
High efficiency of mechanosynthesized Zn-based metal–organic frameworks in photodegradation of congo red under UV and visible light†
Mohammad Yaser Masoomi‡
,
Minoo Bagheri‡ and
Ali Morsali*
Department of Chemistry, Faculty of Sciences, Tarbiat Modares University, Tehran, Islamic Republic of Iran. E-mail: morsali_a@modares.ac.ir; Tel: +98-21-82884416
First published on 22nd January 2016
Abstract
Three zinc(II) metal–organic frameworks, [Zn2(oba)2(4-bpdb)] (TMU-4), [Zn(oba)(4-bpdh)0.5]n (TMU-5) and [Zn(oba)(4-bpmb)0.5]n (TMU-6), (H2oba = 4,4′-oxybisbenzoic acid, 4-bpdb = 1,4-bis(4-pyridyl)-2,3-diaza-1,3-butadiene, 4-bpdh = 2,5-bis(4-pyridyl)-3,4-diaza-2,4-hexadiene and 4-bpmb = N1,N4-bis((pyridin-4-yl)methylene)benzene-1,4-diamine), containing azine-functionalized pores have successfully been synthesized via mechanosynthesis as a convenient, rapid, solventless and green process. These MOFs were studied for degradation of congo red from aqueous solutions under UV and/or visible light irradiation without auxiliary oxidants such as H2O2. TMU-6 could decolorize 100 ppm of congo red readily and quickly within 90 min, exhibiting about 68.4% in COD reduction after 72 h of UV irradiation. These results suggest that TMU-6/UV photocatalysis may be envisaged as a method for treatment of diluted colored waste waters not only for decolorization, but also for detoxification in textile industries.
Introduction
Currently, there is growing interest regarding a clean environment and human health issues, so technologies with high efficiency and low cost to reduce the pollutant contents of wastewater are urgently needed.1Organic synthetic dyes serving as pigments and chemical auxiliaries for industrial coloring purposes and discharge of wastes containing these toxic compounds into aquatic systems are considered as environmental threats among the researchers in this field.2,3 Among versatile physical, chemical, and biological technologies in pollution control, the advanced oxidation processes (AOPs), especially photocatalysis, have been gradually developed for the effective degradation of various environmental pollutants due to their high efficiency, simplicity, good reproducibility, and easy handling.4 Despite the traditional technologies, the photo-catalysis process can convert the contaminants into harmless substances directly in the wastewater.5 In a photocatalytic procedure, the adsorbed photons with suitable energy levels create hole–electron pairs in the valance band (VB) and conduction band (CB). Positive hole could either oxidize the adsorbed organic contaminants directly, or produce very reactive hydroxyl radicals (OH˙). The electron in the CB reduces the adsorbed oxygen on photocatalyst that result in degradation of the adsorbed contaminants.6,7
ZnO and TiO2 have been recognized to be preferable materials for photocatalytic processes due to their high photo-sensitivity, fantastic safety, low cost and chemical stability.8,9 However, their band gap energy limits the absorption ability of the high-energy portion (UV) of sunlight which together with their low surface area results in relatively small efficiency.
Metal–organic frameworks (MOFs) have emerged as an interesting class of multifunctional inorganic–organic hybrid porous crystalline materials, resulting from self-assembly of inorganic secondary building units (SBUs) and organic linkers,10–12 owing to their promising applications in gas storage, separation, ion exchange, sensor, catalysis, molecular recognition, and drug delivery.13–19
In the past few years, several studies of the semiconducting properties of MOFs have been reported. Recent studies have shown that MOFs can act as attractive semiconducting material with band gaps between 1.0 and 5.5 eV.20 Photoactive MOF systems have advantages in solar harnessing more efficiently in comparison with conventional semiconductor photocatalytic systems due to their structural features of tunable active sites (i.e., metal-oxo clusters and organic linkers), desirable porosity and high surface area.21,22 These features allow not only for rational design and fabrication of the photocatalytic system at the molecular level, but also for fast transportation and good accommodation of guest molecules.23 In other words, MOFs can serve as an ideal choice for light harvesting to achieve photocatalytic degradation of organic pollutions. In 2006, an MOF was used for the first time as an efficient photocatalyst for degradation of organic pollutants24,25 and other types of MOFs including MOF-5, isoreticular MOFs (IRMOF-1, IRMOF-2, IRMOF-7, IRMOF-8, IRMOF-9), MIL-53 (M = Al, Cr, Fe), UiO-66 and UiO-673 were subsequently reported with the assistance of a co-photocatalyst such as Ir, Re or Ru complexes, or under UV-light irradiation.26–29 In fact, desirable porosity, band gap and high surface area of Zn-based MOFs make them a better candidate for photocatalytic activity compared with their similar metal oxide photocatalysts.
In this study, the photocatalytic efficiency of mechanosynthesized Zn-based MOFs [Zn2(oba)2(4-bpdb)]n·2(DMF) (TMU-4), [Zn(oba)(4-bpdh)0.5]n·1.5(DMF) (TMU-5) and [Zn(oba)(4-bpmb)0.5]n·1.5(DMF) (TMU-6) for degradation of 100 ppm congo red under UV/vis light irradiation was investigated.
