
Metal-free amidation of carboxylic acids with tertiary amines†
Wong Phakhodee*,
Sirilak Wangngae and
Mookda Pattarawarapan
Department of Chemistry and Center of Excellence for Innovation in Chemistry, Faculty of Science, Chiang Mai University, Chiang Mai 50200, Thailand. E-mail: wongp2577@gmail.com; Fax: +66 53892277; Tel: +66 53943341
First published on 17th June 2016
Abstract
A direct amidation of carboxylic acids with tertiary amines could be carried out in the presence of the Ph3P–I2 activator. With an appropriate reagent addition sequence, a range of carboxylic acids including aliphatic, allylic, and aromatic acids could be converted into their corresponding tertiary amides under mild conditions without requirement of metal catalysis.
Amide C–N bond formation through nucleophilic acyl substitutions constitutes one of the most fundamental functional group transformations in organic synthesis.1–3 The classical methods involve activation of carboxylic acids to generate reactive intermediates such as acyl halides, active esters or anhydrides prior to subsequent reaction with reactive amine sources such as primary or secondary amines. Since tertiary amines are considerably less nucleophilic, amidation with these amines is a challenging task generally gives low yields, requires metal catalysis, long reaction times, and/or harsh conditions.
In the classic Polonovski reaction, N,N-disubstituted acetamides were prepared by converting tertiary amines into amine N-oxides before subjecting to thermal cleavage in the presence of acetic acid anhydride.4 Alternatively, anhydrides,5 acyl chloride,6–8 and acyl iodide9,10 have been reported to undergo amide bond formation with tertiary amines through acylation, followed by N-dealkylation of the generated quaternary ammonium salts. Other recently developed methods involve metal-catalyzed oxidative cleavage of the N–C bond of the tertiary amines to provide secondary amines before reaction with acylating agents.11–19 To the best of our knowledge, a direct amidation of carboxylic acids with tertiary amines has never been achieved in the presence of any existing coupling reagents.
According to our previous study, during the synthesis of acid anhydrides promoted by the Ph3P–I2/Et3N system,20 we have observed an unexpected formation of N,N-diethylamides derived from the amidation of some carboxylic acids with triethylamine. While anhydride formation was rapid for benzoic acid and other electron-rich derivatives such as those containing –Me, –OMe group, the electron-deficient nitro-containing substrates gave rise to exclusive formation of N,N-diethylamides without detectable anhydride formation.
This observation prompts us to further investigate the amide bond formation reaction using triethylamine as well as other tertiary amines as nitrogen sources. The reagent addition sequence was carefully reexamined, while various synthetic parameters were evaluated.
To establish the optimum conditions, amidation between 2-chloro-5-nitrobenzoic acid and triethylamine was chosen as a model reaction. Various set of reaction conditions were screened as summarized in Table 1.
