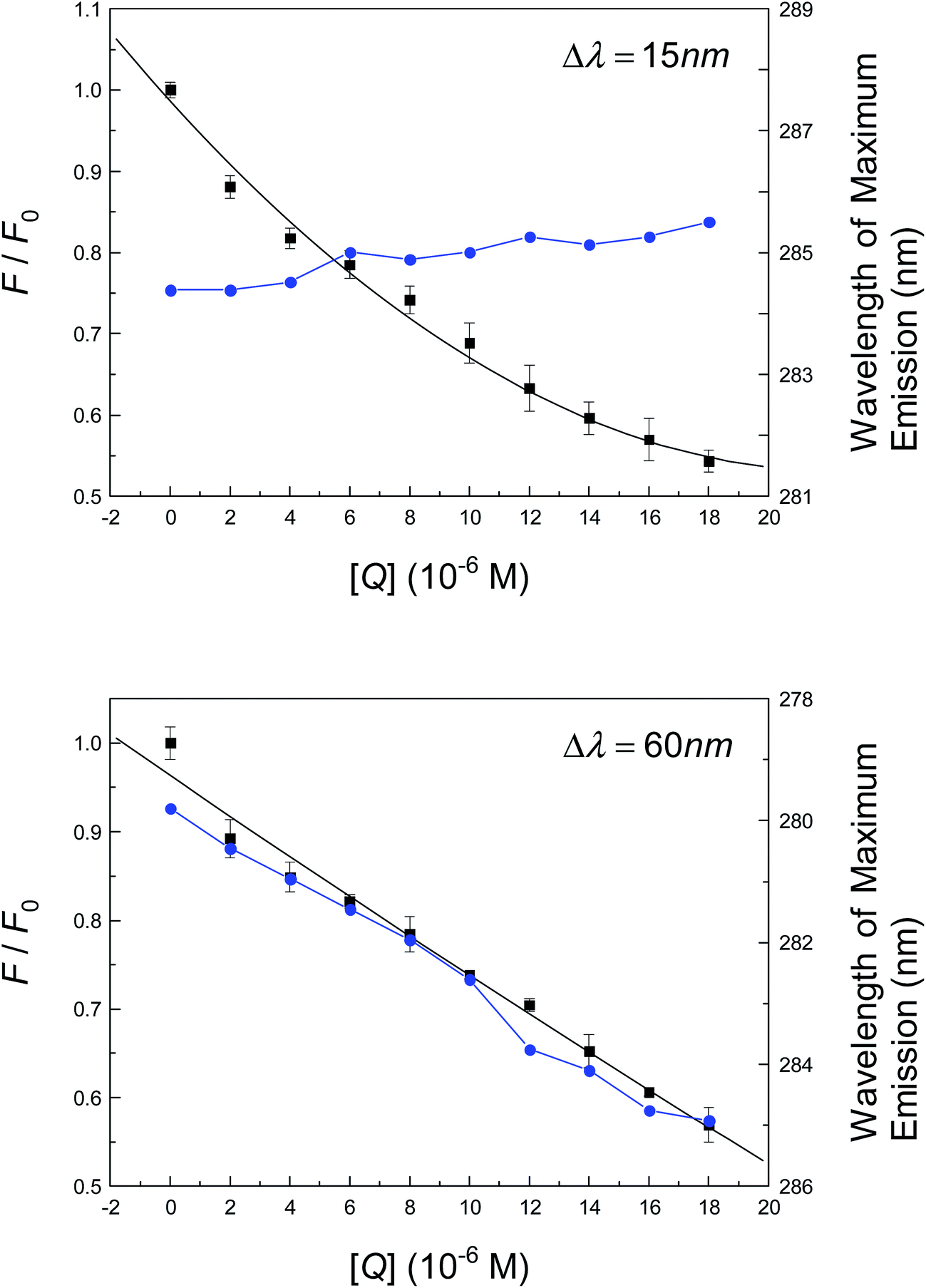 | ||
| Fig. 5 Synchronous fluorescence intensity of albumin (1.0 μM) at pH = 7.4, T = 298 K, plotted as extinction of albumin Tyr and Trp residues (F/F0) versus imidacloprid concentration (black square). And the blue circle reveals the relationships between the wavelength of maximum emission and the concentration of imidacloprid. Each data was the mean of three autonomous detections ±S.D. ranging 0.46–2.86%. | ||
Circular dichroism (CD), in particular far-UV CD, is an important tool in structural biology for examining folding and kinetics and whether protein–protein or protein–ligand interactions alter the conformation of a protein. If there are any conformational changes, this event can lead to a spectrum that will differ from the sum of the individual portions. To quantitatively analyze the structural alterations of a globular protein, experiments that recorded CD spectra of the protein in the absence and presence of an insecticide are illustrated in Fig. S3† and the components of secondary structures that were deduced based on raw CD data are also illustrated as follows. The CD curve of albumin shows two negative bands in the far-UV CD area at 208 nm and 222 nm, which are a feature of the α-helical configuration of a globular protein. The sensible deduction is that the negative peaks between 208 nm and 209 nm and between 222 nm and 223 nm both originate from π → π* and n → π* transitions of the peptide bond of α-helix.83 The free protein possesses 55.9% α-helix, 8.1% β-sheet, 11.6% turn and 24.4% random coil; upon interaction with the neonicotinoid, a decline in the α-helix structure was observed from 55.9% for free albumin to 48.5% for the albumin–neonicotinoid complex, whereas increases in β-sheet, turn and random coil were observed from 8.1%, 11.6% and 24.4% for free albumin to 9.2%, 14.2% and 28.1% for albumin–neonicotinoid, respectively, at a molar ratio of albumin to pesticide of 1:8. The decrease in α-helix with a growth in β-sheet, turn and random coil implies that the neonicotinoid interacted with some residues of the peptide chain and eventually produced a disturbance in the three-dimensional structure of albumin, e.g., some degree of destabilization of the biomacromolecule occurred upon complexation with the neonicotinoid.84 These experimental facts may further corroborate the previous speculation by Mikhailopulo et al.36 that the conformation of the protein would be disrupted by non-covalent protein–ligand recognition.
All the abovementioned measurements and illustrations confirm that biorecognition of the neonicotinoid by albumin caused conformational perturbations in the protein, which could probably be related to its physiological function. It is worth noting that the unfolding of albumin in this region does not signify that the pesticide caused widespread destruction of the three-dimensional structure of the protein. Although albumin in solution might usually be considered as possessing a single shape overall, it is possibly more accurate to consider it as an assembly of contractile, flexible parts that frequently alter in conformation via the opening and closing of large fissures.85,86 This mode of alteration, together with the fact that many of its amino acid side chains are incessantly in motion on the microscale, make albumin well adapted to absorb or remove the many substances such as insecticides that it carries in the human body.
Molecular docking
Molecular docking can suitably be used to investigate the complexation of a ligand at the active site of a receptor. In the current study, this method was applied to examine the binding of neonicotinoids to the active domain of the protein. Albumin is one of the most studied model proteins, because its high-resolution tertiary structure was solved by X-ray diffraction crystallography by He and Carter87 in 1992. According to the atomic analysis of crystallized albumin, it is a heart-shaped tri-domain protein, which consists of 585 amino acid residues, and each domain is a combination of two subdomains that comprise common structural motifs. The three domains, although they are homologous, have dissimilar ligand-binding functions. Prior to explaining the results of docking, the influences of the two force fields, i.e., Gasteiger–Hückel partial charges and AM1-BCC charges, on the outcomes of molecular docking have been interpreted using a superposition pattern in this study, and the data are clearly shown in Fig. 6. The results indicate that the addition of Gasteiger–Hückel partial charges and AM1-BCC charges has little impact on the docking results and the superimposed data also suggest that the optimal conformations are highly comparable. Although by overlaying multiple low-energy conformations in the present four docking systems we could find some subtle differences, undoubtedly the optimal conformations may satisfactorily be superimposed on the conformations with lowest energy that have similar energy to the best conformations.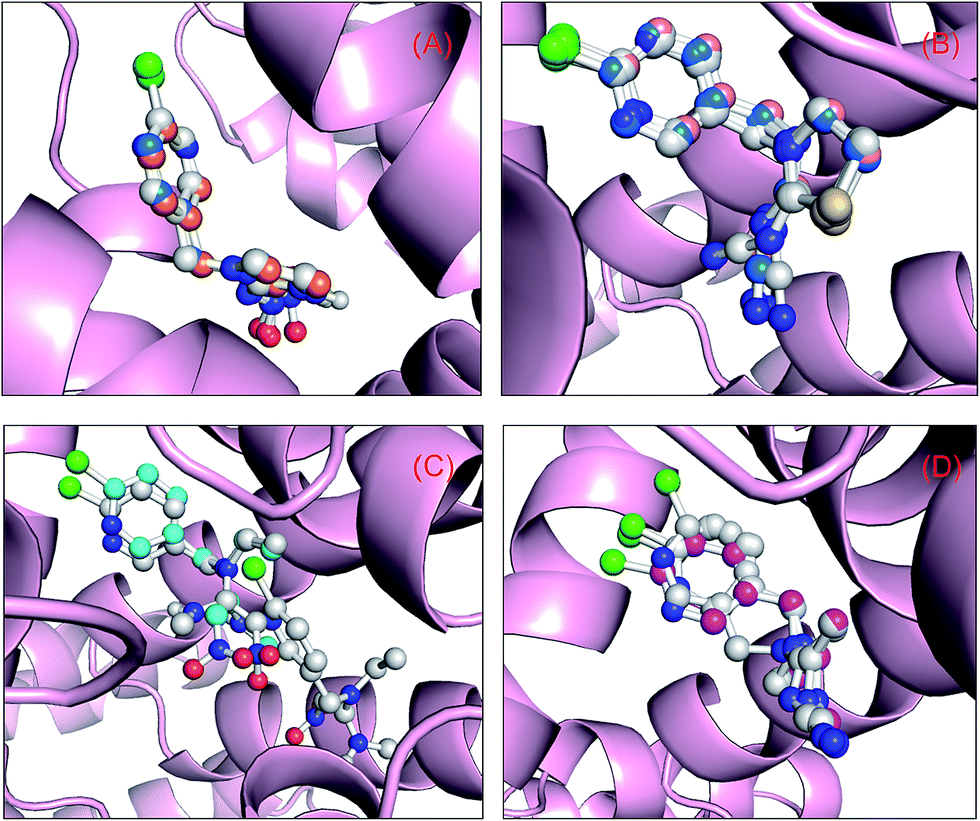 | ||