Experimental
Materials and physical techniques
All reagents for the synthesis and analysis were commercially available from Aldrich and Merck Company and used as received. The bulk ZnO was commercially available from Aldrich company with <5 μm particle size. The ligands 1,4-bis(4-pyridyl)-2,3-diaza-1,3-butadiene (4-bpdb), 2,5-bis(4-pyridyl)-3,4-diaza-2,4-hexadiene (4-bpdh) and N1,N4-bis((pyridin-4-yl)methylene)benzene-1,4-diamine (4-bpmb) were synthesized according to previously reported methods.30 Fig. S1† shows the chemical structures of these ligands.Melting points were measured on an Electrothermal 9100 apparatus. IR spectra were recorded using Thermo Nicolet IR 100 FT-IR. The thermal behavior was measured with a PL-STA 1500 apparatus with the rate of 10 °C min−1 in a static atmosphere of argon. X-Ray powder diffraction (XRD) measurements were performed using a Philips X'pert diffractometer with monochromated Cu-Kα radiation. Elemental analyses were collected on a CHNS Thermo Scientific Flash 2000 elemental analyzer. The samples were characterized with a field emission scanning electron microscope (FE-SEM) ZEISS SIGMA VP (Germany) with gold coating. The ultraviolet-visible diffuse reflectance spectra were recorded on an Avantes avaspec 2048tec reflection probe (fcr-7uv400) using BaSO4 as a reference.
Synthesis of TMU-4, TMU-5 and TMU-6
[Zn2(oba)2(4-bpdb)]n (TMU-4), [Zn(oba)(4-bpdh)0.5]n (TMU-5) and [Zn(oba)(4-bpmb)0.5]n (TMU-6) were mechanochemically synthesized according previously papers.31,32Evaluation of photocatalytic activity
The CR (C.I. Direct Red 28C32H24N6O6S2·2Na) was chosen as model pollutant to evaluate the photocatalytic activity of the MOFs. The irradiation experiments of CR were carried out on stirred aqueous solutions in a cylindrical quartz UV reactor, containing about 50 mL of a 100 ppm aqueous CR solution, in presence of about 25 mg of the MOF photocatalyst without auxiliary oxidant such as H2O2. The suspension was sonicated for 5 min and subsequently stirred in the dark for 35–50 min (depending on the type of samples and their darkness time found based on the absorption experiments), to establish the adsorption/desorption equilibrium on the MOF surface before UV and/or visible light irradiation (Fig. S2, ESI†). The irradiation was done with a UV lamp (30 W, UV-C, λ = 253.7 nm, 4.89 eV, Philips (The Netherlands)) and/or a 500 W lamp (high-pressure mercury-vapor lamp, Yaming Company, Shanghai). The temperatures of the samples did not exceed 20 °C, owing to the circulation of tap water in the jacket of the UV reactor. Perpendicular UV irradiation was applied when the distance between the UV source and the reaction mixture was almost 15 cm. Air was blown into the reaction mixture by an air compressor, to maintain the oxygen saturation in the solution during the course of reaction. Analytical samples were taken from the reaction suspension after specified reaction times, immediately centrifuged at 6000 rpm for 10 min to remove the particles, and further analyzed through monitoring their absorbance at 497 nm using an ultraviolet-visible (UV-Vis) spectrophotometer (Shimadzu UV 2100). The concentration of dye in each degraded sample was determined at λmax = 497 nm, using the calibration curve. With this method, the percent removal of CR could be attained at different intervals. The percent removal was calculated by the following equation:| % removal = (Ci − Ct)/Ci × 100 | (1) |
The chemical oxygen demand (COD) test is extensively employed as an effective technique to measure the organic strength of wastewater. This test allows measurement of waste in terms of the total quantity of oxygen required for oxidation of organic matter to CO2 and water. In the present work, the open reflux method was used for COD determination.33 The photocatalyst stability tests were carried out in the same way as the photocatalytic activity tests, but repeated four times.
Results and discussion
TMU-4, TMU-5 and TMU-6 were synthesized by mechanochemical reaction of a mixture of Zn(OAc)2·2H2O, H2oba and N-donor ligand (4-bpdb, 4-bpdh or 4-bpmb, respectively) for approximately 15 min. Fig. S3† shows the morphologies of mechanosynthesized MOFs. Comparison between the simulated and experimental (resulting from the mechanosynthesized powder) powder X-ray diffraction (PXRD) patterns revealed that the mechanosynthesized TMU-4, TMU-5 and TMU-6 were structurally identical to TMU-4, TMU-5 and TMU-6 prepared via conventional heating.31,32The structure of [Zn2(oba)2(4-bpdb)]n·(DMF)x (TMU-4) consists of three-dimensional honeycomb framework with double interpenetration which is obtained by coordination of the oba and 4-bpdb ligands to Zn(II) nods (Fig. 1a). Although the double interpenetration is formed to avoid an extremely large void space, but still TMU-4 has one-dimensional (1D) open channels (aperture size: 5.3 × 9 Å, taking into account the van der Waals radii; 40% void space per unit cell)34 running along [101] direction (Fig. S4a, ESI†). MOF [Zn(oba)(4-bpdh)0.5]n·(DMF)y (TMU-5) is built up from a Zn2(oba)4 paddle-wheel secondary building unit (SBU) which shows 3D, interconnected, narrow pores31 (aperture size: 4.4 × 6.2 Å, taking into account the van der Waals radii; 34.6% void space per unit cell)34 (Fig. 1b and S4b, ESI†).