|
||||
|---|---|---|---|---|
| Entry | Additive | Et3N (equiv.) | Base (equiv.) | Yield (%) |
| a Reaction conditions: 2-chloro-5-nitrobenzoic acid (0.41 mmol), Ph3P (0.62 mmol), additive (0.62 mmol), CH2Cl2 (2 mL), 0 °C-RT, 30 min. | ||||
| 1 | I2 | 3 | — | 95 |
| 2 | I2 | 1.2 | K2CO3 (2) | 38 |
| 3 | I2 | 1.2 | iPr2NEt (2) | 63 |
| 4 | I2 | 1.2 | DMAP (2) | Trace |
| 5 | I2 | 1.2 | Imidazole (2) | Trace |
| 6 | Br2 | 3 | — | Trace |
| 7 | CCl4 | 3 | — | 21 |
| 8 | NBS | 3 | — | 23 |
| 9 | NCS | 3 | — | 0 |
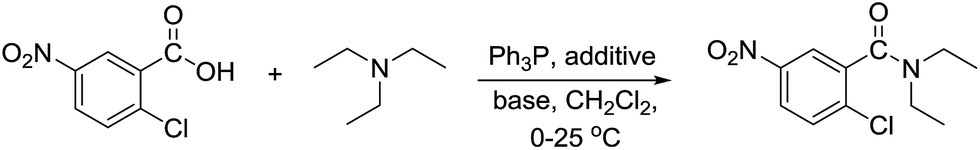
Under our previously reported conditions,20 when a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of iodine and Ph3P (1.5 equiv.) in dichloromethane was added with 2-chloro-5-nitrobenzoic acid (1 equiv.) at 0 °C, followed by treatment with Et3N (3 equiv.), the corresponding diethylamide product was obtained in 75% after 3 h stirring at 25 °C. To our delight, by switching the order of the reagent addition between the carboxylic acid and Et3N, the yield of the respective amide and the reaction rate was significantly improved (entry 1). Following this new reagent addition sequence, attempts to reduce the amount of Et3N to 1.2 equiv. by performing the reaction in the presence of 2 equiv. of other inorganic or organic bases failed to give satisfactory results. While the reaction in the presence of K2CO3 or Hünig's base gave the product in low to moderate yields (entries 2 and 3), no amide could be detected in the reaction using DMAP or imidazole (entries 4 and 5). Interestingly, the corresponding N-acylimidazole (18%) was isolated from the reaction using imidazole as base (entry 5) suggesting that imidazole and possibly DMAP could react with the formed phosphonium iodide species leading to the formation of reactive intermediate that rapidly decompose under the applied conditions. The yields of the reaction also decreased dramatically when replacing iodine with other halogenated additives indicating that the reaction is more effective under the Ph3P–I2 system (entries 6–9).
:1 mixture of iodine and Ph3P (1.5 equiv.) in dichloromethane was added with 2-chloro-5-nitrobenzoic acid (1 equiv.) at 0 °C, followed by treatment with Et3N (3 equiv.), the corresponding diethylamide product was obtained in 75% after 3 h stirring at 25 °C. To our delight, by switching the order of the reagent addition between the carboxylic acid and Et3N, the yield of the respective amide and the reaction rate was significantly improved (entry 1). Following this new reagent addition sequence, attempts to reduce the amount of Et3N to 1.2 equiv. by performing the reaction in the presence of 2 equiv. of other inorganic or organic bases failed to give satisfactory results. While the reaction in the presence of K2CO3 or Hünig's base gave the product in low to moderate yields (entries 2 and 3), no amide could be detected in the reaction using DMAP or imidazole (entries 4 and 5). Interestingly, the corresponding N-acylimidazole (18%) was isolated from the reaction using imidazole as base (entry 5) suggesting that imidazole and possibly DMAP could react with the formed phosphonium iodide species leading to the formation of reactive intermediate that rapidly decompose under the applied conditions. The yields of the reaction also decreased dramatically when replacing iodine with other halogenated additives indicating that the reaction is more effective under the Ph3P–I2 system (entries 6–9).
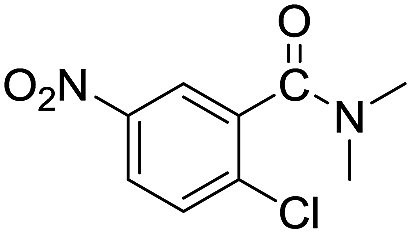
With the optimum conditions in hand (Table 1, entry 1), the scope of the amidation with a variety of tertiary amines was investigated using 2-chloro-5-nitrobenzoic acid as a substrate. According to Table 2, the starting acid was found to react smoothly with most of the applied amines to provide the N,N-disubstituted amides in moderate to excellent yields. The reaction proceeded readily with aliphatic amines, whereas amidation using aromatic amine was sluggish.