| Fig. 6 Superposition of the results of molecular docking. Albumin is shown as a surface colored in light pink and the ball-and-stick models depict neonicotinoids colored according to their atoms: (A) the orange and blue stick model displays the optimal skeletal structure of the binding conformation of imidacloprid with Gasteiger–Hückel partial charges and AM1-BCC charges, respectively, whereas the blue stick model depicts the skeletal structure of the optimal conformation obtained using the ligand from the crystal structure (entry code 3WTL) as the initial conformation; (B) the wheat and blue stick model depicts the optimal skeletal structure of the binding conformation of thiacloprid with Gasteiger–Hückel partial charges and AM1-BCC charges, respectively, whereas the blue stick model shows the skeletal structure of the optimal conformation obtained by utilizing the ligand from the crystal structure (entry code 3WTJ) as the initial conformation; (C) the cyan stick model depicts the optimal skeletal structure of the binding conformation of nitenpyram with Gasteiger–Hückel partial charges; and (D) the magenta stick model shows the optimal skeletal structure of the binding conformation of acetamiprid with Gasteiger–Hückel partial charges, whereas the white stick model depicts the skeletal structures of the two low-energy conformations that have the closest energy to the optimal conformation. (For clarification of the references to color in this figure legend, the reader is referred to the web version of the article.). | ||
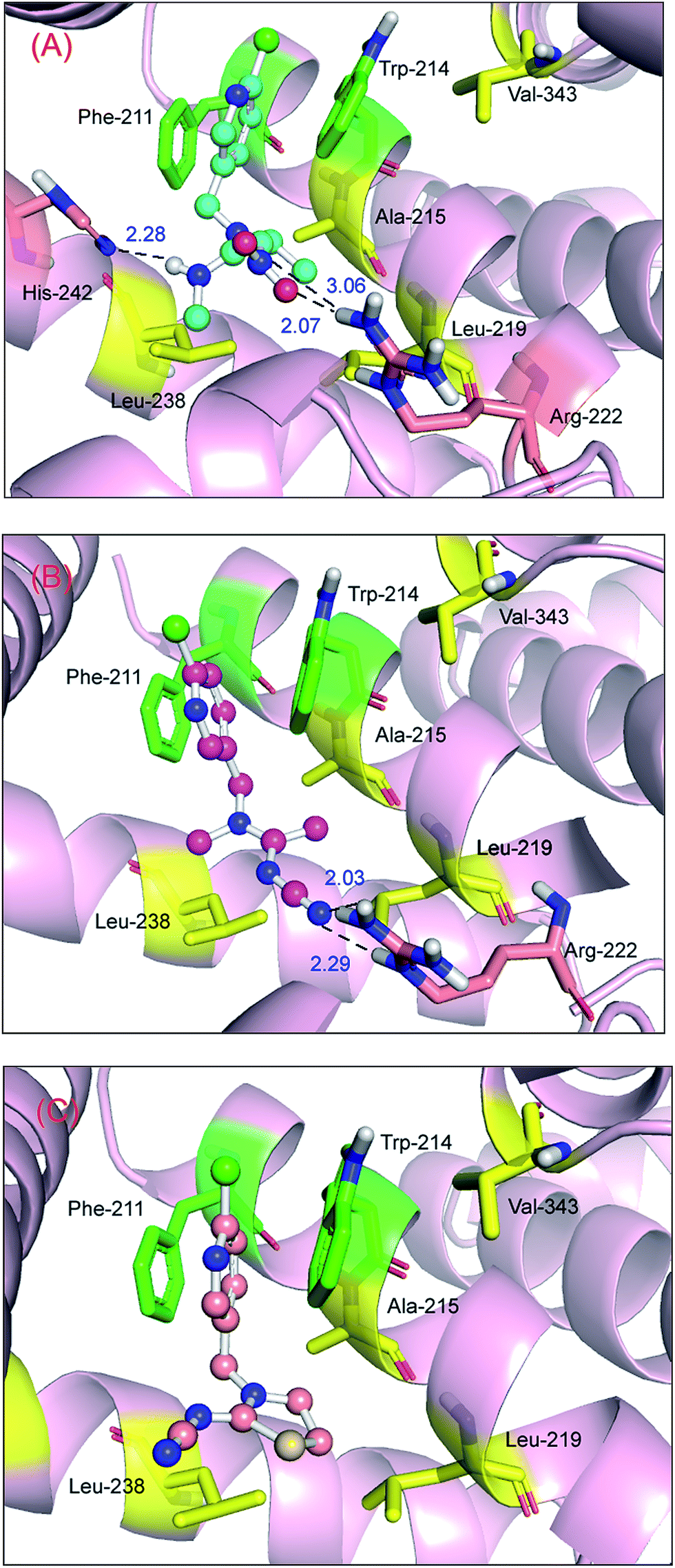
The best outcome for the docking energy (ΔG° = −5.87 kcal mol−1) of the protein–imidacloprid adduct is shown in Fig. 7. As can be seen in Fig. 7, the two oxygen atoms of the nitro group in imidacloprid can form hydrogen bonds with the hydrogen atom of the amino group and the hydrogen atom of the secondary amine in the Arg-222 residue, and the bond lengths are 2.05 Å and 2.47 Å, respectively. Furthermore, the distances between the center of the pyridine ring in the ligand and the center of the indole ring in the Trp-214 residue and that of the benzene ring in the Phe-211 residue are 3.14 Å and 3.27 Å, respectively, accordingly indicating that distinct π–π stacking, which looks like a “sandwich”, also occurred between albumin and imidacloprid. In the light of surface modification of the protein, we perceived that the entire neonicotinoid is oriented towards the hydrophobic pocket that is composed of Phe-211, Trp-214, Ala-215, Leu-219, Leu-238 and Val-343 residues, which confirms that hydrophobic interactions took place between them. Moreover, the optimal docking results of other protein–neonicotinoid (thiacloprid, nitenpyram and acetamiprid) complexes are shown in Fig. 13, and the critical non-covalent interactions are described in the following section on structure–activity relationships.
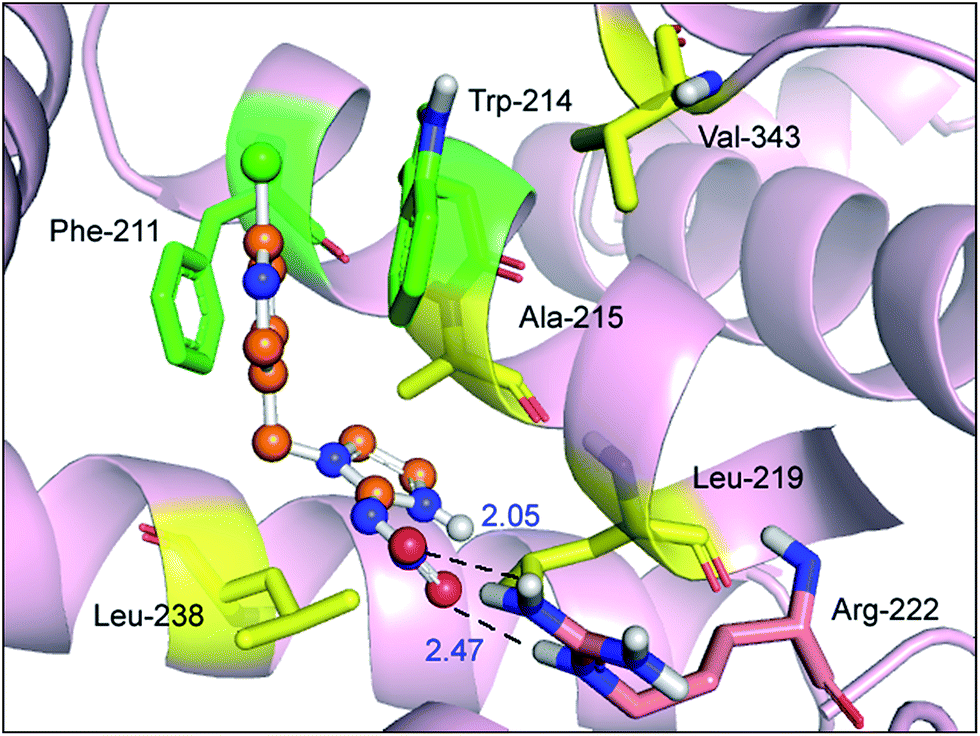 | ||
| Fig. 7 Molecular docking of imidacloprid docked onto albumin. Albumin is shown as a surface colored in light pink and the ball-and-stick model shows imidacloprid colored according to the atoms. The key amino acid residues around imidacloprid have been indicated in the stick model; the salmon stick model shows hydrogen bonds between the Arg-222 residue and imidacloprid; the green stick model represents π–π stacking between Phe-211 and Trp-214 residues and imidacloprid; the yellow stick model represents hydrophobic interactions between Phe-211, Trp-214, Ala-215, Leu-219, Leu-238, and Val-343 residues and imidacloprid. (For an interpretation of the references to color in this figure legend, the reader is referred to the web version of the article). | ||
Site-directed mutagenesis is a powerful research tool that is used to study the structure and function of enzymes and proteins, especially crucial amino acid residues and the main non-covalent interactions generated by these residues in biopolymers. To further confirm the key forces in the non-covalent protein–neonicotinoid interaction, three amino acid residues—Trp-214, Phe-211 and Arg-222—were chosen based on the above explanation, and site-directed mutagenesis experiments on these residues were conducted. In the following section, we describe the mutation of the Trp-214 residue in detail and the phenomena of mutation of the Phe-211 and Arg-222 residues are illustrated in the ESI.† The result of the mutated protein–imidacloprid reaction is displayed in Fig. 8. We noticed that the hydrogen bonds between the oxygen atoms of the polar nitro group in imidacloprid and the hydrogen atom of the hydroxyl group in the Ser-202 residue were clearly weakened and the bond lengths were found to be 2.32 Å and 2.84 Å. However, although the π–π stacking disappeared after the mutation of the Trp-214 residue, weak hydrophobic interactions remained in the mutated system with residues that include Phe-211, Ala-213, Ala-214, Ala-215, Leu-219, Leu-238 and Val-343. Significantly, the non-covalent strength of the whole system exhibits a downward tendency; such an evident change can be attributed to the mutation of the tryptophan (Trp) residue to alanine (Ala), which significantly decreases the non-covalent interactions between the amino acid residues situated within the active region and the neonicotinoid.
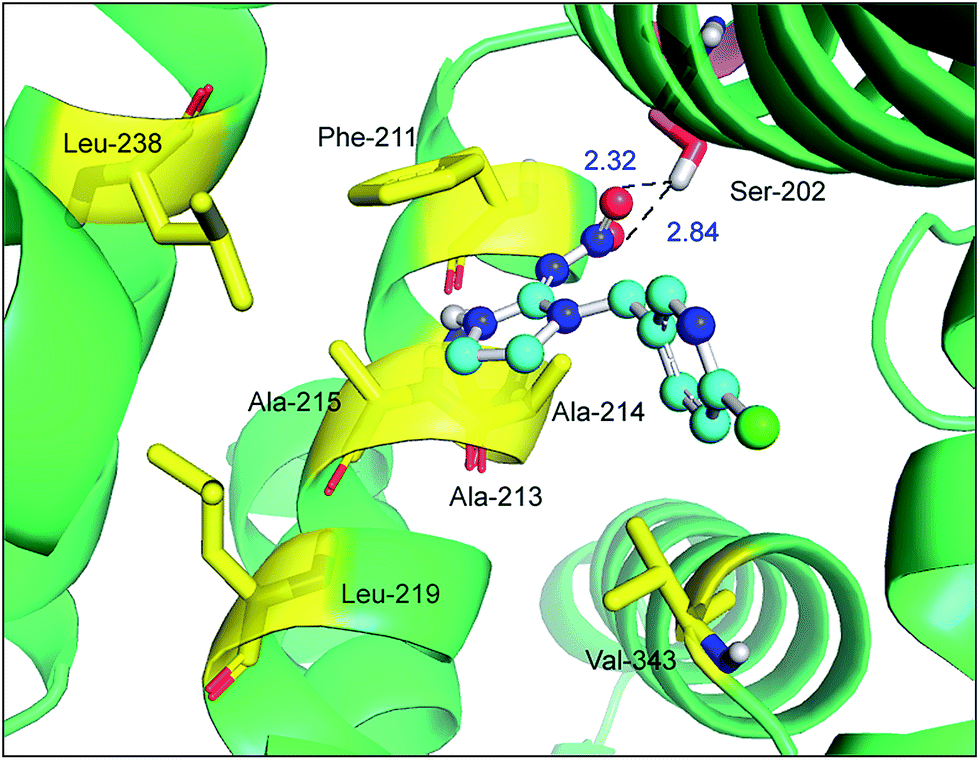 | ||
| Fig. 8 Molecular docking of imidacloprid docked onto mutated albumin (Trp-214 → Ala). The ball-and-stick model portrays imidacloprid, colored according to the atoms and the critical amino acid residues around imidacloprid have been indicated in the stick model; the salmon and yellow stick model shows hydrogen bonds and hydrophobic interactions between mutated albumin and imidacloprid, respectively. (For an explanation of the references to color in this figure legend, the reader is referred to the web version of the article.). | ||
Molecular dynamics simulations and free energy calculations
Molecular dynamics (MD) simulations capture the behavior of biomacromolecules in full atomic detail, and this method could therefore help to substantiate the accuracy of the docking results. Moreover, these dynamics data may assist us in determining the changes in energy in the biopolymer–ligand systems within a short timeframe, and then provide important information for the decomposition of free energies.88,89 To confirm the conformational stability of the protein–neonicotinoid complexes under simulated physiological conditions and further determine the binding free energies of the non-covalent complexes, MD simulations were performed for the four protein–ligand conjugates. In the present attempt, the complexed conformation that was obtained from molecular docking was used as the initial conformation for the MD simulation and the simulation time for the native protein–neonicotinoid complexes was 30 ns. In addition, the pure protein also underwent a simulation process for 10 ns. Frequently, if the fluctuations in the value of the RMSD for a typical dynamic system remain within 0.1 nm, the system can be regarded as achieving a stable state of dynamic equilibrium. It is quite evident from Fig. 9 that the four non-covalent protein–neonicotinoid adducts might be equilibrated within a time period of 5000 ps, whereas the pure protein may reach an equilibrium state at a time point of 2000 ps.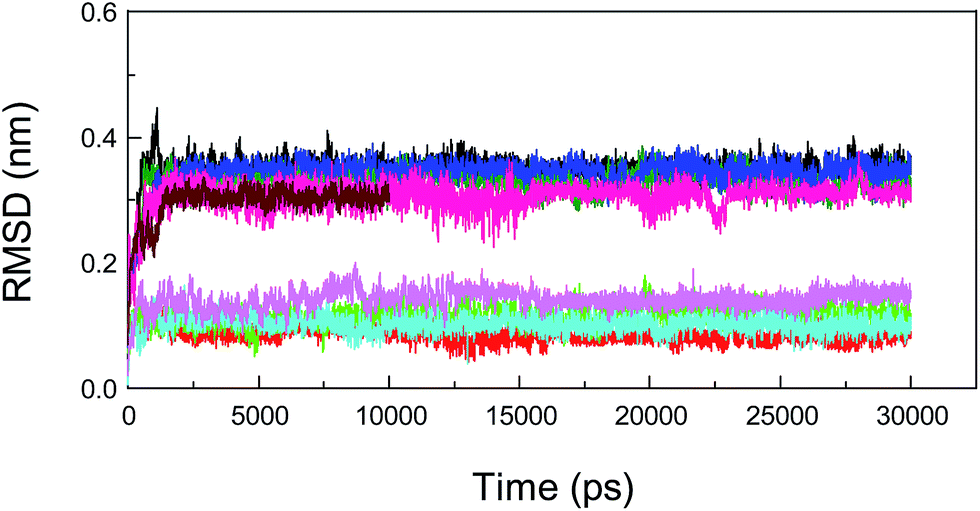 | ||
| Fig. 9 Calculated values of root-mean-square deviation (RMSD) for the neonicotinoids and backbone Cα atoms of albumin from MD simulations at a temperature of 298 K with respect to their docking results as a function of the simulation time. The red and black, green and olive, cyan and blue, and magenta and pink trajectories illustrate values of RMSD for imidacloprid, thiacloprid, acetamiprid, nitenpyram and the backbone Cα atoms of the mutated protein, respectively. The wine trajectory shows RMSD data for the backbone Cα atoms of the pure protein. | ||
In addition, MD simulations of the mutated protein–neonicotinoid complexes were also executed to prove the rationality and stability of the pattern of binding between the mutated amino acid residues (Trp-214, Phe-211 and Arg-222) on the protein and the neonicotinoid. As regards the three mutated systems, that is, protein (Trp-214 → Ala-214)-imidacloprid, protein (Phe-211 → Ala-211)-imidacloprid and protein (Arg-222 → Ala-222)-imidacloprid, simulation processes were studied with a time length of 50 ns. We found that these mutated protein–ligand systems could achieve a state of dynamic equilibrium before 8000 ps. The variations in the tendency of non-covalent interactions between the mutated protein and the neonicotinoid under physiological conditions might be explained via dynamics data. Fig. 10 illustrates the changes in RMSD for conformations with respect to the results of molecular docking for the mutated protein–imidacloprid adducts in the MD simulations. Apparently, if we mutate the Trp-214 residue to an Ala residue in the polypeptide chain, the mutated protein–imidacloprid system begins to stabilize after 1500 ps. The RMSD for the backbone Cα atoms of the mutated protein (black) fluctuates stably at 0.4 nm and the amplitude is within a range of 0.1 nm, whereas the RMSD for imidacloprid (red) fluctuates at about 0.15 nm and the range should be within 0.05 nm. As for the other mutated protein–imidacloprid adducts, i.e., those with Phe → Ala and Arg → Ala residues, the MD simulations of these conjugates and a careful explanation of the dynamics results are given in the ESI.†
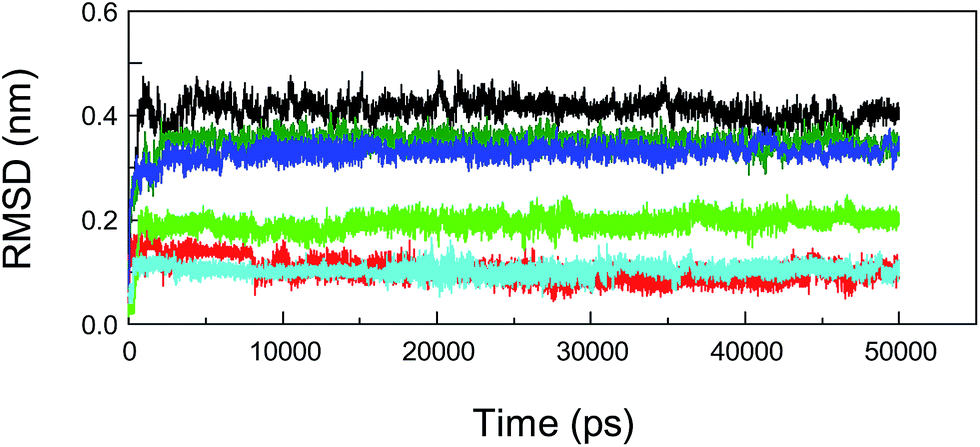 | ||
| Fig. 10 Calculated values of root-mean-square deviation (RMSD) for imidacloprid and the backbone Cα atoms of mutated albumin from MD simulations at a temperature of 298 K with respect to their docking results as a function of the simulation time. The red and black (Trp-214 → Ala), green and olive (Phe-211 → Ala), and cyan and blue (Arg-222 → Ala) trajectories symbolize values of RMSD for imidacloprid and the backbone Cα atoms of the mutated protein, respectively. | ||
To determine whether the binding conformation in dynamic equilibrium can match the results of molecular docking, the average conformation in the timeframe between 2000 ps and 8000 ps was selected and superimposed on the initial conformation of the MD simulation and the outcome is presented in Fig. 11. It is very clear that the original conformation of the mutated protein–neonicotinoid adduct overlaps the equilibrium conformation completely and the alterations in the binding mode between the mutated protein and the neonicotinoid are rather small; the fluctuation in the RMSD mainly originates in the overall translation of the complex. Although there are no obvious changes in the binding style and conformation, it is noteworthy that a downward trend is observable in hydrogen bonding between the Ser-202 residue and imidacloprid. The equilibrium conformation from MD simulations shows that the oxygen atoms of the nitro group in imidacloprid could form hydrogen bonds with the hydrogen atom of the hydroxyl group in the Ser-202 residue, and the bond lengths are 2.41 Å and 3.15 Å, respectively. This means that the mutation of the Trp-214 residue should be a trigger for a decrease in affinity between the protein and neonicotinoids, or rather that the Trp-214 residue is extremely important in the protein–neonicotinoid reactions.
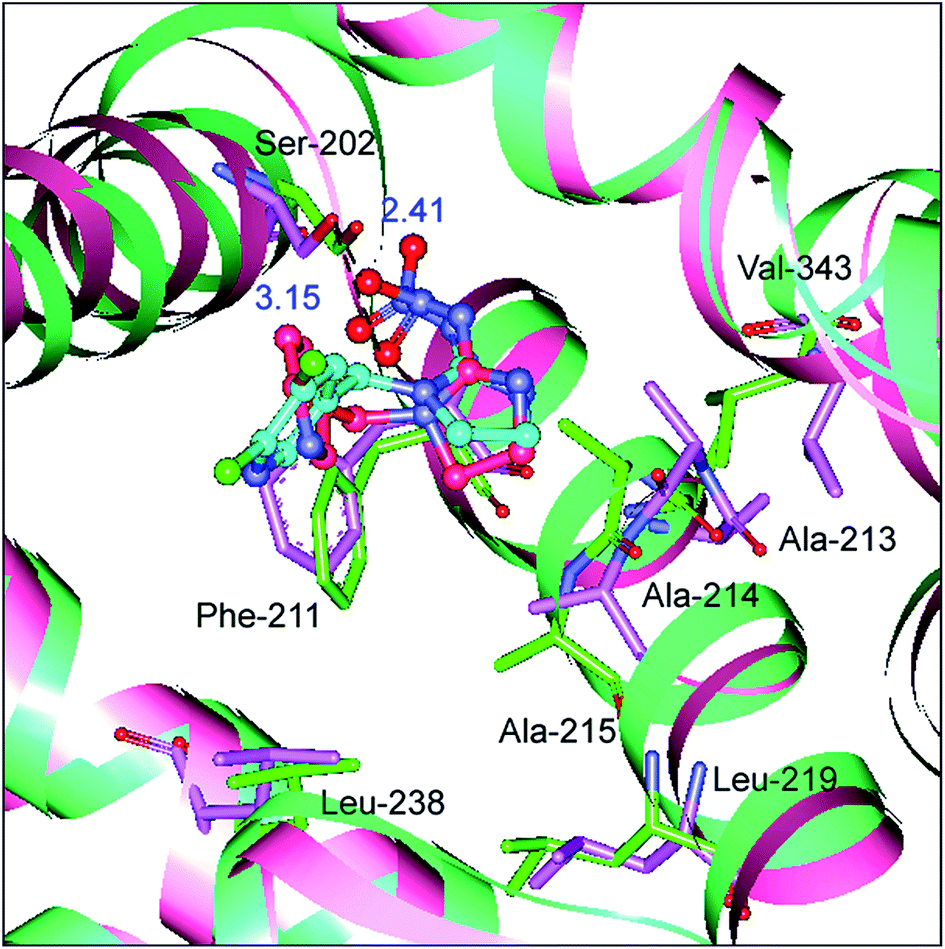 | ||
| Fig. 11 Superposition of the mean conformation from MD simulations on the original conformation from the results of molecular docking for the mutated albumin–imidacloprid complex. The protein is indicated by a surface colored in blue-green (initial) and pink (average) and the original and average conformations of imidacloprid are indicated in the cyan and hot pink ball-and-stick model, respectively. The green and pink stick model shows, respectively, the initial and average conformations of the crucial amino acid residues involved in the mutated albumin–imidacloprid reaction process. (For an interpretation of the references to color in this figure legend, the reader is referred to the web version of the article). | ||
Considering the contrast in the characteristics of recognition between native and mutated proteins, we may reasonably draw the conclusion that the mutation of some crucial amino acid residues, such as Trp-214, Phe-211 and Arg-222, will not only cause changes in hydrogen bonds but also cause the decrease and disappearance of conjugation effects and hydrophobic interactions. These issues would remarkably reduce the strength of non-covalent bonding between the protein and neonicotinoid. Accordingly, there is no doubt that the Trp-214, Phe-211 and Arg-222 residues play an essential role in the molecular recognition of neonicotinoids by plasma albumin.
At the same time, we might assign the secondary structural constituents of the protein in the native and mutated states by a combination of DSSP and GROMACS programs and the results for α-helix, β-sheet and turn are displayed in Table 3. In particular, assignments of secondary structure based on dynamics data suggest that the free protein has relatively high contents of 54.1% α-helix, 10.3% β-sheet and 10.7% turn; upon complexation with imidacloprid, a major reduction in α-helix was observed from 54.1% in the free protein to 44.3% in protein–imidacloprid, and increases in the β-sheet and turn structures were also detected, from 10.3% and 10.7% in the free protein to 12.7% and 13.5% in protein–imidacloprid, respectively. According to wet experiments and far-UV CD, the free protein in solution contains 55.9% α-helix, 8.1% β-sheet and 11.6% turn, whereas the secondary structures of the protein changed to 48.5% α-helix, 9.2% β-sheet and 14.2% turn after interaction with imidacloprid. It is found that the secondary structures that are estimated from both far-UV CD spectra and MD simulations are very similar and consequently this phenomenon confirms that the results of molecular modeling are fully reliable in the current context.
| Samples | Secondary structure elements (%) | RMSD (nm) | |||
|---|---|---|---|---|---|
| α-Helix | β-Sheet | Turn | Backbone | Ligand | |
| Albumin | 54.1 | 10.3 | 10.7 | 0.305 | — |
| Albumin + imidacloprid | 44.3 | 12.7 | 13.5 | 0.352 | 0.086 |
| Albumin + thiacloprid | 43.5 | 13.8 | 14.1 | 0.337 | 0.112 |
| Albumin + nitenpyram | 48.2 | 15.6 | 11.8 | 0.321 | 0.144 |
| Albumin + acetamiprid | 46.1 | 14.2 | 12.2 | 0.346 | 0.108 |
| Albumin (Trp-214 → Ala-214) + imidacloprid | 41.8 | 13.3 | 15.6 | 0.409 | 0.095 |
| Albumin (Phe-211 → Ala-211) + imidacloprid | 44.0 | 11.9 | 14.2 | 0.363 | 0.201 |
| Albumin (Arg-222 → Ala-222) + imidacloprid | 42.7 | 15.1 | 12.8 | 0.339 | 0.114 |
Free energy is a basic quantity that corresponds to the stability of a system because the free energy of a system is minimized if the system is in equilibrium with its environment; therefore, determining the free energy is highly useful in simulations of biological systems.90 Frequently, the method of molecular mechanics/generalized Born surface area (MM/GBSA) has been applied to a variety of computational problems with biomolecules, including receptor-ligand recognition.91,92 According to previous data from MD simulations, calculations of the free energy in the last 10 ns of dynamic processes in an equilibrium state have proceeded by employing the MM/GBSA approach and the time interval is 2.0 ps. As indicated distinctly in Table 4, the energies that are derived from MM/GBSA show some discrepancies with the results from molecular docking, but the trends in the variation of energy are in agreement with the analyses from molecular docking, and the sequence is found to be thiacloprid < imidacloprid < acetamiprid < nitenpyram. The values of binding free energy indicate that the most favorable interaction energies are found in the protein–nitenpyram system (ΔGbind = −7.24 kcal mol−1), and the differences in van der Waals energies (ΔEvdW) for the four non-covalent systems are relatively small, whereas the electrostatic energies (ΔEele) show some notable disparities. This may well be the crucial reason that leads to the generation of differences in free energy for the neonicotinoid agents. By comparison with the discrepancies in hydrophobicity between the non-polar solvation and ligand molecules, it can be noticed that the stronger the hydrophobicity of the ligand, the lower is the ΔGSA value. However, for the mutated protein–neonicotinoid systems, the free energies obtained from molecular docking and MD simulations are slightly higher than for the native protein–neonicotinoid adducts. These phenomena further support the former view that the Trp-214, Phe-211 and Arg-222 residues are vitally important for biopolymer-neonicotinoid recognition.
| Systems | ΔEele | ΔEvdW | −TΔS | ΔGSA | ΔGGB | ΔGbind | ΔGbind (docking) |
|---|---|---|---|---|---|---|---|
| Albumin + imidacloprid | −36.32 ± 0.27 | −22.91 ± 0.12 | 16.39 ± 0.11 | −3.44 ± 0.05 | 38.15 ± 0.57 | −6.13 | −5.87 |
| Albumin + thiacloprid | −33.41 ± 0.15 | −21.08 ± 0.03 | 15.57 ± 0.78 | −4.61 ± 0.03 | 35.31 ± 1.06 | −4.22 | −5.13 |
| Albumin + nitenpyram | −40.73 ± 0.33 | −23.15 ± 0.27 | 23.21 ± 1.21 | −3.08 ± 0.10 | 36.51 ± 0.63 | −7.24 | −6.69 |
| Albumin + acetamiprid | −37.94 ± 0.29 | −23.63 ± 0.06 | 21.02 ± 1.17 | −2.77 ± 0.04 | 36.60±0.52 |
−6.50 | −6.11 |
| Albumin (Trp-214 → Ala-214) + imidacloprid | −34.22 ± 0.21 | −20.02 ± 0.05 | 14.99 ± 0.38 | −3.13 ± 0.04 | 36.68 ± 0.25 | −5.70 | −5.28 |
| Albumin (Phe-211 → Ala-211) + imidacloprid | −34.37 ± 0.19 | −19.30 ± 0.13 | 18.27 ± 0.24 | −3.06 ± 0.10 | 33.14 ± 0.39 | −5.32 | −4.99 |
| Albumin (Arg-222 → Ala-222) + imidacloprid | −31.48 ± 0.12 | −21.36 ± 0.48 | 18.45 ± 0.21 | −3.50 ± 0.22 | 31.88 ± 0.46 | −6.01 | −5.54 |
Structure–activity relationships
According to the preceding explanations, commercial and potential neonicotinoids are commonly composed of three structural elements (structures are shown in Fig. 12). Among these, imidacloprid, thiacloprid, nitenpyram and acetamiprid display some similarity as they all have a p-substituted chloropyridyl group in their structure.2,93,94 However, imidacloprid and thiacloprid contain a heterocyclic spacer in the B component; in contrast, nitenpyram and acetamiprid include an acyclic spacer in this area. Chemically, the modification of the substituent group in the B component might have the potential to affect biorecognition between a protein and the neonicotinoid. To confirm this point, three neonicotinoids, viz., thiacloprid, nitenpyram and acetamiprid, were selected for performing ligand-docking studies and the best results are shown in Fig. 13.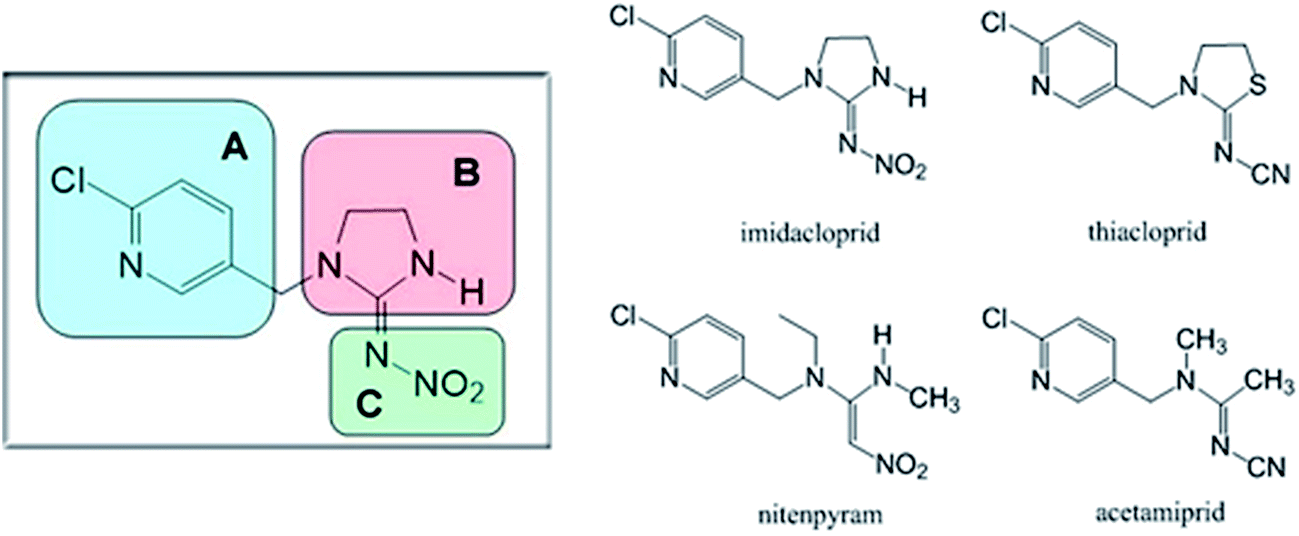 | ||
| Fig. 12 Commercial neonicotinoids consisting of three structural components (A, B and C) and the molecular structures of imidacloprid, thiacloprid, nitenpyram and acetamiprid. | ||
 | ||
| Fig. 13 Molecular docking of nitenpyram (A), acetamiprid (B) and thiacloprid (C) docked onto albumin. Albumin is shown as a surface colored in light pink and the ball-and-stick models show neonicotinoids colored according to the atoms. The key amino acid residues around the neonicotinoids have been indicated in the stick model; the salmon stick model shows hydrogen bonds between Arg-222, His-242 (Panel (A)) and Arg-222 (Panel (B)) residues and nitenpyram and acetamiprid, respectively; the green stick model represents π–π stacking between Phe-211 and Trp-214 residues and neonicotinoids; and the yellow stick model shows hydrophobic interactions between Phe-211, Trp-214, Ala-215, Leu-219, Leu-238, and Val-343 residues and neonicotinoids. (For an interpretation of the references to color in this figure legend, the reader is referred to the web version of the article). | ||
Obviously, the free energies of the protein–nitenpyram adduct (ΔGbind/ΔGdocking = −7.24/−6.69 kcal mol−1) and the protein–acetamiprid adduct (ΔGbind/ΔGdocking = −6.50/−6.11 kcal mol−1) are higher than those of the protein–imidacloprid complex. One logical explanation is that the critical non-covalent bonds such as hydrogen bonds in the two protein–neonicotinoid complexes are stronger than those in the protein–imidacloprid adduct. The oxygen atoms and nitrogen atom in the nitro group and the hydrogen atom of the secondary amine in the acyclic spacer of nitenpyram could form hydrogen bonds with a hydrogen atom of the amino group in the Arg-222 residue and the nitrogen atom in the imidazole ring in the His-242 residue, and the bond lengths are 2.07 Å, 3.06 Å and 2.28 Å (Fig. 13(A)), respectively. This pattern will make the conformation of nitenpyram more stable in the active cavity of the protein molecule. As a result, the recognition ability for acetamiprid with protein is lower than that for nitenpyram, but still better than that for imidacloprid.
With regard to the protein–acetamiprid adduct (Fig. 13(B)), the nitrogen atom in the cyano group in acetamiprid may form two hydrogen bonds with the hydrogen atoms of the amino group and the secondary amine in the Arg-222 residue, and the bond lengths are 2.03 Å and 2.29 Å, respectively. Both bond lengths and affinity support the deduction that the toxicity of acetamiprid is greater than that of its analog imidacloprid. However, for the protein–thiacloprid adduct (ΔGbind/ΔGdocking = −4.22/−5.13 kcal mol−1), which has a similar constituent heterocyclic group to imidacloprid in the B spacer, the reaction of thiacloprid with the protein is worse compared with that of the other neonicotinoids, and the best recognition profile is also illustrated in Fig. 13(C). It is evident that thiacloprid cannot form hydrogen bonds with the biomacromolecule; however, π–π stacking exists between the pyridine ring in thiacloprid and the benzene ring in the Phe-211 residue and the indole ring in the Trp-214 residue. This phenomenon should enable the neonicotinoid to remain at the functional domain. On the basis of structure–activity discussions, we might conclude that the two neonicotinoids—nitenpyram and acetamiprid—with an acyclic spacer possess higher affinity and stronger non-covalent interactions than imidacloprid and thiacloprid, which have heterocyclic spacers.
Probably, these disparities in recognition arise from the molecular flexibility of the neonicotinoids. Actually, the flexibility of the heterocyclic segment is less than that of the acyclic segment in insecticides; the polar functional groups, that is, the nitro and cyano groups in nitenpyram and acetamiprid, respectively, would form excellent non-covalent bonds with the surrounding amino acid residues, and in consequence this feature could lead to more powerful association interactions compared with imidacloprid and thiacloprid. Such a phenomenon can explain discrepancies in the biomolecular recognition of neonicotinoids by biopolymers, and we also believe that the properties of substituents in neonicotinoids may play a fundamental role in macromolecule-pesticide biorecognition. Furthermore, these conclusions provide a theoretical foundation for our previous opinion; a neonicotinoid with a high binding strength to a protein has a longer half-life, which can increase the toxicity of the agrochemical to human health.
Toxicological relevance
To investigate the relationships between the molecular structures of the four typical neonicotinoid insecticides and their possible noxious effects in great detail, the physicochemical and toxicological data for these neonicotinoids have been assessed based upon authoritative tools such as VEGA,95 the Estimation Program Interface (EPI) Suite and the Toxicity Estimation Software Tool (TEST),96,97 which were developed by the European Environment Agency and the US Environmental Protection Agency, and the results are presented in Table 5. Evidently, nitenpyram possesses strong hydrophilicity; this quality might largely be attributed to the ring-open structure and multiple hydrophilic groups of the neonicotinoid. However, in the case of acetamiprid, this compound retains a ring-open structure but hydrophilic groups are few; thus, acetamiprid has a relatively high hydrophobicity.98 In addition, nitenpyram has characteristics of both a carcinogen and a developmental toxicant, whereas the other neonicotinoids, that is, acetamiprid, imidacloprid and thiacloprid, do not possess both carcinogen and developmental toxicant characteristics simultaneously; conversely, these neonicotinoids could be classed as either a carcinogen or a developmental toxicant. Such facts are closely associated with the structural features of neonicotinoids. As we discussed in the structure–activity studies, the non-covalent forces between the protein and nitenpyram clearly outweigh those for the other three neonicotinoids, because nitenpyram contains a representative ring-open structure and more polar groups. This research finding should also indicate that nitenpyram may have greater toxicity and carcinogenicity. However, it is worth mentioning that there are no very strong correlations between acute toxicity, carcinogenicity and mutagenicity, which is why nitenpyram has a lower LC50 and LD50, but the chronic toxicity of this neonicotinoid is far higher than those of acetamiprid, imidacloprid and thiacloprid.| Biochemical parameter | Neonicotinoid | |||
|---|---|---|---|---|
| Imidacloprid | Nitenpyram | Acetamiprid | Thiacloprid | |
| a From Estimation Program Interface (EPI) suite.b From VEGA.c From Toxicity Estimation Software Tool (TEST), consensus method.d From VEGA.e From Toxicity Estimation Software Tool (TEST), consensus method.f From Toxicity Estimation Software Tool (TEST), consensus method.g From VEGA.h Experimental data from Estimation Program Interface (EPI) Suite and Toxicity Estimation Software Tool (TEST). | ||||
| log Kow (293 K)a | (0.57)h | (−0.66)h | (0.80)h | (1.26)h |
| Carcinogenicityb | Non-carcinogen | Carcinogen | Carcinogen | Carcinogen |
| Developmental toxicityc | Developmental toxicant | Developmental toxicant | Developmental non-toxicant | Developmental non-toxicant |
| Fathead minnow LC50 (96 h) (mg L−1)d | 108.89 | 71.47 | 27.64 | 4.13 |
| Mutagenicitye | Positive | Positive | Positive | Positive |
| Oral rat LD50 (mg kg−1)f | 369.01 (409.93)h | 954.59 (1576.04)h | 678.93 | 1232.67 (444.32)h |
| Ready biodegradabilityg | Not readily biodegradable | Not readily biodegradable | Not readily biodegradable | Not readily biodegradable |
Furthermore, there are several exact evidence that imidacloprid might not be the most toxic analog among the commercial neonicotinoids, whereas nitenpyram and acetamiprid may have a greater negative impact on the human body than imidacloprid. Haemato-biochemical and histopathological examinations in male Wistar rats have demonstrated that the administration of acetamiprid for nearly one month will result in significant increases in levels of alanine transaminase, aspartate transaminase, lactate dehydrogenase and creatinine kinase in serum and obvious decreases in hemoglobin and total erythrocyte count.99 Moreover, individual cell necrosis and karyomegaly were observed in liver, and mild glomerular oedema, congestion and desquamated epithelial cells were also detected in the kidney. In female Wistar rats, an in vivo hematological study suggested that acetamiprid has an adverse effect on hemopoietic organs in animals via subacute exposure.100 Moreover, a toxicological evaluation of imidacloprid noted similar changes in male rats; the result also implied that imidacloprid can lead to a reduction in acetylcholinesterase activity in the brain. Furthermore, Ford and Casida93 concluded that the chloropyridyl neonicotinoid insecticides are readily metabolized and excreted in male albino Swiss-Webster mice. The value of t1/2 relative to the maximum level is considerably higher for acetamiprid (>240 min) than for imidacloprid (80 min) and thiacloprid (50 min) in plasma.
In mammals, neonicotinoids will chiefly form a complex with neuronal nAChR, and such biomolecular interactions may produce several pathological symptoms, for instance, neuronal apoptosis, differentiation, migration, proliferation and synapse formation.101 Recent scientific achievements show that neonicotinoids, including imidacloprid and acetamiprid, are absorbed and transported by functional biomolecules (primarily albumin) in the organism, which then pass through the blood–brain barrier, and eventually bind to the target nAChR.93,102 Moreover, the values of IC50 of both imidacloprid and acetamiprid on mammalian neuronal nAChR are 2600 nM and 700 nM, respectively, which signifies that the biological effects of acetamiprid on nAChR should evidently be larger than those of imidacloprid.103 In general, a substance with a high protein-binding affinity might possess a long half-life (t1/2), which would increase its toxicity. In contrast, a compound with a low protein-binding affinity is restricted in its capacity to perfuse tissues and reach its location of action. As previously described, the overall lengths of non-covalent bonds in the protein–acetamiprid reaction are observed to be shorter than those for the protein–imidacloprid reaction. This fact distinctly suggests that the association affinity of protein–acetamiprid is higher than that of protein–imidacloprid; in other words, protein–acetamiprid adducts may exist in the body for quite a long time. Under these circumstances, more acetamiprid molecules can be delivered to neuronal nAChR via the active transporter (albumin) and ultimately give rise to greater toxic activity. These conclusions have aspects in common with our previous comprehensive investigations, and a neonicotinoid with more flexibility would possess greater capacity for recognition by non-target biomacromolecules, which would increase its toxicity.
Aside from the parent compound of neonicotinoids, we should point out that these chemicals might be biodegraded by metabolic attack at different moieties. Maybe several metabolites, in some cases, contribute to overall toxicities such as carcinogenesis and hepatotoxicity in mammals. Recently, an interesting observation has been made by Casida,104 who considered that we may get opportunities for metabolic selectivity and programmed persistence if we take the wide diversity of neonicotinoid substituents into consideration and finally obtain neonicotinoid pesticides that possess selective toxicity against various pests, while being relatively safe to human beings and beneficial organisms.
Conclusions
To sum up, the current study explains the biorecognition of the most widely used neonicotinoids by multifunctional albumin by combining experimental and computational techniques at the molecular scale. Data from fluorescence confirmed that a decrease in the emissions of Trp residues originated from a static reaction at low concentrations of neonicotinoids, whereas both static and dynamic processes operated when the concentration of neonicotinoids exceeded 10 μM. The binding strength of neonicotinoids with the protein decreases within the range of moderate affinity, with a stoichiometry of 1:1, and non-covalent bonds, such as hydrogen bonds, π–π stacking and hydrophobic interactions, are largely responsible for stabilizing protein–neonicotinoid adducts. Moreover, GuHCl-induced denaturation of albumin, extrinsic ANS fluorescence and site-specific competitive binding experiments all suggested that subdomain IIA, Sudlow's site I, has high affinity for the binding of neonicotinoids to the protein. These outcomes are in agreement with those from molecular docking, site-directed mutagenesis, MD simulations and the decomposition of free energy, which locate neonicotinoids at the warfarin–azapropazone site, and several amino acid residues, i.e., Phe-211, Trp-214 and Arg-222, play a major role in non-covalent recognition.
Time-resolved fluorescence decay illustrates that the conformation of the protein may undergo a slight transformation when a neonicotinoid is conjugated to the protein. This phenomenon has been further confirmed by synchronous fluorescence and far-UV CD, which shows that the content of α-helix in the protein was reduced from 55.9% to 48.5%, with increases in the contents of β-sheet, turn and random coil in the protein–neonicotinoid complex. Moreover, the results of MD simulations confirm the trends of conformational alterations in the presence of neonicotinoids. Based on structure–activity relationships, it can be assumed that structural differences in the B component of neonicotinoids could affect the recognition capacity between the protein and neonicotinoids. To be more exact, a ring-open structure will endow neonicotinoids with greater flexibility and is more likely to lead to non-covalent bonds with amino acid residues during protein–neonicotinoid reactions. Perhaps this is the reason why the association abilities of nitenpyram and acetamiprid with the protein are higher than those of ring-closed neonicotinoids, e.g., imidacloprid and thiacloprid. Indeed, the protein–neonicotinoid complexes are found to be closely related to the toxicological actions of these agrochemicals. As neonicotinoids are among the most widely used pesticides, along with the highly controversial current debate regarding the possible toxicity of these compounds to non-target mammals, we hope that this study might offer useful information for evaluating potentially detrimental effects of these insecticides.
Conflict of interest
The authors declare no competing financial interest.Abbreviations
| Ala | Alanine |
| ANS | 8-Anilino-1-naphthalenesulfonic acid |
| Arg | Arginine |
| CD | Circular dichroism |
| DNA | Deoxyribonucleic acid |
| DSSP | Dictionary of protein secondary structure |
| EPI | Estimation program interface |
| GuHCl | Guanidine hydrochloride |
| HCl | Hydrochloric acid |
| His | Histidine |
| IRF | Instrument response function |
| LCPO | Linear combination of pairwise overlaps |
| Leu | Leucine |
| LGA | Lamarckian genetic algorithm |
| Lys | Lysine |
| MD simulation | Molecular dynamics simulation |
| MM/GBSA | Molecular mechanics/generalized Born surface area |
| nAChRs | Nicotinic acetylcholine receptors |
| NPT | Isothermal-isobaric |
| Phe | Phenylalanine |
| PME | Particle mesh Ewald |
| R | Correlation coefficient |
| RCSB | Research collaboratory for structural bioinformatics |
| RMSD | Root-mean-square deviation |
| RNA | Ribonucleic acid |
| SASA | Solvent-accessible surface area |
| S.D. | Standard deviation |
| Ser | Serine |
| TEST | Toxicity estimation software tool |
| Tris | Tris(hydroxymethyl)aminomethane |
| Trp | Tryptophan |
| Tyr | Tyrosine |
| UV/vis | Ultraviolet-visible spectroscopy |
| Val | Valine |
| VEGA | Virtual models for property evaluation of chemicals within a global architecture |
Acknowledgements
We are greatly indebted to Professor Ulrich Kragh-Hansen of the Department of Biomedicine, University of Aarhus, for the precious gift of his doctoral dissertation. We particularly appreciate Dr Peter Ertl of Novartis Institutes for BioMedical Research (Basel, Switzerland) for the kind supply of JME Molecular Editor. We thank Editors Jessie Morgan and Allison Holloway of the Royal Society of Chemistry, for their warm support during the manuscript processing. We are also grateful to the reviewers of this manuscript for their constructive and insightful suggestions.References
- S. C. Kessler, E. J. Tiedeken, K. L. Simcock, S. Derveau, J. Mitchell, S. Softley, J. C. Stout and G. A. Wright, Nature, 2015, 521, 74–76 CrossRef CAS PubMed.
- J. E. Casida and K. A. Durkin, Annu. Rev. Entomol., 2013, 58, 99–117 CrossRef CAS PubMed.
- M. R. Douglas and J. F. Tooker, Environ. Sci. Technol., 2015, 49, 5088–5097 CrossRef CAS PubMed.
- T. T. Talley, M. Harel, R. E. Hibbs, Z. Radić, M. Tomizawa, J. E. Casida and P. Taylor, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 7606–7611 CrossRef CAS PubMed.
- P. Jeschke, R. Nauen and M. E. Beck, Angew. Chem., Int. Ed., 2013, 52, 9464–9485 CrossRef CAS PubMed.
- G. Di Prisco, V. Cavaliere, D. Annoscia, P. Varricchio, E. Caprio, F. Nazzi, G. Gargiulo and F. Pennacchio, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 18466–18471 CrossRef CAS PubMed.
- A. D. Ozsahin, R. Bal and O. Yılmaz, Toxicol. Res., 2014, 3, 324–330 RSC.
- V. Doublet, M. Labarussias, J. R. de Miranda, R. F. A. Moritz and R. J. Paxton, Environ. Microbiol., 2015, 17, 969–983 CrossRef CAS PubMed.
- D. Goulson, E. Nicholls, C. Botías and E. L. Rotheray, Science, 2015, 347 DOI:10.1126/science.1255957.
- S. L. Carmichael, W. Yang, E. Roberts, S. E. Kegley, A. M. Padula, P. B. English, E. J. Lammer and G. M. Shaw, Environ. Res., 2014, 135, 133–138 CrossRef CAS PubMed.
- K. V. Vinod, S. Srikant, G. Thiruvikramaprakash and T. K. Dutta, Am. J. Emerg. Med., 2015, 33, 310 CrossRef PubMed.
- P. Bagri, V. Kumar and A. K. Sikka, Drug Chem. Toxicol., 2015, 38, 342–348 CrossRef CAS PubMed.
- S. Bhardwaj, M. K. Srivastava, U. Kapoor and L. P. Srivastava, Food Chem. Toxicol., 2010, 48, 1185–1190 CrossRef CAS PubMed.
- R. Bal, G. Türk, M. Tuzcu, O. Yilmaz, T. Kuloglu, R. Gundogdu, S. Gür, A. Agca, M. Ulas, Z. Çambay, Z. Tuzcu, H. Gencoglu, M. Guvenc, A. D. Ozsahin, N. Kocaman, A. Aslan and E. Etem, J. Environ. Sci. Health, Part B, 2012, 47, 434–444 CrossRef CAS PubMed.
- M.-L. Jugan, Y. Levi and J.-P. Blondeau, Biochem. Pharmacol., 2010, 79, 939–947 CrossRef CAS PubMed.
- N. Hoshi, T. Hirano, T. Omotehara, J. Tokumoto, Y. Umemura, Y. Mantani, T. Tanida, K. Warita, Y. Tabuchi, T. Yokoyama and H. Kitagawa, Biol. Pharm. Bull., 2014, 37, 1439–1443 CAS.
- L. Gawade, S. S. Dadarkar, R. Husain and M. Gatne, Food Chem. Toxicol., 2013, 51, 61–70 CrossRef CAS PubMed.
- R. K. S. Devan, P. C. Prabu and S. Panchapakesan, Drug Chem. Toxicol., 2015, 38, 328–336 CrossRef PubMed.
- W. J. Rea, J. Nutr. Environ. Med., 1996, 6, 55–124 CrossRef.
- F. Sánchez-Bayo, Science, 2014, 346, 806–807 CrossRef PubMed.
- J.-L. Brunet, M. Maresca, J. Fantini and L. P. Belzunces, Toxicol. Appl. Pharmacol., 2004, 194, 1–9 CrossRef CAS PubMed.
- C. A. Hallmann, R. P. B. Foppen, C. A. M. van Turnhout, H. de Kroon and E. Jongejans, Nature, 2014, 511, 341–343 CrossRef CAS PubMed.
- M. M. Kandil, C. Trigo, W. C. Koskinen and M. J. Sadowsky, J. Agric. Food Chem., 2015, 63, 4721–4727 CrossRef CAS PubMed.
- G. J. Rocklin, S. E. Boyce, M. Fischer, I. Fish, D. L. Mobley, B. K. Shoichet and K. A. Dill, J. Mol. Biol., 2013, 425, 4569–4583 CrossRef CAS PubMed.
- I. Vayá, V. Lhiaubet-Vallet, M. C. Jiménez and M. A. Miranda, Chem. Soc. Rev., 2014, 43, 4102–4122 RSC.
- S. Tabassum, W. M. Al-Asbahy, M. Afzal, F. Arjmand and R. H. Khan, Mol. BioSyst., 2012, 8, 2424–2433 RSC.
- M. Kallubai, A. Rachamallu, D. P. Yeggoni and R. Subramanyam, Mol. BioSyst., 2015, 11, 1172–1183 RSC.
- M. H. Tarhoni, T. Lister, D. E. Ray and W. G. Carter, Biomarkers, 2008, 13, 343–363 CrossRef CAS PubMed.
- F. Zsila, Mol. Pharmaceutics, 2013, 10, 1668–1682 CrossRef CAS PubMed.
- L. Brülisauer, G. Valentino, S. Morinaga, K. Cam, J. T. Bukrinski, M. A. Gauthier and J.-C. Leroux, Angew. Chem., Int. Ed., 2014, 53, 8392–8396 CrossRef PubMed.
- M. A. Williams, in Protein–Ligand Interactions: Methods and Applications, ed. M. A. Williams and T. Daviter, Humana Press, New York, NY, 2nd edn, 2013, vol. 1008, pp. 3–34 Search PubMed.
- K. Shanmugaraj, S. Anandakumar and M. Ilanchelian, RSC Adv., 2015, 5, 3930–3940 RSC.
- J. Wang, Q. Li, L. J. Yang, Y. J. Zhang, J. Yu, X. F. Zhao, J. B. Zheng, Y. Y. Zhang and X. H. Zheng, Anal. Methods, 2015, 7, 3340–3346 RSC.
- P. C. Hebert and L. A. MacManus-Spencer, Anal. Chem., 2010, 82, 6463–6471 CrossRef CAS PubMed.
- C. E. Tinberg, S. D. Khare, J. Y. Dou, L. Doyle, J. W. Nelson, A. Schena, W. Jankowski, C. G. Kalodimos, K. Johnsson, B. L. Stoddard and D. Baker, Nature, 2013, 501, 212–216 CrossRef CAS PubMed.
- K. I. Mikhailopulo, T. S. Serchenya, E. P. Kiseleva, Y. G. Chernov, T. M. Tsvetkova, N. V. Kovganko and O. V. Sviridov, J. Appl. Spectrosc., 2008, 75, 857–863 CrossRef CAS.
- Y.-Q. Wang, B.-P. Tang, H.-M. Zhang, Q.-H. Zhou and G.-C. Zhang, J. Photochem. Photobiol., B, 2009, 94, 183–190 CrossRef CAS PubMed.
- F. Ding, W. Peng, J.-X. Diao, L. Zhang and Y. Sun, J. Agric. Food Chem., 2013, 61, 4497–4505 CrossRef CAS PubMed.
- F. Ding and W. Peng, J. Photochem. Photobiol., B, 2015, 147, 24–36 CrossRef CAS PubMed.
- O. H. Lowry, N. J. Rosebrough, A. L. Farr and R. J. Randall, J. Biol. Chem., 1951, 193, 265–275 CAS.
- S. Sugio, A. Kashima, S. Mochizuki, M. Noda and K. Kobayashi, Protein Eng., 1999, 12, 439–446 CrossRef CAS PubMed.
- A. Jakalian, D. B. Jack and C. I. Bayly, J. Comput. Chem., 2002, 23, 1623–1641 CrossRef CAS PubMed.
- M. Ihara, T. Okajima, A. Yamashita, T. Oda, T. Asano, M. Matsui, D. B. Sattelle and K. Matsuda, Mol. Pharmacol., 2014, 86, 736–746 CrossRef PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. J. Olson, J. Comput. Chem., 1998, 19, 1639–1662 CrossRef CAS.
- S. Pronk, S. Páll, R. Schulz, P. Larsson, P. Bjelkmar, R. Apostolov, M. R. Shirts, J. C. Smith, P. M. Kasson, D. van der Spoel, B. Hess and E. Lindahl, Bioinformatics, 2013, 29, 845–854 CrossRef CAS PubMed.
- L. D. Schuler, X. Daura and W. F. van Gunsteren, J. Comput. Chem., 2001, 22, 1205–1218 CrossRef CAS.
- A. W. Schüttelkopf and D. M. F. van Aalten, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 1355–1363 CrossRef PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- A. D. Nola, H. J. C. Berendsen and O. Edholm, Macromolecules, 1984, 17, 2044–2050 CrossRef.
- R. Edberg, D. J. Evans and G. P. Morriss, J. Chem. Phys., 1986, 84, 6933–6939 CrossRef CAS.
- A. Baranyai and D. J. Evans, Mol. Phys., 1990, 70, 53–63 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- T. Darden, L. Perera, L. P. Li and L. Pedersen, Structure, 1999, 7, R55–R60 CrossRef CAS PubMed.
- J. A. Snyman, Practical Mathematical Optimization: an Introduction to Basic Optimization Theory and Classical and New Gradient-based Algorithms, Springer Science + Business Media, New York, NY, 2005 Search PubMed.
- M. R. Hestenes and E. Stiefel, J. Res. Natl. Bur. Stand., 1952, 49, 409–436 CrossRef.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- W. Kabsch and C. Sander, Biopolymers, 1983, 22, 2577–2637 CrossRef CAS PubMed.
- W. G. Touw, C. Baakman, J. Black, T. A. H. Te Beek, E. Krieger, R. P. Joosten and G. Vriend, Nucleic Acids Res., 2015, 43, D364–D368 CrossRef PubMed.
- M. R. Shirts and D. L. Mobley, in Biomolecular Simulations: Methods and Protocols, ed. L. Monticelli and E. Salonen, Humana Press, New York, NY, 2013, vol. 924, pp. 271–311 Search PubMed.
- J. E. Kerrigan, in In Silico Models for Drug Discovery, ed. S. Kortagere, Humana Press, New York, NY, 2013, vol. 993, pp. 95–113 Search PubMed.
- J. Weiser, P. S. Shenkin and W. C. Still, J. Comput. Chem., 1999, 20, 217–230 CrossRef CAS.
- M. R. Eftink, in Topics in Fluorescence Spectroscopy: Protein Fluorescence, ed. J. R. Lakowicz, Kluwer Academic Publishers, New York, NY, 2002, vol. 6, pp. 1–15 Search PubMed.
- M. Zolfagharzadeh, M. Pirouzi, A. Asoodeh, M. R. Saberi and J. Chamani, J. Biomol. Struct. Dyn., 2014, 32, 1936–1952 CAS.
- L. Bekale, P. Chanphai, S. Sanyakamdhorn, D. Agudelo and H. A. Tajmir-Riahi, RSC Adv., 2014, 4, 31084–31093 RSC.
- O. J. Rolinski, A. Martin and D. J. S. Birch, Ann. N. Y. Acad. Sci., 2008, 1130, 314–319 CrossRef CAS PubMed.
- L. Brancaleon, Adv. Protein Chem. Struct. Biol., 2013, 93, 95–152 CAS.
- J. M. Beechem and L. Brand, Annu. Rev. Biochem., 1985, 54, 43–71 CrossRef CAS PubMed.
- S. L. C. Moors, A. Jonckheer, M. D. Maeyer, Y. Engelborghs and A. Ceulemans, Curr. Protein Pept. Sci., 2008, 9, 427–446 CrossRef CAS PubMed.
- O. K. Abou-Zied, N. Al-Lawatia, M. Elstner and T. B. Steinbrecher, J. Phys. Chem. B, 2013, 117, 1062–1074 CrossRef CAS PubMed.
- S. Agatonovic-Kustrin, D. W. Morton, L. Truong and S. Razic, Comb. Chem. High Throughput Screening, 2014, 17, 879–890 CrossRef CAS PubMed.
- C. Dufour and O. Dangles, Biochim. Biophys. Acta, Gen. Subj., 2005, 1721, 164–173 CrossRef CAS PubMed.
- N. A. Kratochwil, W. Huber, F. Müller, M. Kansy and P. R. Gerber, Biochem. Pharmacol., 2002, 64, 1355–1374 CrossRef CAS PubMed.
- P. Sevilla, J. M. Rivas, F. García-Blanco, J. V. García-Ramos and S. Sánchez-Cortés, Biochim. Biophys. Acta, Proteins Proteomics, 2007, 1774, 1359–1369 CrossRef CAS PubMed.
- H. N. Bischel, L. A. MacManus-Spencer and R. G. Luthy, Environ. Sci. Technol., 2010, 44, 5263–5269 CrossRef CAS PubMed.
- A. Bolli, M. Marino, G. Rimbach, G. Fanali, M. Fasano and P. Ascenzi, Biochem. Biophys. Res. Commun., 2010, 398, 444–449 CrossRef CAS PubMed.
- E. J. Olson and P. Bühlmann, J. Org. Chem., 2011, 76, 8406–8412 CrossRef CAS PubMed.
- J. B. F. Lloyd, Nature-Phys. Sci., 1971, 231, 64–65 CrossRef CAS.
- J. N. Miller, Analyst, 1984, 109, 191–198 RSC.
- E. A. Burstein, N. S. Vedenkina and M. N. Ivkova, Photochem. Photobiol., 1973, 18, 263–279 CrossRef CAS PubMed.
- N. J. Greenfield, Methods Enzymol., 2004, 383, 282–317 CAS.
- A. J. S. Jones, Adv. Drug Delivery Rev., 1993, 10, 29–90 CrossRef CAS.
- F. R. N. Gurd and T. M. Rothgeb, Adv. Protein Chem., 1979, 33, 73–165 CrossRef CAS PubMed.
- A. D. Vogt and E. D. Cera, Biochemistry, 2013, 52, 5723–5729 CrossRef CAS PubMed.
- X. M. He and D. C. Carter, Nature, 1992, 358, 209–215 CrossRef CAS PubMed.
- A. Cavalli, A. Spitaleri, G. Saladino and F. L. Gervasio, Acc. Chem. Res., 2015, 48, 277–285 CrossRef CAS PubMed.
- R. Buonfiglio, M. Recanatini and M. Masetti, ChemMedChem, 2015, 10, 1141–1148 CrossRef CAS PubMed.
- J. Wereszczynski and J. A. McCammon, Q. Rev. Biophys., 2012, 45, 1–25 CrossRef CAS PubMed.
- I. Slynko, M. Scharfe, T. Rumpf, J. Eib, E. Metzger, R. Schüle, M. Jung and W. Sippl, J. Chem. Inf. Model., 2014, 54, 138–150 CrossRef CAS PubMed.
- M. Lundborg and E. Lindahl, J. Phys. Chem. B, 2015, 119, 810–823 CrossRef CAS PubMed.
- K. A. Ford and J. E. Casida, Chem. Res. Toxicol., 2006, 19, 944–951 CrossRef CAS PubMed.
- M. Tomizawa, D. Maltby, T. T. Talley, K. A. Durkin, K. F. Medzihradszky, A. L. Burlingame, P. Taylor and J. E. Casida, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1728–1732 CrossRef CAS PubMed.
- http://www.vega-qsar.eu/.
- http://www.epa.gov/opptintr/exposure/pubs/episuite.htm.
- http://www.epa.gov/nrmrl/std/qsar/qsar.html.
- E. Taillebois, Z. Alamiddine, C. Brazier, J. Graton, A. D. Laurent, S. H. Thany and J.-Y. le Questel, Bioorg. Med. Chem., 2015, 23, 1540–1550 CrossRef CAS PubMed.
- http://www2.epa.gov/sites/production/files/documents/rmpp_6thed_final_lowresopt.pdf.
- S. Mondal, R. C. Ghosh, M. Mate and C. K. Ghosh, Environ. Ecol., 2009, 27, 1767–1769 CAS.
- J. Kimura-Kuroda, Y. Komuta, Y. Kuroda, M. Hayashi and H. Kawano, PLoS One, 2012, 7, e32432 CAS.
- M. Tomizawa, Adv. Insect Physiol., 2013, 44, 63–99 CrossRef.
- L. P. Sheets, A. A. Li, D. J. Minnema, R. H. Collier, M. R. Creek and R. C. Peffer, Crit. Rev. Toxicol., 2015 DOI:10.3109/10408444.2015.1090948.
- J. E. Casida, Environ. Health Perspect., 2012, 120, 487–493 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed protocols of time-resolved fluorescence and extrinsic ANS displacement, site-specific ligand binding and CD spectra, principles of fluorescence quenching, evaluation of association ability, discussions of the ligand-binding domain and mutations of Phe-211 and Arg-222 residues, and images of time-resolved fluorescence decays, association constant plot, far-UV CD spectra, pictures of fluorescence quenching of albumin and ANS-albumin adduct as well as colored pictures of the mutations of Phe-211 and Arg-222 residues. See DOI: 10.1039/c5ra14661e |
| This journal is © The Royal Society of Chemistry 2016 |