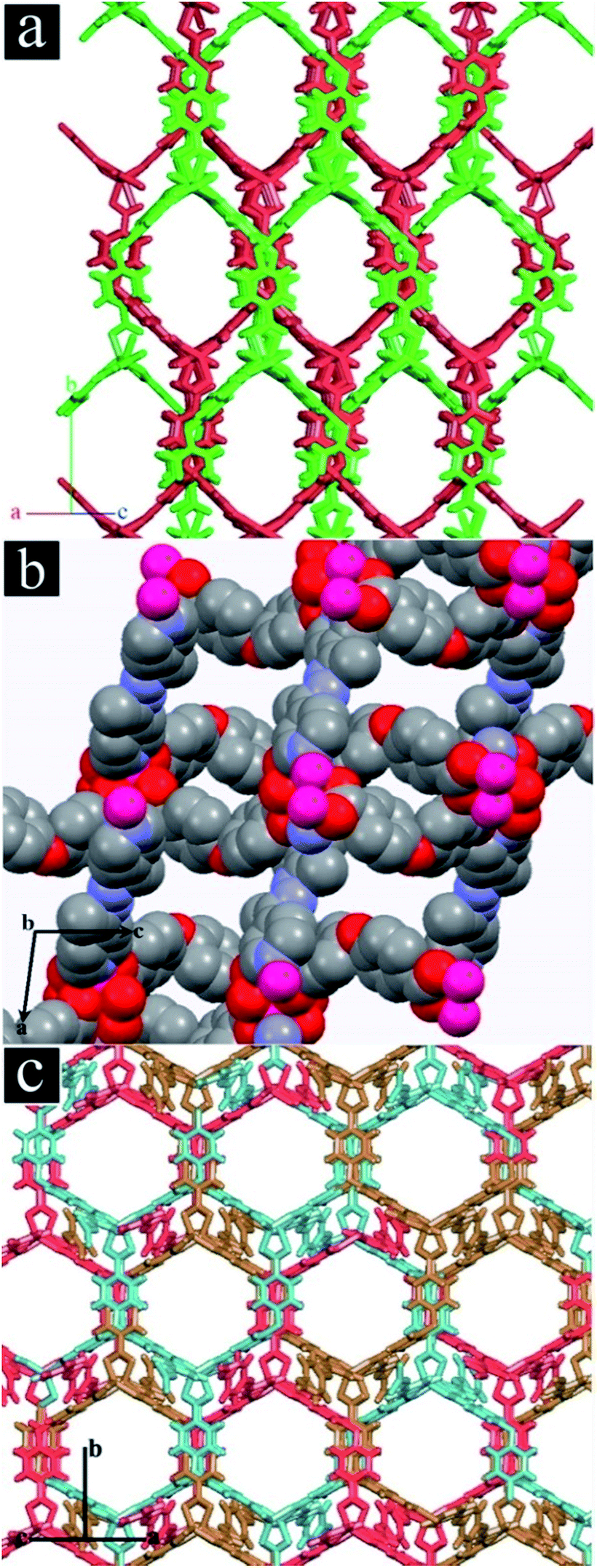 | ||
| Fig. 1 (a) Representation showing the pore channels and that the network is doubly interpenetrated (in red and green) in TMU-4. (b) Representation of the network and pores in TMU-5. (c) Representation showing the pore channels and that the network is threefold interpenetrated (in red, blue and brown). Hydrogen atoms and DMF molecules are omitted for clarity. | ||
By introducing phenyl ring in the pillar ligand, 4-bpdb, higher conjugated length N-donor ligand, that is 4-bpmb, has been obtained which leads to formation of [Zn(oba)(4-bpmb)0.5]n·(DMF)z (TMU-6) with a structure similar to TMU-4 but different from TMU-5. TMU-6 is also based on a binuclear Zn(II) unit in which linear 4-bpmb connects 2D sheets formed by dicarboxylate oba ligand and Zn(II) centres and extends the structure in three dimensions with threefold interpenetration (Fig. 1c). TMU-6 shows 1D, large pores (aperture size: 9.1 × 8.9 Å, taking into account the van der Waals radii; 34.2% void space per unit cell)34 running along the [101] direction (Fig. S4c, ESI†).32
The TGA curves of mechanosynthesized TMU-4, TMU-5 and TMU-6 show a plateau in the range of 30 to 310 °C, 30 to 290 °C and 30 to 380 °C respectively, revealing that their pore channels were devoid of any guest molecules (Fig. S5, ESI†). Above these points these MOFs begin to decompose.
The synthesized TMU-4, TMU-5 and TMU-6 were soaked in water for 24 h to see whether they are stable in water. The XRD patterns indicate that the structures are stabile in water (Fig. S6, ESI†).