|
|||
|---|---|---|---|
| Entry | NR1R2R3 | Product | Yield (%) |
| a The reaction was carried out by adding tertiary amine (1.23 mmol) into a mixture of I2 (0.62 mmol) and Ph3P (0.62 mmol) in CH2Cl2 (2 mL) at 0 °C. After stirring for 5–10 min, 2-chloro-5-nitrobenzoic acid (0.41 mmol) was added. The mixture was further stirred at room temperature until completion of the reaction (0.5–2 h).b 1,2-Dichloroethane was used as the solvent and the reaction was carried out at 80 °C for 16 h. | |||
| 1 | 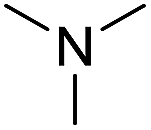 |
 |
92 |
| 2 | 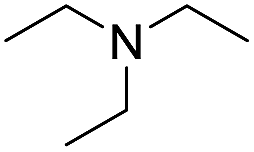 |
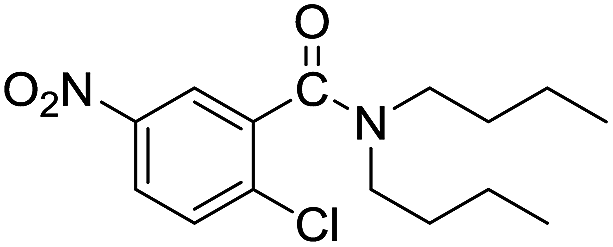
 |
95 |
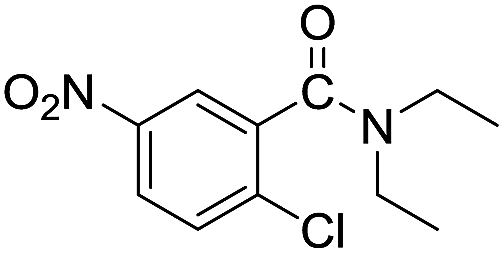
| 3 | 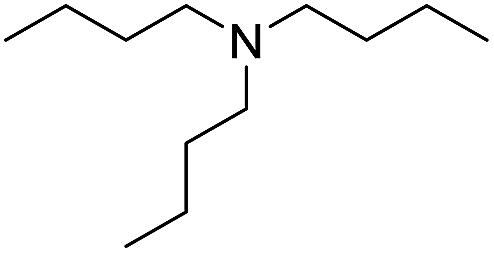 |
 |
52 |
| 4 | 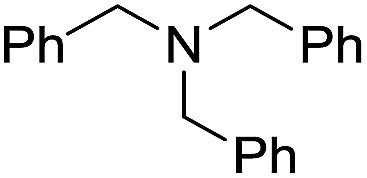 |
 |
94 |
| 5 | 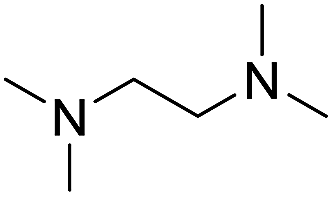 |
 |
46 |
 |
24 | ||
| 6b | 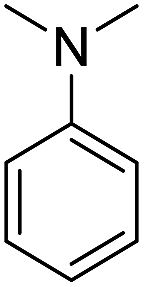 |
 |
25 |
| 7 | 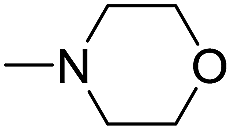 |
 |
73 |
| 8 |  |
 |
96 |
| 9 | 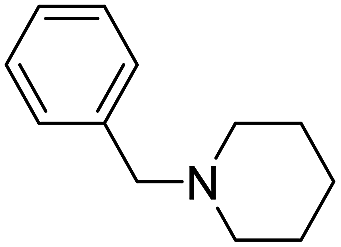 |
 |
75 |
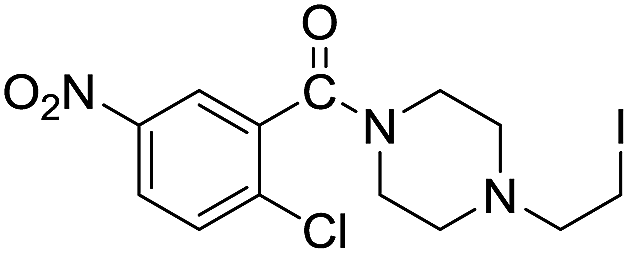
| 10 | 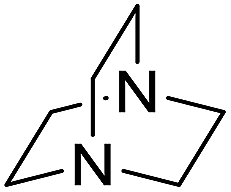 |
 |
52 |
When using symmetrical trialkyl acylic amines, the reaction rates and yields were found to depend on the alkyl chain lengths and steric effect. While the reaction with trimethylamine gave comparable yields with that using triethylamine, amidation using tributylamine was significantly less effective (entries 1–3). Amidation with tribenzylamine, however, proceeded without difficulty to provide the desired amide in high yield (entry 4).
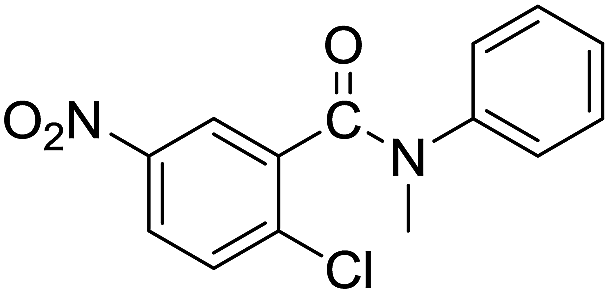
In the reaction with unsymmetrical amine such as N,N,N′,N′-tetramethylethylenediamine, all the amide products derived from two possible N-dealkylations could be observed (entry 5). Nevertheless, the reaction with the less nucleophilic N,N-dimethylaniline did not proceed at room temperature. Prolonged heating of the reaction mixture in 1,2-dichloroethane (80 °C, 16 h) only gave the corresponding N-methyl-N-phenylamide in low yield without detectable product from N-dephenylation (entry 6). In the case of the non-nucleophilic N,N-diisopropylethyamine, no product formation was observed even under more forcing conditions.
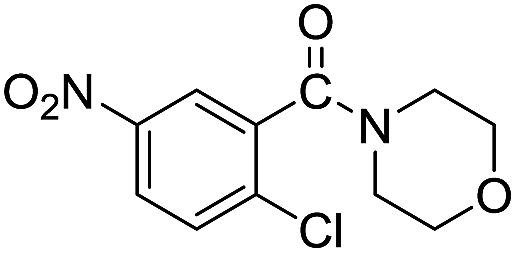
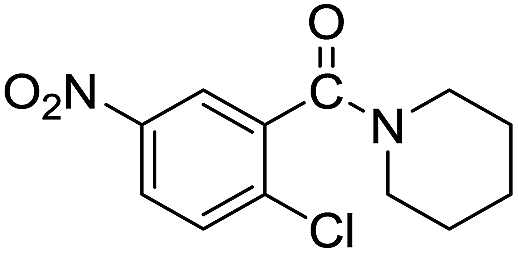
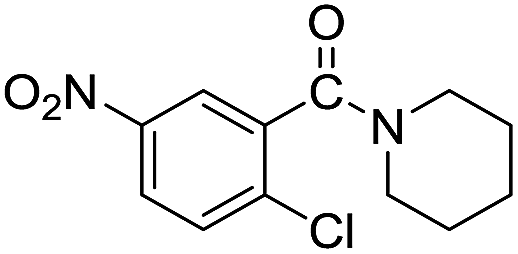
The reaction using cyclic aliphatic amines generally proceeded without cleavage of the endocyclic C–N bond. Both N-methylmorpholine and N-methylpiperidine underwent N-demethylation to provide the respective amide in good to excellent yields (entries 7 and 8). Likewise, the reaction with N-benzylpiperidine proceeded exclusively via N-debenzylation (entry 9). Interestingly, when using bicyclic tertiary amine as a nitrogen source, 1,4-diazabicyclo[2.2.2]octane (DABCO) underwent endocyclic C–N bond cleavage to provide iodo-substituted amide product in moderate yield. This data indicated that N-dealkylation occurred via elimination of alkyl iodide (entry 10). Remarkably, although 2-chloro-5-nitrobenzoic acid is prone toward SNAr displacement,21 no product from the substitution of the chlorine atom by the nitrogen nucleophile was observed under the applied conditions. Such selectivity is ideal for the synthesis of complex molecular structures obviated the need for multistep reactions.