The diffuse reflectance UV-vis spectroscopy of the MOFs was recorded. The onset of the main absorption edges of TMU-4, TMU-5 and TMU-6 are located at 488, 585 and 569 nm, respectively (Fig. S7, ESI†). Based on the relation Eg = 1240/λ,35 the calculated optical band gap of the MOFs are listed in Table 1. This is due to the absorption of light caused by the excitation of electrons from the VB to the CB of MOFs.20,35 As expected, the prominent optical properties in UV-vis region of the MOFs could contribute to an efficient photoactivity.
| MOF | Tdark (min) | % adsorption | Visible | UV | Band gap (eV) | Void space (%) | BET | ||
|---|---|---|---|---|---|---|---|---|---|
| Tremoval (min) | % removala | Tremoval (min) | % removala | ||||||
| a % removal = the % removal at which the photocatalyst shows a maximum removal after adsorption. | |||||||||
| TMU-4 | 35 | 37.5 | 120 | 33 | 120 | 93.8 | 2.6 | 40 | 518 (ref. 31) |
| TMU-5 | 35 | 33.3 | 120 | 28 | 120 | 87.8 | 2.1 | 34 | 503 (ref. 31) |
| TMU-6 | 35 | 39.4 | 120 | 48.4 | 90 | 98.3 | 2.2 | 34.2 | 650 (ref. 42) |
| Bulk ZnO | 30 | 1.7 | 300 | 0.98 | 180 | 5 | 3.37 (ref. 43) | — | — |
| Degussa P25 | 30 | 3.5 | 300 | 1.0 | 180 | 11 | 3.0–3.2 (ref. 44) | — | — |
The removal of CR was used to evaluate the photocatalytic performance of three MOFs, where control experiments were conducted to compare the removal efficiencies of CR by various MOFs at natural pH (ca. 5.5) with initial CR concentration (100 ppm) in presence 0.5 mg L−1 of photocatalyst under UV light irradiation (Fig. 2a and S8–S10a, ESI†).
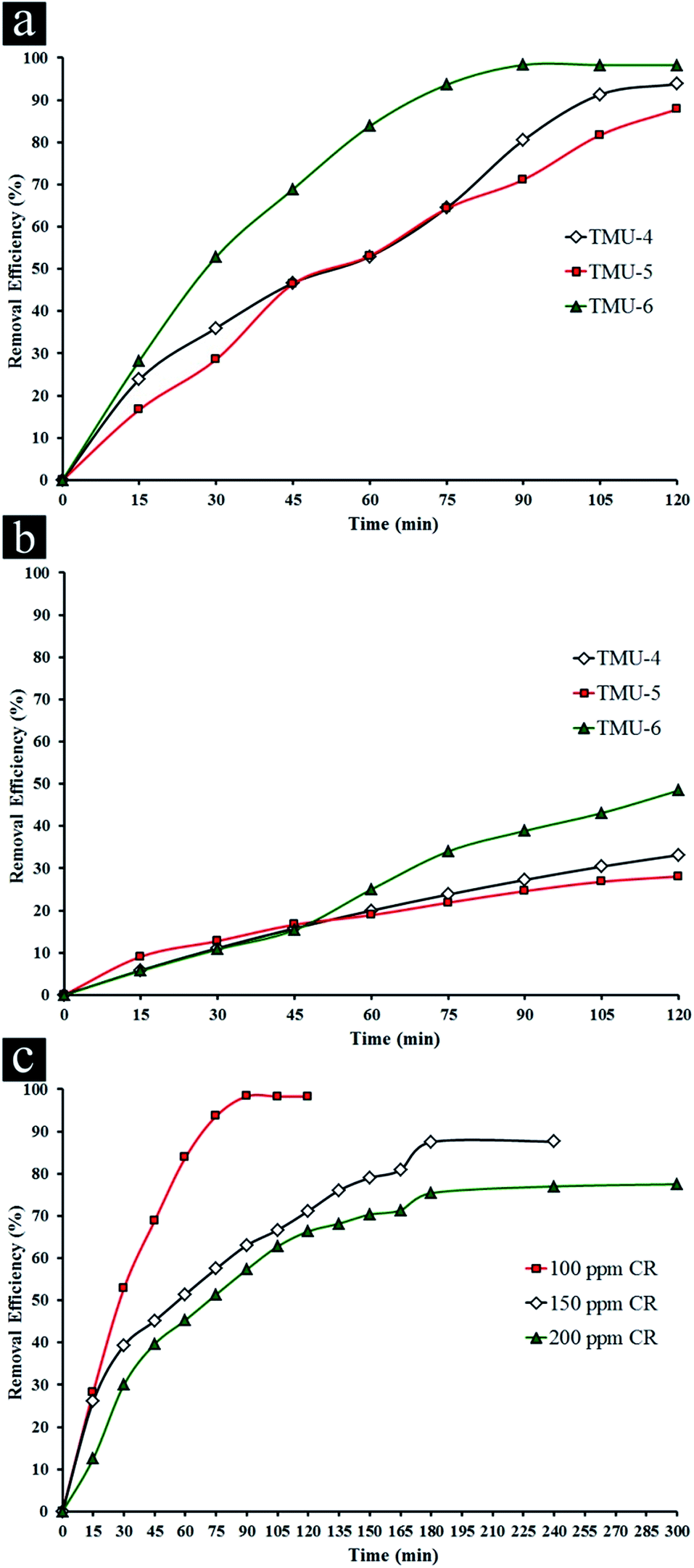 | ||
| Fig. 2 Removal efficiency of three MOFs in presence of 100 ppm CR dye under (a) UV light irradiation and (b) visible light irradiation. (c) Removal efficiency of TMU-6 in presence of various concentrations of CR. | ||
The experiments related to blank (without catalyst in presence of UV light) showed no change in the concentration of CR whereas the concentration of CR decreased obviously with exposure time in presence of three MOFs, indicating the photocatalytic removal of the organic dye was attributed to the photolysis by these MOFs. It was noticed that under visible light (589 nm, cut-off filter), these MOFs had little activity under similar conditions. For this reason, in the experiments, the activity of catalysts was evaluated under UV light. For the TMU-4 and TMU-5, the maximum percent removal of CR is roughly 93.8 and 87.8% under UV light irradiation within 120 min. On