To further demonstrate the synthetic utility of the method, other carboxylic acids including aliphatic, allylic, and aromatic acids were screened in the reaction with tribenzylamine. As shown in Scheme 1, most of the tested carboxylic acids could be converted into the corresponding N,N-dibenzylamides in satisfactory yields. While aliphatic acids required longer times for completion of the reaction (see compounds 1–5), amidation of crotonic acid and cinnamic acid proceeded more rapidly to afford the products 6 and 7 in high yields. When using 2-iodobenzoic acid as the substrate, apart from the formation of 8 in relatively low yield, the corresponding anhydride was also obtained as a side product in 38% yield. On the contrary, the reaction with 2-nitrobenzoic acid provided the amide 9 in high yield without detectable anhydride formation. Other benzoic acid derivatives containing strong electron-withdrawing nitro group were also favorable substrates in comparing with the more electron-rich 4-chlorobenzoic acid (see compounds 10–13). Heterocylic acid such as 2-chloronicotinic acid also underwent amidation giving the respective amide 14 in high yield.
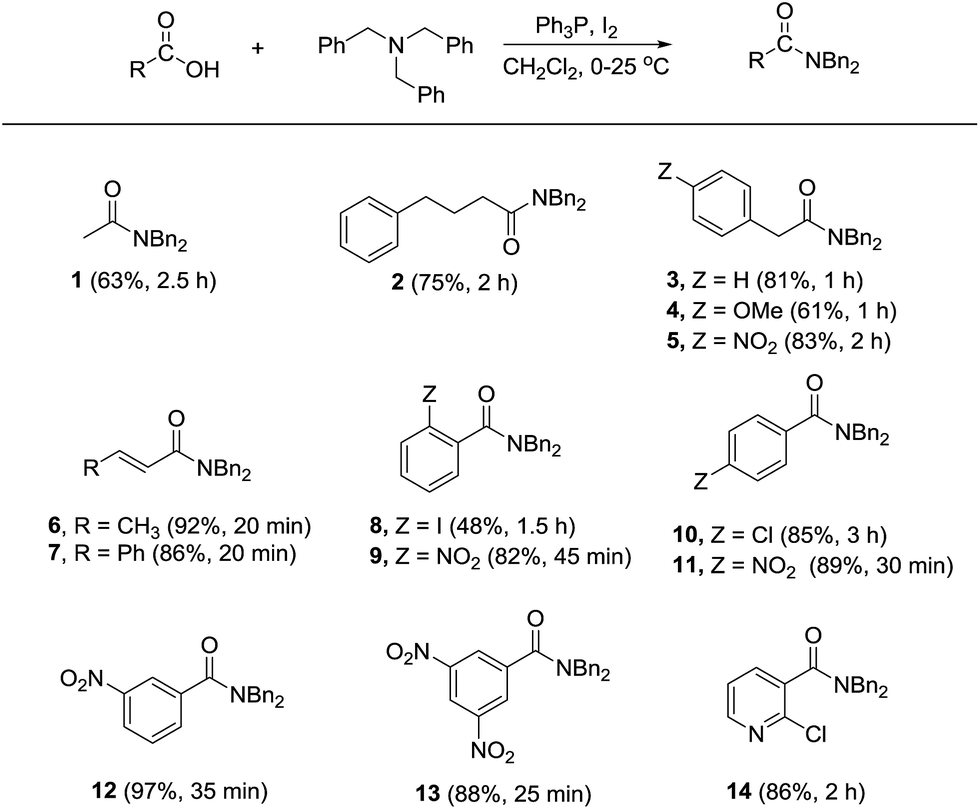 | ||
| Scheme 1 Screening for carboxylic acid scope. | ||
Unlike aliphatic acids or the electron-deficient aromatic substrates, competitive anhydride formation was pronounced when using benzoic acid and the more electron-rich systems such as 2-methylbenzoic acid, 4-methylbenzoic acid, and 4-methoxybenzoic acid. In these cases, the respective anhydride formed exclusively giving no amide product.