the other hand, there is a significant change in the CR concentration in the solution with the TMU-6 after 30 min. Also, the maximum efficiency in presence of TMU-6 is 98.3% after 90 min and remains constant afterwards. Therefore, TMU-6 shows the highest removal efficiency among other MOFs. Meanwhile, removal efficiency of bulk ZnO and Degussa P25 in presence of 100 ppm CR shows negligible changes in initial concentration of dye under UV and visible light (Table 1).
As reported in the literature, MIL-100(Fe) showed the photocatalytic performance of 99% after 40 min of UV light irradiation to remove 40 ppm of methylene blue in presence of H2O2.7 Also, UTSA-38 indicated the photocatalytic performance of 65% in presence of 20 ppm methyl orange aqueous solution after 120 min of UV irradiation.26
The photocatalytic performance could be ascribed to high surface area, value of band gap and differences in the rate of recombination, crystallinity of a photocatalyst, morphology, low degree of agglomeration, defects and adsorptive affinity.36–38 The general mechanism involved in presence of the three MOFs for degradation of CR could be explained by considering the HOMO, filled d-orbital (d10 in Zn2+), and the lowest LUMO, free s-orbital as well as LUMOs of the organic linkers.39
Upon light irradiation, electrons on the VB composed of O, C and N 2p orbitals jump to the CB, and produces holes (h+) in the VB. Positive hole could either oxidize the adsorbed organic contaminants directly, or produce very reactive hydroxyl radicals (OH˙). The electron in the CB reduces the adsorbed oxygen on photocatalyst (Scheme 1). The formed superoxide radicals further react with pollutant, which leads to the final products.5
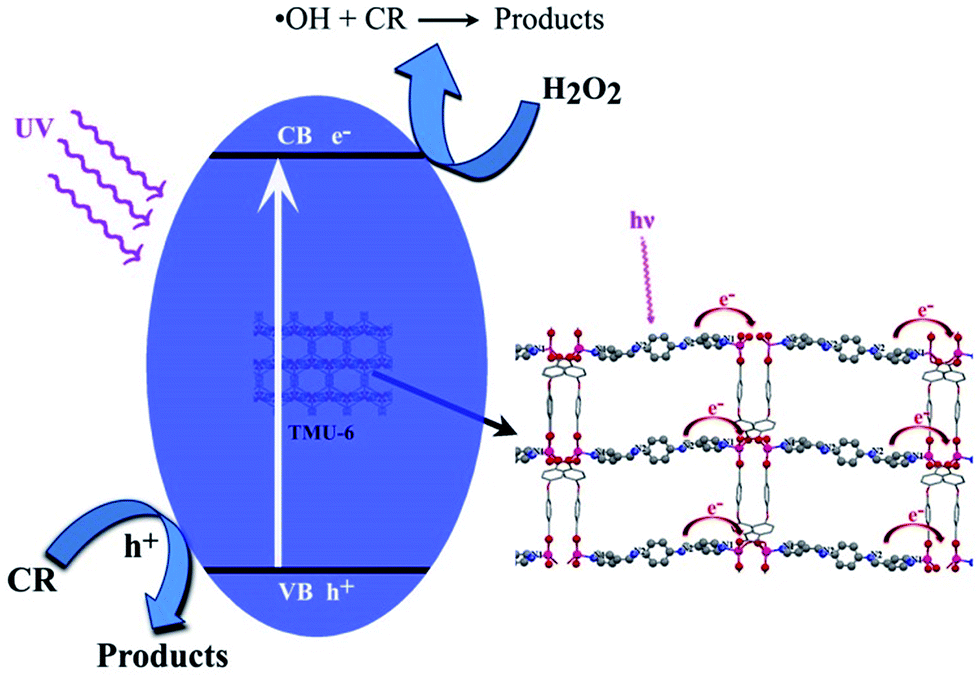 | ||
| Scheme 1 A schematic illustration of CR degradation over TMU-6 photocatalyst under UV irradiation. | ||
In this study, according to DRS UV-Vis results, probably the proper band gap value in TMU-6, decreases the recombination rate of electron–hole pair. The use of different N-donor ligands has led to formation of frameworks with different topologies and electron donating ability to metal centers and therefore different photocatalytic activities. Higher conjugated length of N-donor ligand in TMU-6 causes the HOMO and LUMO to move closer. As a result, electron transfer to the metal center is easier.40 Moreover, higher surface area in TMU-6 leads to more dye adsorption and thus causes an increase in photocatalytic performance.