It should be noted that according to our previously reported conditions,22 amidation between carboxylic acids and primary or secondary amines under the Ph3P–I2/Et3N system was found to provide the corresponding amides in good to excellent yields without detectable formation of N,N-diethylamide side-products. In this study, the same Ph3P–I2 combination could enable an effective synthesis of tertiary amides from tertiary amines used as base and nucleophile. Thus, the precise reagent addition sequence is believed to be a key toward these transformations. Indeed, when 2-chloro-5-nitrobenzoic acid was added into a stirring mixture containing Ph3P, I2 and Et3N, followed by treatment with cyclohexylamine, the corresponding diethylamide product could be isolated in 71% yield (Scheme 2a). On the other hand, when cyclohexylamine was added first to the Ph3P–I2 mixture, followed by the addition of the acid, then Et3N, the respective N-cyclohexylamide was obtained in 76% (Scheme 2b) indicating the presence of tertiary amine prior to the addition of other more reactive nucleophile is necessary for the formation of tertiary amide products.
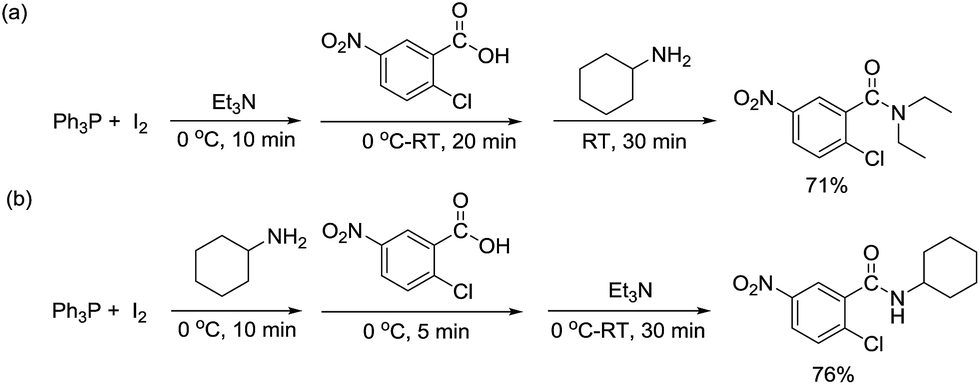 | ||
| Scheme 2 Effect of reagent addition sequence in the Ph3P–I2 mediated amidation. | ||
On the basis of the observed results, the possible mechanism for the reaction is proposed as shown in Scheme 3. Initially, the reaction between Ph3P and iodine generates triphenylphosphonium iodide I along with a pentavalent triphenylphosphinediiodide II as the reactive intermediates.22–25 Nucleophilic displacement of iodine in II by the nitrogen of tertiary amine would lead to a more reactive species III that could undergo substitution with a carboxylate ion to give an intermediate IV. Elimination of an alkyl iodide then produces a phosphorane V20,26 which undergoes subsequent intramolecular acyl transfer to give the tertiary amide product.27,28
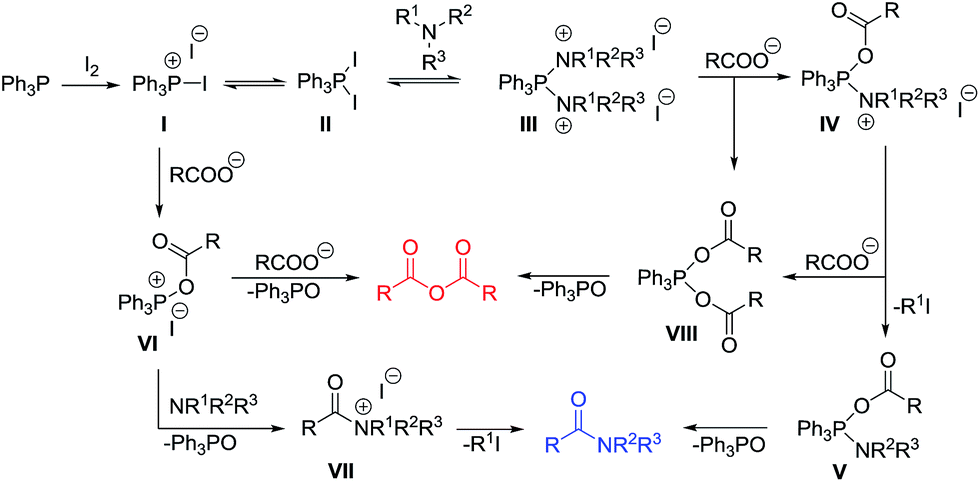 | ||
| Scheme 3 Proposed mechanism for the Ph3P/I2-mediated amidation with tertiary amine. | ||
Alternatively, an attack of I with a carboxylate ion could give rise to the formation of an acyloxyphosphonium salt VI.23,25 This species could also undergo acyl substitution with a tertiary amine to provide a quaternary ammonium salt VII. Subsequent C–N bond cleavage through an elimination of alky iodide then provides the amide product.