The influence of initial concentration of the dye solution on the photocatalytic degradation is a significant aspect of the study. The effect of different concentrations of CR was investigated in presence of TMU-6 under UV light irradiation (Fig. 2c and S10, ESI†). The experiments related to blank (without MOF in presence of light) showed negligible change in the different concentrations of CR (Table S1†). For 50 ppm CR only adsorption takes place. Upon increasing in CR concentration, the maximum percentage removal of dye decreases with an increase in time irradiation. One possible explanation of such circumstances is that as initial concentration increases, more and more organic substances are adsorbed on the surface of sample. Scarcity of active sites in the system causes little adsorption of hydroxyl ions which in turn leads to a decrease in generation of hydroxyl radicals. Further, as the concentration of dye increases, the photons get intercepted before they can reach the catalyst surface, thus the photon adsorption by the catalyst decreases. Consequently, percentage removal is reduced.41
The photocatalytic efficiency of TMU-4, TMU-5 and TMU-6 MOFs in presence of 100 ppm CR under visible light irradiation for 2 h was investigated. TMU-6 has the best efficiency among the two other samples at about 48.2% for 2 h (Fig. S11† and Table 1). As mentioned above, the highest percentage removals of 98.3% and 48.2% are observed for TMU-6 photocatalyst after 90 min under UV and 120 min under visible irradiations, respectively.
Kinetic of the total mineralization of the dye has been followed using COD technique, the disappearance of the chemical oxygen demand, for the best photocatalyst (TMU-6) being complementary for expressing the detoxification level of water (Fig. 3a).
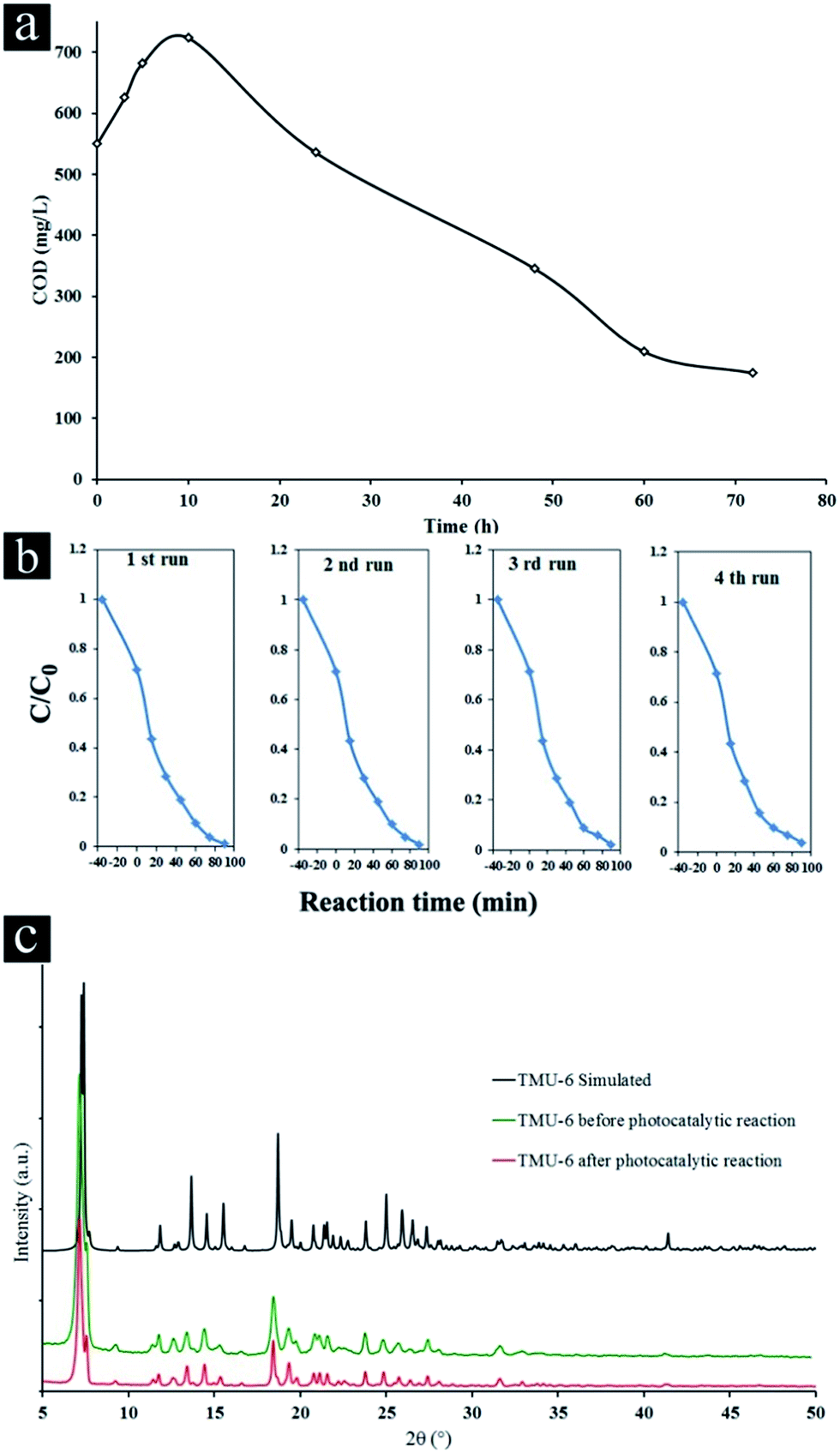 | ||
| Fig. 3 (a) Change of COD removal efficiency of TMU-6 photocatalyst in presence of 100 ppm CR. (b) Cycling run in the removal of CR in presence of TMU-6 under UV light irradiation and (c) XRD pattern of TMU-6 before and after photocatalytic test. | ||
The COD value of the initial color solution initially increases due to cleavage of rings in dye molecule and then significantly decreases after 72 h (68.4% of the initial CR), indicating the high potential of TMU-6 photodegradation process for the removal of CR from wastewater.