Although it is unclear why switching the order of the addition between a carboxylic acid and a tertiary amine could give rise to a significant difference in the reaction outcomes, it is possible that the formation of intermediate IV could be promoted when tertiary amine was added first to the Ph3P–I2 mixture before treatment with carboxylic acid. Using the less reactive acid substrate, IV would be more likely to survive leading to predominant formation of the amide product. However, with other reactive carboxylic acid derivatives such as benzoic acid and 4-methoxybenzoic acid, anhydride formation is most likely to occur through VIII derived from substitution of III or IV with a carboxylate ion. Alternatively, when tertiary amine was added last in the reagent addition sequence, VI could be predominantly formed leading to a rapid formation of acid anhydride in preferable to amide-bond formation. This side reaction would be highly competitive, especially when reactive carboxylic acids are used as the substrates.20,29
In summary, we have reported, for the first time, the use of Ph3P–I2 combination in promoting an amidation reaction between various carboxylic acids and tertiary amines. Through the right sequence of the reagents addition, the method enables the preparation of a series of tertiary amides in good to excellent yields under mild reaction conditions. The developed protocol is also complement well with the metal-catalyzed procedure which only work best with substituted aniline derivatives.15,17,19 Study regarding the chemoselectivity and stereoselectivity of the reaction with more challenging substrates are underway which will be reported in due course.
Acknowledgements
Financial support from the Thailand Research Fund through the Royal Golden Jubilee Ph.D. Program (Grant No. PHD/0206/2556) to S. W. is gratefully acknowledged.Notes and references
- C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405–3415 RSC.
- C. A. G. N. Montalbetti and V. M. Falque, Wiley Encycl. Chem. Biol., 2009, 4, 436–452 CAS.
- V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS PubMed.
- D. Grierson, Org. React., 1990, 39, 85–295 CrossRef CAS.
- R. P. Mariella and K. H. Brown, Can. J. Chem., 1971, 49, 3348–3351 CrossRef CAS.
- J. H. Cooley and E. J. Evain, Synthesis, 1989, 1–7 CrossRef CAS.
- N. O. V. Sonntag, Chem. Rev., 1953, 52, 237–416 CrossRef CAS.
- O. Hess, Ber. Dtsch. Chem. Ges., 1885, 685 CrossRef CAS.
- M. G. Voronkov, I. P. Tsyrendorzhieva and V. I. Rakhlin, Russ. J. Org. Chem., 2008, 44, 481–484 CrossRef CAS.
- M. G. Voronkov, I. P. Tsyrendorzhieva and V. I. Rakhlin, Russ. J. Org. Chem., 2010, 46, 794–797 CrossRef CAS.
- B. T. Khai and A. Arcelli, J. Organomet. Chem., 1983, 252, C9–C13 CrossRef CAS.
- Y. Li, F. Jia and Z. Li, Chem.–Eur. J., 2013, 19, 82–86 CrossRef CAS PubMed.
- Y. Zhang, H. Fu, Y. Jiang and Y. Zhao, Org. Lett., 2007, 9, 3813–3816 CrossRef CAS PubMed.
- S. Murata, K. Suzuki, A. Tamatani, M. Miura and M. Nomura, J. Chem. Soc., Perkin Trans. 1, 1992, 1387–1392 RSC.
- Y.-S. Bao, Z. Bao, A. Bao, M. Baiyin and M. Jia, J. Org. Chem., 2014, 79, 803–808 CrossRef CAS PubMed.
- Y.-S. Bao, M. Baiyin, A. Bao, M. Jia and Z. Bao, J. Org. Chem., 2014, 79, 6715–6719 CrossRef CAS PubMed.
- H.-C. Cheng, W.-J. Hou, Z.-W. Li, M.-Y. Liu and B.-T. Guan, Chem. Commun., 2015, 51, 17596–17599 RSC.
- X. Chen, T. Chen, Q. Li, Y. Zhou, L.-B. Han and S.-F. Yin, Chem.–Eur. J., 2014, 20, 12234–12238 CrossRef CAS PubMed.
- Y. Li, L. Ma, F. Jia and Z. Li, J. Org. Chem., 2013, 78, 5638–5646 CrossRef CAS PubMed.
- W. Phakhodee, C. Duangkamol, S. Wangngae and M. Pattarawarapan, Tetrahedron Lett., 2016, 57, 325–328 CrossRef CAS.
- Y. Baqi and C. E. Mueller, J. Org. Chem., 2007, 72, 5908–5911 CrossRef CAS PubMed.
- S. Wangngae, C. Duangkamol, M. Pattarawarapan and W. Phakhodee, RSC Adv., 2015, 5, 25789–25793 RSC.
- A. Kumar, H. K. Akula and M. K. Lakshman, Eur. J. Org. Chem., 2010, 2709–2715 CrossRef CAS PubMed.
- C. Duangkamol, S. Wangngae, M. Pattarawarapan and W. Phakhodee, Eur. J. Org. Chem., 2014, 2014, 7109–7112 CrossRef CAS.
- S. P. Morcillo, L. Alvarez de Cienfuegos, A. J. Mota, J. Justicia and R. Robles, J. Org. Chem., 2011, 76, 2277–2281 CrossRef CAS PubMed.
- A. D. Kosal, E. E. Wilson and B. L. Ashfeld, Angew. Chem., Int. Ed., 2012, 51, 12036–12040 CrossRef CAS PubMed.
- P. Froyen, Tetrahedron Lett., 1997, 38, 5359–5362 CrossRef CAS.
- S. Schenk, J. Weston and E. Anders, J. Am. Chem. Soc., 2005, 127, 12566–12576 CrossRef CAS PubMed.
- P. J. Harvey, M. von Itzstein and I. D. Jenkins, Tetrahedron, 1997, 53, 3933–3942 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure and spectroscopic data. See DOI: 10.1039/c6ra12801g |
| This journal is © The Royal Society of Chemistry 2016 |