Additionally, TMU-6 was very stable during four repeated experiments (Fig. 3b). The XRD patterns of the TMU-6 photocatalyst before and after repeating the reaction for four cycles obviously proves that the structure of photocatalyst remains intact and no adsorption takes place on its surface (Fig. 3c and S12, ESI†). However, the diffraction intensity is reduced due to a decrease in the crystallinity of the sample. A similar observation has been reported by others.45,46
Conclusions
Photocatalytic activity of three mechanosynthesized zinc(II) metal–organic frameworks, [Zn2(oba)2(4-bpdb)]·(DMF)x (TMU-4), [Zn(oba)(4-bpdh)0.5]n·(DMF)y (TMU-5) and [Zn(oba)(4-bpmb)0.5]n·(DMF)z (TMU-6) in presence of 100 ppm congo red was investigated. The photocatalysts were successful not only in decolorization, but also in degradation and mineralization without auxiliary oxidants such as H2O2. The organic part was converted into mineral compounds as testified by the elimination of COD. Photocatalytic performance depended on band gap value of MOFs and electron donating ability of N-donor ligands to metal centres. TMU-6 photocatalyst showed maximum removal of congo red compared with two other MOFs at shorter UV irradiation. Our findings may provide some insight into the preparation of photocatalysts with superior performance in practical applications.Acknowledgements
Support of this investigation by Tarbiat Modares University and Iran National Science Foundation are gratefully acknowledged.Notes and references
- K. Melghit, M. S. Al-Rubaei and I. Al-Amri, J. Photochem. Photobiol., A, 2006, 181, 137–141 CrossRef CAS.
- H. Lachheb, E. Puzenat, A. Houas, M. Ksibi, E. Elaloui, C. Guillard and J.-M. Herrmann, Appl. Catal., B, 2002, 39, 75–90 CrossRef CAS.
- V. A. Sakkas, M. A. Islam, C. Stalikas and T. A. Albanis, J. Hazard. Mater., 2010, 175, 33–44 CrossRef CAS PubMed.
- W. Liu, Z. Ai and L. Zhang, J. Hazard. Mater., 2012, 243, 257–264 CrossRef CAS PubMed.
- G. Crini, Bioresour. Technol., 2006, 97, 1061–1085 CrossRef CAS PubMed.
- B. Palanisamy, C. M. Babu, B. Sundaravel, S. Anandan and V. Murugesan, J. Hazard. Mater., 2013, 252–253, 233–242 CrossRef CAS PubMed.
- C.-F. Zhang, L.-G. Qiu, F. Ke, Y.-J. Zhu, Y.-P. Yuan, G.-S. Xu and X. Jiang, J. Mater. Chem. A, 2013, 1, 14329–14334 CAS.
- M. L. Marin, L. Santos-Juanes, A. Arques, A. M. Amat and M. A. Miranda, Chem. Rev., 2012, 112, 1710–1750 CrossRef CAS PubMed.
- X. Bai, L. Wang, R. Zong, Y. Lv, Y. Sun and Y. Zhu, Langmuir, 2013, 29, 3097–3105 CrossRef CAS PubMed.
- S. Hasegawa, S. Horike, R. Matsuda, S. Furukawa, K. Mochizuki, Y. Kinoshita and S. Kitagawa, J. Am. Chem. Soc., 2007, 129, 2607–2614 CrossRef CAS PubMed.
- M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319–330 CrossRef CAS PubMed.
- N. Stock and S. Biswas, Chem. Rev., 2011, 112, 933–969 CrossRef PubMed.
- O. M. Yaghi, H. Li, C. Davis, D. Richardson and T. L. Groy, Acc. Chem. Res., 1998, 31, 474–484 CrossRef CAS.
- A. Corma, H. García and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed.
- X. Zhao, B. Xiao, A. J. Fletcher, K. M. Thomas, D. Bradshaw and M. J. Rosseinsky, Science, 2004, 306, 1012–1015 CrossRef CAS PubMed.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- K. A. Cychosz, R. Ahmad and A. J. Matzger, Chem. Sci., 2010, 1, 293–302 RSC.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2011, 112, 1105–1125 CrossRef PubMed.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2011, 112, 1232–1268 CrossRef PubMed.
- A. Kuc, A. Enyashin and G. Seifert, J. Phys. Chem. B, 2007, 111, 8179–8186 CrossRef CAS PubMed.
- T. Toyao, M. Saito, Y. Horiuchi, K. Mochizuki, M. Iwata, H. Higashimura and M. Matsuoka, Catal. Sci. Technol., 2013, 3, 2092–2097 CAS.
- C.-B. Liu, Y. Cong, H.-Y. Sun and G.-B. Che, Inorg. Chem. Commun., 2014, 47, 71–74 CrossRef CAS.
- H. Yang, X.-W. He, F. Wang, Y. Kang and J. Zhang, J. Mater. Chem., 2012, 22, 21849–21851 RSC.
- P. Mahata, G. Madras and S. Natarajan, J. Phys. Chem. B, 2006, 110, 13759–13768 CrossRef CAS PubMed.
- F. X. Llabrés i Xamena, A. Corma and H. Garcia, J. Phys. Chem. C, 2006, 111, 80–85 CrossRef.
- M. C. Das, H. Xu, Z. Wang, G. Srinivas, W. Zhou, Y.-F. Yue, V. N. Nesterov, G. Qian and B. Chen, Chem. Commun., 2011, 47, 11715–11717 RSC.
- C. Wang, Z. Xie, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 13445–13454 CrossRef CAS PubMed.
- M. Bagheri, A. R. Mahjoub and B. Mehri, RSC Adv., 2014, 4, 21757–21764 RSC.
- J. R. Choi, T. Tachikawa, M. Fujitsuka and T. Majima, Langmuir, 2010, 26, 10437–10443 CrossRef CAS PubMed.
- D. M. Ciurtin, Y.-B. Dong, M. D. Smith, T. Barclay and H.-C. zur Loye, Inorg. Chem., 2001, 40, 2825–2834 CrossRef CAS PubMed.
- M. Y. Masoomi, K. C. Stylianou, A. Morsali, P. Retailleau and D. Maspoch, Cryst. Growth Des., 2014, 14, 2092–2096 CAS.
- M. Y. Masoomi, S. Beheshti and A. Morsali, J. Mater. Chem. A, 2014, 2, 16863–16866 CAS.
- Open Reflux Method, American Public Health Association, 1997, vol. 5220, p. 12 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- J. Long, S. Wang, Z. Ding, S. Wang, Y. Zhou, L. Huang and X. Wang, Chem. Commun., 2012, 48, 11656–11658 RSC.
- Y. Hou, L. Wu, X. Wang, Z. Ding, Z. Li and X. Fu, J. Catal., 2007, 250, 12–18 CrossRef CAS.
- Q. Zhang, Y. Li, E. A. Ackerman, M. Gajdardziska-Josifovska and H. Li, Appl. Catal., A, 2011, 400, 195–202 CrossRef CAS.
- H. Li, S. Yin, Y. Wang and T. Sato, Appl. Catal., B, 2013, 132–133, 487–492 CrossRef CAS.
- J. Gascon, M. D. Hernández-Alonso, A. R. Almeida, G. P. M. van Klink, F. Kapteijn and G. Mul, ChemSusChem, 2008, 1, 981–983 CrossRef CAS PubMed.
- F. A. Carey and R. J. Sundberg, Advanced Organic Chemistry, Springer, US, New York, 2007 Search PubMed.
- S. Erdemoğlu, S. K. Aksu, F. Sayılkan, B. İzgi, M. Asiltürk, H. Sayılkan, F. Frimmel and Ş. Güçer, J. Hazard. Mater., 2008, 155, 469–476 CrossRef PubMed.
- E. Tahmasebi, M. Y. Masoomi, Y. Yamini and A. Morsali, Inorg. Chem., 2015, 54, 425–433 CrossRef CAS PubMed.
- P. X. Gao, Y. Ding and Z. L. Wang, Nano Lett., 2003, 3, 1315–1320 CrossRef CAS.
- D. C. Hurum, A. G. Agrios, K. A. Gray, T. Rajh and M. C. Thurnauer, J. Phys. Chem. B, 2003, 107, 4545–4549 CrossRef CAS.
- J. Zhang, A. V. Biradar, S. Pramanik, T. J. Emge, T. Asefa and J. Li, Chem. Commun., 2012, 48, 6541–6543 RSC.
- J. Park, J.-R. Li, Y.-P. Chen, J. Yu, A. A. Yakovenko, Z. U. Wang, L.-B. Sun, P. B. Balbuena and H.-C. Zhou, Chem. Commun., 2012, 48, 9995–9997 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Other figures, FE-SEM images, TGA, PXRD, DRS, absorption spectra and IR spectra. See DOI: 10.1039/c5ra24238j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |