
Tao-Phos-controlled desymmetrization of succinimide-based bisalkynes via asymmetric copper-catalyzed Huisgen alkyne–azide click cycloaddition: substrate scope and mechanism†
Mu-Yi Chen‡
a,
Tao Song‡a,
Zhan-Jiang Zhenga,
Zheng Xua,
Yu-Ming Cuia and
Li-Wen Xu*ab
aKey Laboratory of Organosilicon Chemistry and Material Technology of Ministry of Education, Hangzhou Normal University, Hangzhou 311121, P. R. China. E-mail: liwenxu@hznu.edu.cn; Fax: +86 2886 5135; Tel: +86 2886 5135
bState Key Laboratory for Oxo Synthesis and Selective Oxidation, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou, P. R. China. E-mail: licpxulw@yahoo.com
First published on 13th June 2016
Abstract
It was found that the catalytic Huisgen cycloaddition reaction of succinimide-derived bisalkynes with azides resulted in useful succinimide-derived triazoles bearing quaternary carbon-stereogenic centers with good to excellent yields as well as good chemoselectivity and moderate to high enantioselectivities (up to 97% ee), in which the CuF2/Tao-Phos complex was proved to be an effective bimetallic catalyst in the presence of triethylamine. The mechanistic studies based on the effect of reaction parameters on stereoselectivity as well as ESI-MS analysis suggested that our ligand (Tao-Phos) and related binuclear copper centers play a crucial role in this asymmetric click chemistry because of the strong catalyst–substrate interaction with the aid of a possibly in situ formed binuclear or multinuclear copper-based transition state. This work not only provides a new example of a Cu(II)-mediated asymmetric AAC reaction involving chelating base and azides, but also suggests the significance of binuclear copper species in catalytic asymmetric click reactions with two copper center-involved enantioselective inductions.
Introduction
Maleimides, and their substituted derivatives, succinimides, are valuable synthetic targets as basic and important structural scaffolds or precursors of synthetically and biologically interesting molecules as well as polymers.1 Especially, their functional derivatives are important backbones and substances in organic systems, biologically active products, key motifs in natural alkaloids, and total syntheses.2,3 Accordingly, the significance of these maleimides or succinimides has inspired researchers to develop new strategies to access optically active maleimide/succinimide derivatives.4 In this regard, we have developed a novel synthetic method for the asymmetric organocatalytic Michael reaction of maleimides with aliphatic aldehydes by the combination of triphenylphosphine and cinchona alkaloid-derived primary amine.5 This novel cooperative catalyst system exhibited high enantioselectivity (up to 99% yield, >99% ee) and could be one of the best examples in the synthesis of chiral succinimides via Michael reactions.6 Very recently, we successfully developed succinimide-derived triazoles bearing quaternary carbon stereogenic centers by catalytic asymmetric Huisgen alkyne–azide [3 + 2] cycloaddition.7 The desired succinimide-based products were achieved in good yields (60–80%) and 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 to >99:1 enantiomeric ratio (e.r.) in this click desymmetrization reaction. Notably, desymmetrizations of bis-functional groups are of great interest in organic synthesis because of its atom-economic construction of chiral compounds.8
:15 to >99:1 enantiomeric ratio (e.r.) in this click desymmetrization reaction. Notably, desymmetrizations of bis-functional groups are of great interest in organic synthesis because of its atom-economic construction of chiral compounds.8
Since the chemistry of Huisgen alkyne–azide cycloaddition, also often referred to concept of “Click” addition was first introduced by the groups of Sharpless and Meldal,9 copper-catalyzed alkyne–azide cycloaddition reaction (CuAAC) has been intensively recognized as a powerful carbon–nitrogen bond-forming reaction for the facile synthesis of a range of structurally diversity triazoles that are prevalent in many research areas of chemistry, materials, biology, pharmaceutical and environmental science.10,11 In particular, the asymmetric version of CuAAC has also been reported in the past decade.7,12 For the first example reported by Fokin and Finn, the use of bis(oxazolinyl)pyridine (pybox) as chiral ligands in the form of kinetic resolution of α-azides and desymmetrization of gem-diazides resulted in low to moderate enantioselectivity and yield.12a In 2013, Zhou and coworkers12b have successfully established a highly enantioselective CuAAC via desymmetrization of oxindole-based 1,6-heptadiynes with up to 98% ee. Thus the previous and limited reports suggested that the enantioselective CuAAC with desymmetrization, or referred to asymmetric click chemistry (ACC) had powerful potential for the enantioselective construction of structurally diversity and optically pure triazoles.
In view of the importance of succinimide-based structures and triazoles in medicinal chemistry, it is highly desirable to explore the highly enantioselective Huisgen cycloaddition reactions for the synthesis of succinimide-derived triazoles via the click desymmetrization. Herein, by considering the versatility of catalytic desymmetrization and click chemistry in organic synthesis, we continue to provide an improved approach for the stereoselective construction of quaternary carbon-stereogenic center on the succinimide-based molecules by Huisgen alkyne–azide cycloaddition. In addition, despite previous achievement on the desymmetrization of succinimide-based bisalkynes by CuAAC have been determined,7 the possible mechanism of this reaction is still not clear. Thus the detailed evaluation of corresponding reaction parameters and substrate scope would be beneficial to the understanding the desymmetrization by copper-catalyzed alkyne–azide cycloaddition.
Because of the distinctive performances of the Ar-BINMOL-Phos ligands bearing multiple stereogenic centers in the desymmetrization of succinimide-based bis-alkyne 1a with azidomethyl acetate or azidomethyl propionate, we expected the multifunctional phosphine ligands could also serve as an effective ligand in the copper-catalyzed Huisgen cycloaddition with much more broad substrate scope. Thus initially, owing to the structural similarity of benzylic azides and azidomethyl acetate, we chose the Huisgen cycloaddition of succinimide-based bisalkyne 1a and benzylic azide 2a as a model reaction.
Results and discussion
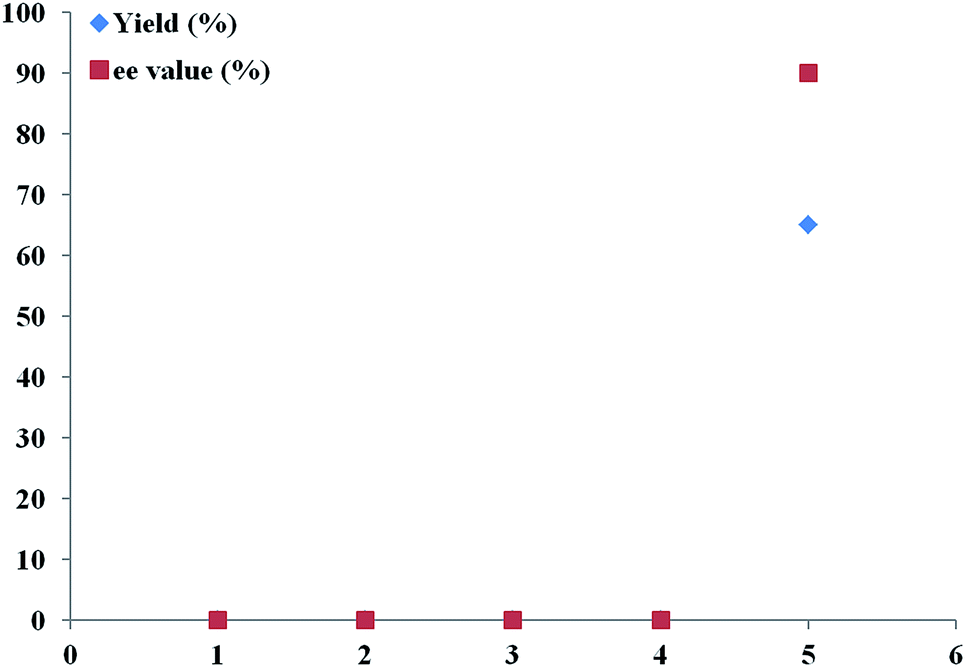
With various Ar-BINMOL-derived phosphine ligands in hand, we first attempted the conditions previously developed for the Huisgen alkyne–azide cycloaddition reaction.7 The Ar-BINMOL-Phos ligand with various substituents in acetonitrile gave moderate to good isolated yields with varied enantioselectivities from 5–90% ee. Similarly to previous work, the Tao-Phos with a silicon-based bulky group had a beneficial effect and was found to the best ligand in this reaction. As shown in Scheme 1, the simplest Ar-BINMOL-derived phosphine ligand without additional substituent on the benzylic phenyl ring gave moderate enantioselectivity, while as methyl (L3), methoxyl (L5), phenyl (L6)-substituted groups on the ortho-position of benzylic phenyl ring did not lead to the improvement of enantioselectivity (40–50% ee with L3, L5, and L6). Notably, when the methyl group on benzylic phenyl ring was changed to meta-position showed as Ar-BINMOL-derived ligand L4, an unexpectedly poor enantioselectivity (only 5% ee) was detected in this reaction. These experimental results described above suggested that the steric repulsion-induced enantioselectivity played important role in this reaction, which further confirmed the privileged action of Tao-Phos with possible enzyme-like reaction space.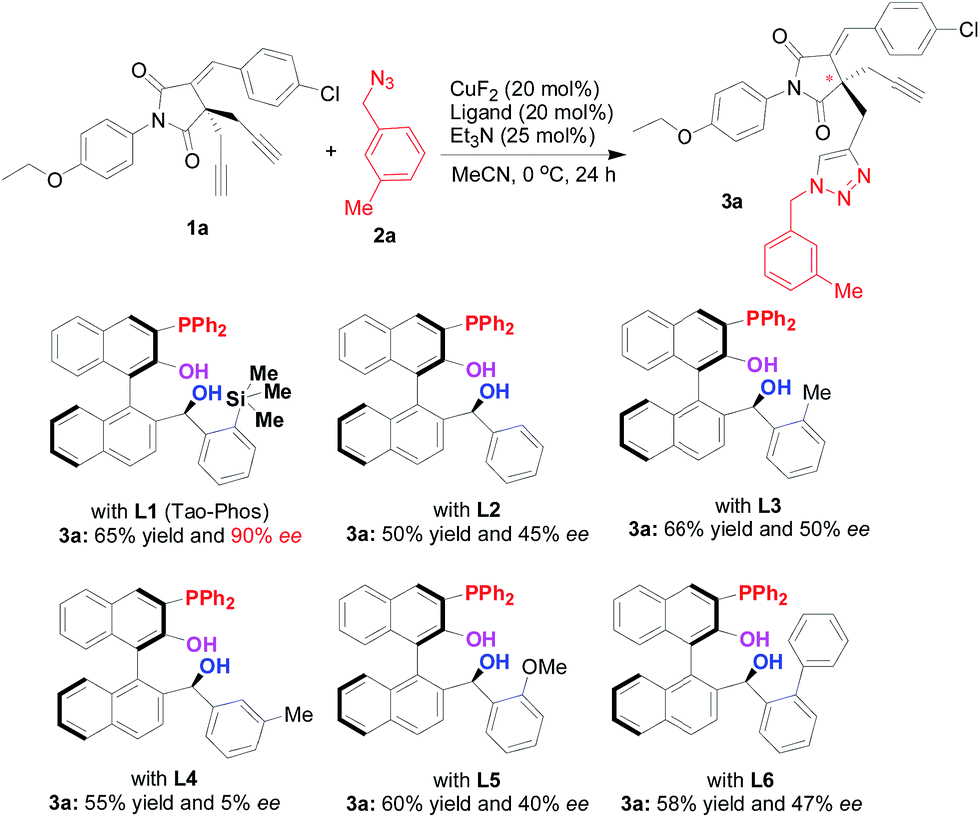 | ||
| Scheme 1 The confirmation of Tao-Phos (L1) with high enantioselectivity in desymmetrization of bisalkyne 1a with azide 2a via screening of Ar-BINMOL-derived phosphine ligands. | ||
Therefore, an extensive optimization of reaction parameters was undertaken (Scheme 2), and these results would be useful for the understanding of combinational and optimized use of copper/Tao-Phos catalyst system with triethyl amine in acetonitrile. Some of results are presented in Fig. 1–3. Variation of copper salts (Fig. 1) showed the CuF2 was an irreplaceable copper source because of its conspicuous enantioselectivity in this reaction. There are no rule for the enantioselectivity or activity of Cu(I) salts in this reaction. More interestingly, and the use of CuCl led to the inversed configuration for the product, which supported the importance of anion in this reaction. The nature of copper(II) difluoride is critical for the stereochemical outcome of the reaction. We hypothesized that the stable Cu(II) catalyst is more better that than Cu(I) salts in this reaction. A further evaluation of solvent effect showed that acetonitrile is a better solvent than methanol and tetrahydrofuran (THF), and surprisingly, the use of toluene, or DCM led to poor enantioselectivity, slightly better than diethyl ether, acetone, and DMF with almost no enantioselectivity (Fig. 2).
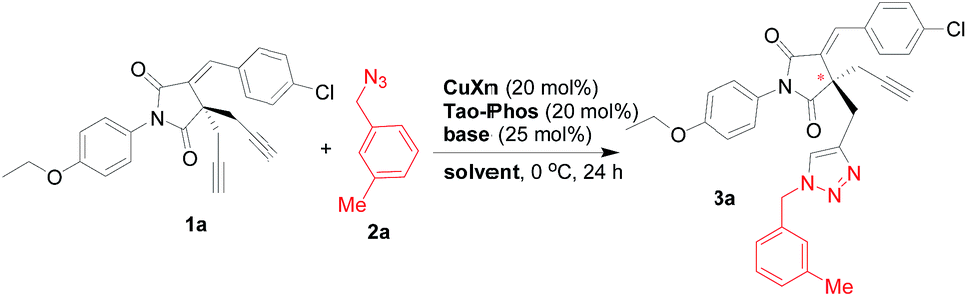 | ||
| Scheme 2 The screening of reaction parameters with Tao-Phos in desymmetrization of bisalkyne 1a with azide 2a. | ||
 | ||
| Fig. 1 Optimization of reaction parameters with copper effect in the presence of Et3N in CH3CN: 1, Cu(MeCN)4PF6 (32% yield, 50:50 er); 2, CuBr2 (41% yield, 50:50 er); 3, CuCl (28% yield, 47.5:52.5 er); 4, CuI (58% yield, 54:46 er); 5, CuBr (66% yield, 55:45 er); 6, CuF2 (65% yield, 95:5 er). | ||
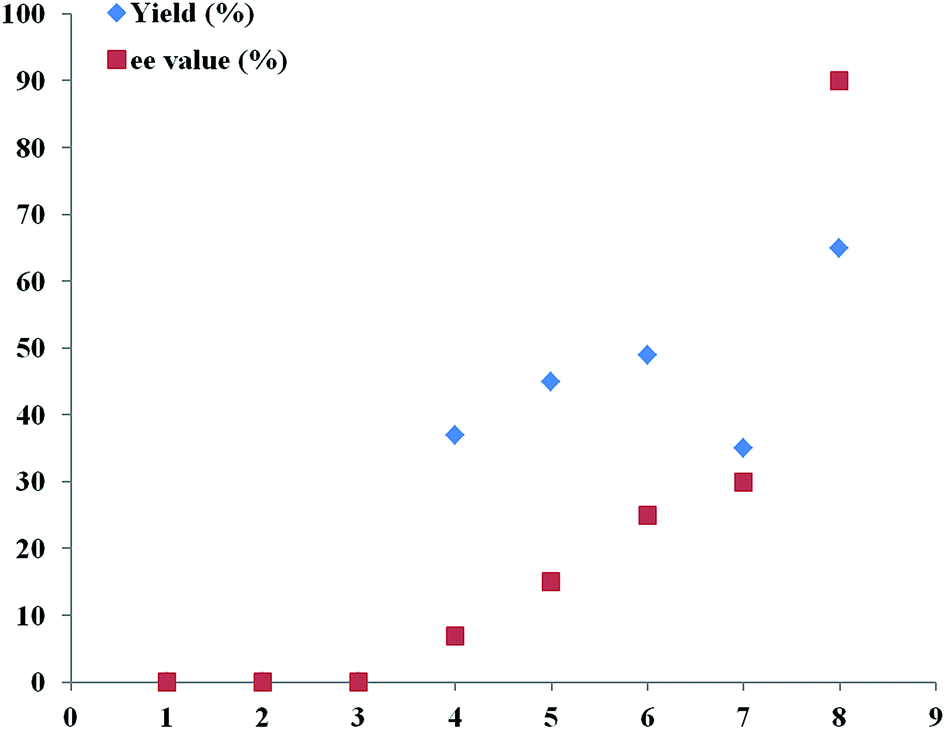 | ||
| Fig. 2 Optimization of reaction parameters with solvent effect in the presence of CuF2 and Et3N: 1, acetone (no reaction, n.r); 2, DMF (no reaction, n.r); 3, Et2O (no reaction, n.r); 4, PhMe (37% yield, 53.5:46.5 er); 5, DCM (45% yield, 57.5:42.5 er); 6, THF (49% yield, 62.5:37.5 er); 7, MeOH (35% yield, 67.5:32.5 er); 8, MeCN (65% yield, 95:5 er). | ||
 | ||
| Fig. 3 Optimization of reaction parameters with base effect in the presence of CuF2 in CH3CN: 1, DIPEA (no reaction, n.r); 2, DMEDA (no reaction, n.r); 3, K3PO4 (no reaction, n.r); 4, K2CO3 (no reaction, n.r); 5, Et3N (65% yield, 95:5 er). | ||
More interestingly and notably, the triethyl amine (Et3N) could not be replaced by other bases in term of stereoselectivity, because other organic bases, such as DIPEA, DMEDA, and inorganic bases (K2CO3 and K3PO4) gave almost no enantioselectivity in this reaction (Fig. 3). The significant impact of triethyl amine on the copper/Tao-Phos catalyst that exhibited with good enantioselectivity in this reaction could be deduced to its dual role in this case, one is possible coordination between copper and nitrogen center of Et3N, and another function might be aroused from suitable base-promoted deprotonation of alkyne and transfer of copper catalyst in the catalytic cycle.
Encouraged by these results described above, we concluded that the copper(II) difluoride was a distinctively active catalyst in this reaction. Accordingly, with the highly enantioselective catalyst system based on CuF2 and Tao-Phos ligand in hand, we hypothesized that the effect of fluoride anion or cation could not be ignored on the desymmetrization of bisalkyne 1a with azide 2b. As shown in Table S4 (see ESI†), we examined various fluorine-containing additives in this reaction. It turned out that the addition of most of fluoride-based reagents to copper-catalyzed Huisgen cycloaddition led to a promisingly negative affect. For example, although the same level of enantioselectivity was achieved by MgF2 or CaF2, the use of BaF2 or others as an additive inhibited the Huisgen alkyne–azide cycloaddition. Notably, the use of Et3N·HF instead of Et3N in this reaction gave rise to no conversion, which further supported the importance of Et3N and CuF2 in the copper-catalyzed Huisgen cycloaddition. Therefore, on the basis of the evaluation of different reaction parameters as well as various additives, the optimized reaction conditions with suitable basic additive and solvent have been established for further synthetic application.
In order to demonstrate the versatility of the CuF2/Tao-Phos-catalyzed Huisgen cycloaddition, we examined some succinimide-based bisalkynes and benzylic azides to provide corresponding chiral succinimide-derived triazoles. Notably, it seems that this Tao-Phos and copper-based catalytic system was sensitive to both the alkyne and azide substrate, thus as well as examining the results for a range of differently substituted substrates, the order in which the alkyne and the amine were added to the reaction mixture was also varied (see ESI,† simplified as method A and method B respectively). Meanwhile, the reaction results that came from method A and method B would provide sufficient and direct data to clarify corresponding mechanism controlled by the catalyst–substrate interaction.
Under the optimized reaction conditions as determined in Fig. 1–3 for the enantioselective synthesis of succinimide-derived triazoles, the desymmetrization reaction of bisalkynes was proved to be highly selective, in which only mono-triazoles were achieved and trace amount of bis-triazole (<5%) was detected in this reaction. And the desired product of mono-triazole came from desymmetrization of succinimide-based bisalkynes could be isolated easily from the reaction mixture by flash column chromatography. As shown in Table 1, the use of most of succinimide-derived bis-alkynes as well as method A for the asymmetric click addition resulted in good enantioselectivities, chemoselectivities, and yields (61–81% yields, 67–97% ees, the ratio of mono-triazole/bis-triazole is >95:5). In fact, the isolation of bis-triazoles from the reaction mixtures was unsuccessful because there is almost no side reaction for the double Huisgen cycloaddition. We envisaged the chiral mono-triazole product synthesized in this protocol mentioned above would act as a substrate for the synthesis of bis-triazole under the same reaction conditions, thus we applied the CuF2/Tao-Phos catalyst system in the subsequent synthesis of chiral bis-triazole through Huisgen cycloaddition of 3a with azide 4a. Unexpectedly, no desired bis-triazole product was detected in this reaction (Fig. 4), which indirectly supported the excellent chemoselectivity in the CuF2/Tao-Phos-catalysed desymmetrization reaction of bisalkynes with azides.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Product | Method A | Method B | Δee (A − B) | ||
| Yieldb (%) | erc | Yieldb (%) | erc | |||
| a All reaction is carried with CuF2 (20 mol%), Tao-Phos (20 mol%), Et3N (25 mol%) in CH3CN at 0 °C.b Isolated yield.c Determined by chiral HPLC. Although the configurations of products 3 are not formally proved by X-ray analysis, the absolute configurations of the chiral monotriazole products 3 could be referred to previous report7 by HPLC for the determination of chiral product 5 of Table 2. | ||||||
| 1 | 3a | 61 | 95:5 |
65 | 85:15 |
+20 |
| 2 | 3b | 75 | 91.5:8.5 |
77 | 95.5:4.5 |
−6 |
| 3 | 3c | 61 | 91:9 |
65 | 91:9 |
0 |
| 4 | 3d | 67 | 98.5:1.5 |
77 | 95:5 |
+7 |
| 5 | 3e | 72 | 91.5:8.5 |
73 | 83.5:16.5 |
+16 |
| 6 | 3f | 73 | 93.5:6.5 |
70 | 88.5:11.5 |
+10 |
| 7 | 3g | 80 | 88:12 |
78 | 70.5:29.5 |
4 |
| 8 | 3h | 81 | 84:16 |
75 | 92.5:7.5 |
−17 |
| 9 | 3i | 62 | 93.5:6.5 |
60 | 83.5:16.5 |
+20 |
| 10 | 3j | 66 | 91.5:8.5 |
70 | 91.5:8.5 |
0 |
| 11 | 3k | 70 | 90.5:9.5 |
72 | 88.5:11.5 |
+4 |
| 12 | 3l | 70 | 83.5:16.5 |
73 | 89.5:10.5 |
−12 |
| 13 | 3m | 69 | 90.5:9.5 |
73 | 88.5:11.5 |
+4 |
| 14 | 3n | 74 | 95:5 |
74 | 82.5:17.5 |
+25 |
| 15 | 3o | 75 | 85:15 |
74 | 77:23 |
+16 |
| 16 | 3p | 77 | 90.5:0.5 |
80 | 92:8 |
−3 |

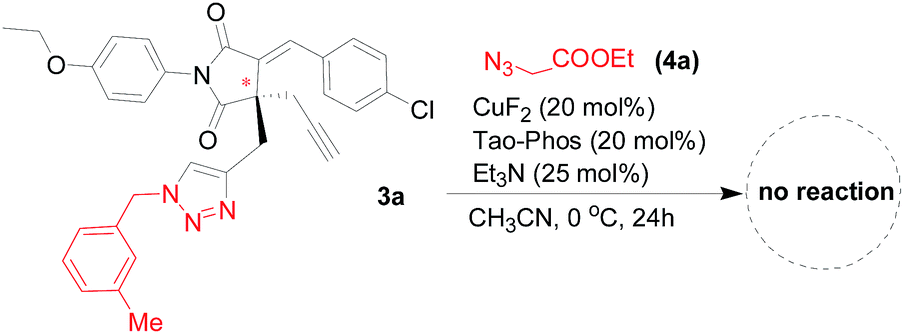 | ||
| Fig. 4 The Huisgen cycloaddition of 3a with azide 4a under the optimized reaction conditions (Method A): an indirect evidence for the excellent chemoselectivity in the CuF2/Tao-Phos-promoted desymmetrization reaction of bisalkynes. | ||
As shown in Table 1, both the phenyl-substituted succinimide substrates and the substituted succinimide-based bis-alkynes were proved to suitable material in the enantioselective Huisgen alkyne–azide cycloaddition. However, the variation of aryl groups on these alkynes or azides were subjected to enantioselective Huisgen cycloaddition with good chemoselectivity (>95:5 mono-triazole/bis-triazole) and yields. Furthermore, the use of method B instead of method A also afforded the desired products with moderate to good enantioselectivities and yields. Unexpectedly, the enantioselective Huisgen cycloaddition with method B is inferior to that with method A. As shown in Table 1, the deviations between method A and method B were varied from Δee = 0–25, and the better enantioselectivity was obtained with method A. As a hypothesis, we suggested the catalyst–substrate interaction was impacted by the base (Et3N) and the basic substrate (azide) largely. It might be aroused from that the base-promoted protonation of terminal alkyne as well as the azide-promoted regeneration of the copper catalyst respectively in the catalytic cycle.
Remarkably, to highlight the catalytic characteristics of Tao-Phos/copper complex with method A in the catalytic asymmetric Huisgen cycloaddition and desymmetrization of bisalkenes with azides, we then explored the Huisgen alkyne–azide [3 + 2] cycloaddition of these succinimide-based bisalkynes with azidomethyl acetate or azidomethyl propionate 4 for the comparative study of method A and method B (Table 2). As expected, the Huisgen alkyne–azide [3 + 2] cycloaddition of succinimide-based bisalkynes with azidomethyl acetate or azidomethyl propionate was also sensitive to the order of addition of the alkyne and the amine under the optimization reaction conditions, which further confirmed the superiority of method A in this reaction. As shown in Table 2, similarly to the catalytic asymmetric Huisgen cycloaddition of benzylic azides showed in Table 1, the copper-catalyzed alkyne–azide [3 + 2] cycloaddition of azidomethyl acetate with Tao-Phos as the ligand resulted in the desired product 5 with inferior enantioselectivity in the most cases when method B was applied in this reaction. The difference between ee values for method A and method B could be considered as important information for the obvious interaction between substrates and copper/Tao-Phos catalyst system.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Product | Procedure A (ref. 7) | Procedure Ba | Δee (A − B) | ||
| Yieldb (%) | erc | Yieldb (%) | erc | |||
| a All reaction is carried with CuF2 (20 mol%), Tao-Phos (20 mol%), Et3N (25 mol%) in CH3CN at 0 °C.b Isolated yield.c Determined by chiral HPLC, and the absolute configurations of the chiral monotriazole products 5 have been reported in the previous work.7 | ||||||
| 1 | 5a | 68 | 93.5:6.5 |
68 | 78:22 |
+31 |
| 2 | 5b | 69 | 87:13 |
65 | 95.5:4.5 |
−17 |
| 3 | 5c | 71 | 91:9 |
70 | 87.5:12.5 |
+7 |
| 4 | 5d | 74 | 88:12 |
74 | 82.5:17.5 |
+11 |
| 5 | 5e | 72 | 89:11 |
68 | 82.5:17.5 |
+13 |
| 6 | 5f | 71 | 90:10 |
70 | 75.5:24.5 |
+29 |
| 7 | 5g | 69 | 85:15 |
65 | 73.5:26.5 |
+23 |
| 8 | 5h | 72 | 91.5:8.5 |
75 | 77.5:22.5 |
+28 |
| 9 | 5i | 74 | 90:10 |
67 | 77:23 |
+26 |
| 10 | 5j | 70 | 88.5:11.5 |
75 | 82:18 |
+13 |
| 11 | 5k | 69 | 86.5:13.5 |
75 | 90.5:9.5 |
−8 |
| 12 | 5l | 72 | 87:13 |
63 | 84.5:55 |
+5 |

Notably, in the past decades, the use of various additives in catalytic asymmetric organic transformations to improve enantioselectivity has been reported.13 However, it is not an easy task to improve the enantioselectivity by the optimization of reaction conditions with the aid of additives. In this context, we have ever reported that the atropisomeric amide-derived olefins could be used as a reactive additive to promote the palladium-catalyzed hydrogenation/reductive decarbonylation of olefins and acyl chlorides respectively.14 Although the concept of springboard chemistry with high-reactive substrate as a “reactive tractor” to drive another transformation of low-reactive substrates (HDL catalysis) was not suitable for all the transformations, the related mechanism might be aroused from strong catalyst–substrate interaction. Thus previous reaction results on the HDL catalysis revealed that the in situ generated14b catalyst–substrate complex was probably a key catalyst in the presence of a high-reactive substrate. In fact, a good example with HDL catalysis was also reported by Pfaltz and coworkers,15 in which they disclosed firstly the high-reactive substrate (aryl alkyl-N-arylketimines)-promoted Ir-phosphinooxazoline (PHOX) complex-catalyzed hydrogenation of low-reactive dialkyl ketimines substrate with improved reactivity and enantioselectivity. It should be noted that it is generally suffer from low yields for dialkyl ketimines but give excellent enantioselectivities with high turnover numbers for the hydrogenation of structurally similar aryl alkyl-N-arylketimines.16 Pfaltz's work provides an excellent example on the substrate-type additive-activated transition-metal-catalysis as well as the utilization of pronounced catalyst–substrate interaction for the reactivity enhancement of asymmetric catalysis. Similarly and contrary to previous examples,14,15 the evaluation of HDL catalysis in the copper-catalyzed Huisgen cycloaddition would gave useful information to support the substrate-chelating click addition.
Guided by above results on the application of Tao-Phos in this alkyne–azide cycloaddition reaction and the discussion on HDL catalysis, we hypothesized that the chiral azide with high ee could be used as an effective additive to improve the substrate with inferior enantioselectivity in this reaction. Additionally, the competitive Huisgen cycloaddition reaction with two different azides could also be a useful probe for the understanding of catalyst–substrate interaction. As shown in Table 1, the use of 1-(azidomethyl)-3-methylbenzene (2a) and 1-(azidomethyl)-4-methylbenzene (2b) led to the corresponding product 3a and 3c with good ee value respectively (entries 2 and 3). Thus as a model reaction, we investigated the copper/Tao-Phos-catalyzed Huisgen cycloaddition with mixed azides 2a and 2b, in which 1.1 eq. 2a was used as a substrate and 20 mol% of 2b was utilized as a reactive additive. Unexpectedly, both the major product 3b and the minor product 3c was obtained with the same level of enantioselectivity but with decreased ee value (Scheme 3, 44% ee). Having observed that competed copper-catalyzed Huisgen cycloaddition of two different azides led to poor stereoselective control in the presence of Tao-Phos (Scheme 3), we suggested that the azide was possibly involved the formation of catalyst system similarly to that of Pfaltz's iridium catalysis.
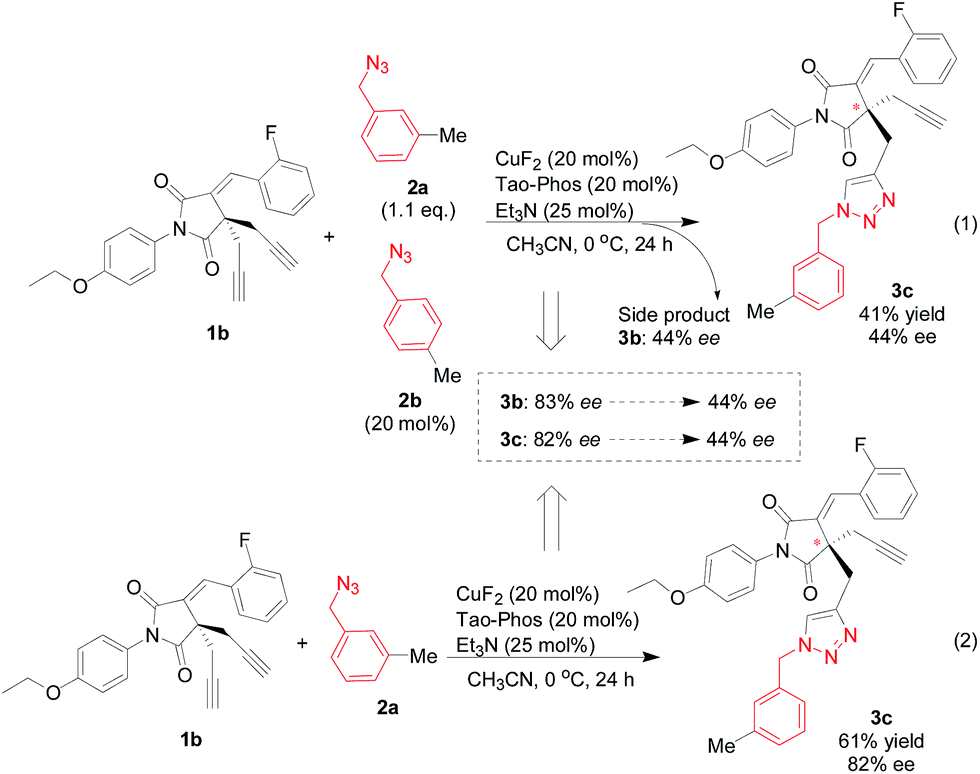 | ||
| Scheme 3 The impact of purity of azide on the enantioselective copper-catalyzed Huisgen cycloaddition: unexpected findings with decreased enantioselectivity in the desymmetrization reaction of bisalkyne 1b with the mixture of two azides 2a and 2b. | ||
With the copper/Tao-Phos catalyst system, and inspired by our previous findings on the preparation of sugar- or BINOL (1,1′-binaphthalene-2,2′-diol)-derived atropisomeric bistriazole by CuAAC,17 we also evaluated the Huisgen cycloaddition of sugar-derived azide 6 with the succinimide-derived bisalkyne 1l under the optimized reaction conditions. However, the diastereoselectivity of the click reaction showed in Scheme 4 (eqn (1)) is not good, but with only moderate diastereoselectivity (66:34 de). Notably, the yield of the desired product is quite good, in which it could highlight the importance of the quaternary carbon-stereogenic core of sugar-based triazole 7, and could be potentially used as a biologically active candidate in the near future. Notably, without the Tao-Phos as chiral ligand, the chiral sugar-based azide was also worked as a chiral auxiliary in this reaction, and the diastereoselectivity could be reached up to 60.5:39.5 de with the reversal configuration that detected by HPLC analysis (eqn (2) of Scheme 4). Thus, this sugar-based Huisgen cycloaddition reaction supported the powerful catalyst–substrate interaction in this reaction, which would be useful information for the mechanistic study in this reaction.
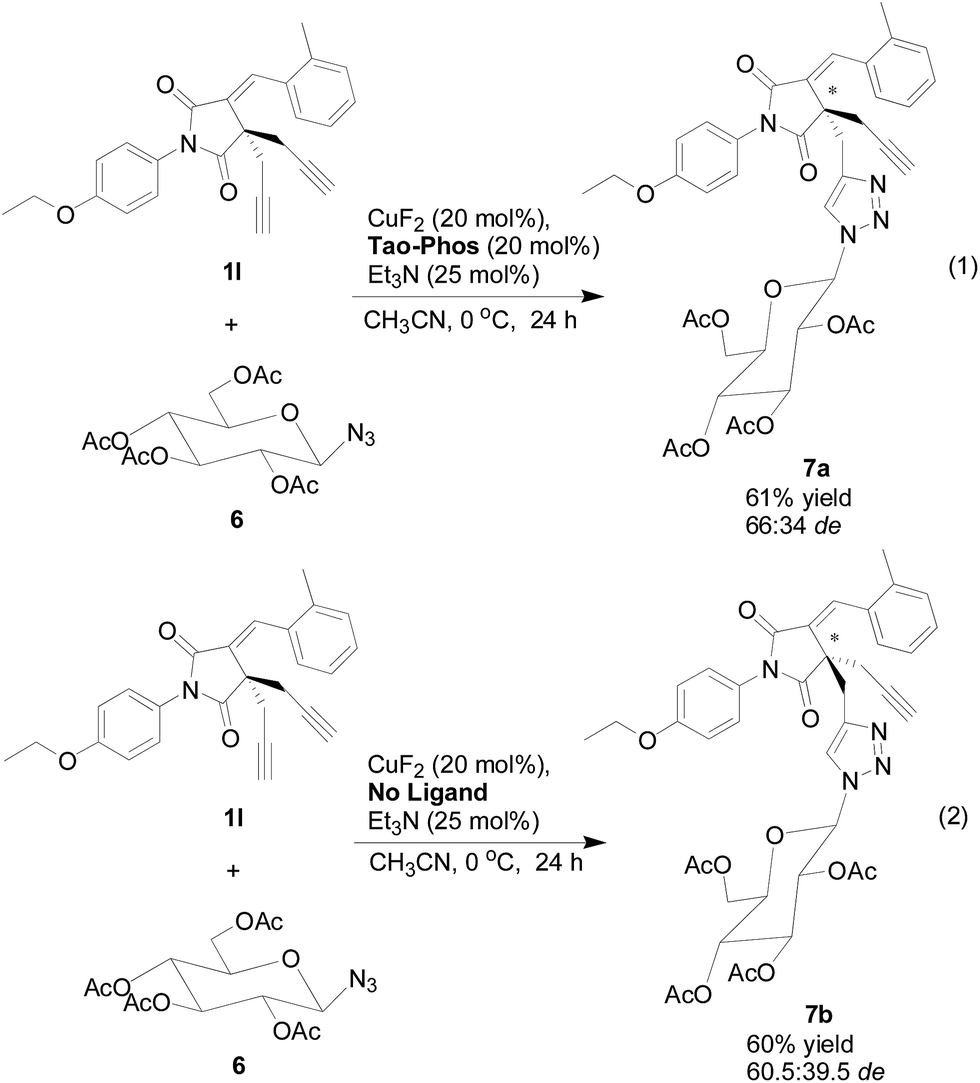 | ||
| Scheme 4 The synthesis of quaternary carbon-stereogenic and sugar-based triazole 7 by copper/Tao-Phos-catalyzed Huisgen cycloaddition. | ||
Although a full mechanistic picture should be awaited with further investigation for such copper-catalyzed Huisgen cycloaddition reaction, it is interesting to clarify the role of phosphorous atom in the multifunctional Tao-Phos-controlled copper-catalyzed alkyne–azide cycloaddition reaction under the optimized reaction conditions. As expected, the oxidized Tao-Phos was proved to be an invalid ligand in this reaction (Scheme 5). Notably, it was also found that deuteration of Tao-Phos led to poor conversion (<5% yield). Thus it showed privileged role of both the phosphine and phenol/alcohol moieties on this Tao-Phos ligand in this reaction, especially the intermolecular hydrogen-bonding of Tao-Phos with succinimide-based bisalkyne or azide was a favourable factor for the enantioselective Huisgen azide–alkyne cycloaddition reaction.18
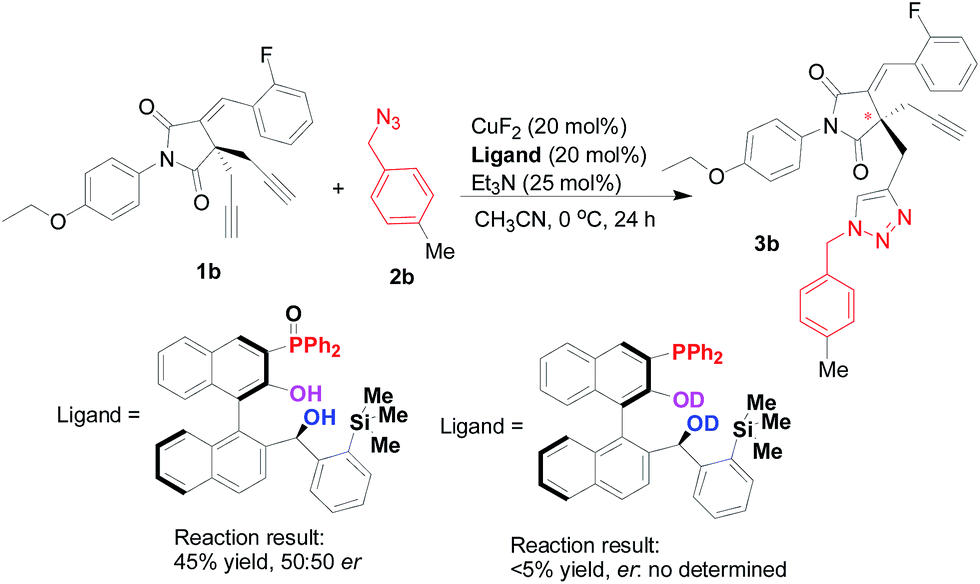 | ||
| Scheme 5 A comparative study: the elimination of phosphine oxide as true ligand in this reaction. | ||
The experimental results described in this work, as well as the nonlinear effect (NLE)19 study (Fig. S1†) and the electrospray ionization mass spectrometry (ESI-MS) analysis (Fig. S2–7 in ESI†), supported that a binuclear copper complex with at least two ligands worked as the active species in this reaction. Notably, the electrospray ionization mass spectrometry was proved to be powerful tool to clarify the mechanism of copper-catalyzed azide–alkyne cycloaddition reaction. In this context, Angelis and co-workers have revealed the existence of dinuclear copper intermediates in general copper-catalyzed Huisgen cycloaddition reaction by ESI-MS analysis.20 Thus on the basis of ESI-MS analysis, our experimental work further supported the reliable findings on the two copper centers-controlled azide–alkyne cycloaddition in this reaction. Accordingly, we proposed a possible model and mechanistic procedure for the copper/Tao-Phos-catalyzed Huisgen alkyne–azide cycloaddition reaction (Fig. 5). As a direct evidence by ESI-MS analysis,21 the ion at m/z = 1328 supported the formation of [Cu-(Tao-Phos)2 complex]. And we found an ion at m/z = 1192 (Fig. S2†) that could be deduced as the copper/Tao-Phos complex interacted with Et3N and alkyne (intermediate I in Fig. 5). In fact, the Fig. S2† showed the formation of multinuclear copper complex in the reaction mixture of CuF2, bisalkyne, trimethylamine, and Tao-Phos, is possibly because of the detection of m/z 1215.6 and 1319.1 respectively, in which it could be calculated with m/z 1215.4 and m/z 1319.3 respectively as [Cu(L)(Et3N)(alkyne) + Na] and [Cu3(L)(Et3N)(alkyne)]. More importantly, the major ion peak at m/z 1302.6 provided a direct evidence for the intermediate III of Fig. 5 because the calculated m/z of Cu2L(azide)(alkyne) is 1302.3, in which the Tao-Phos ligand (L) could be oxidized easily to phosphine oxide during the ESI-MS analysis. Therefore the ESI-MS analysis (Fig. S2–S7 in ESI†) provided a powerful evidence for the mechanistic procedure showed in Fig. 5. In fact, as described in the previous reports,22 binuclear copper intermediate was generally recognized as active catalyst in copper(I)-catalyzed Huisgen azide–alkyne cycloaddition reaction. Thus according to previous study on the mechanism of copper-catalyzed click reaction,22,23 binuclear copper-acetylides could be a key intermediate and responsible for the high enantioselectivity in this reaction.
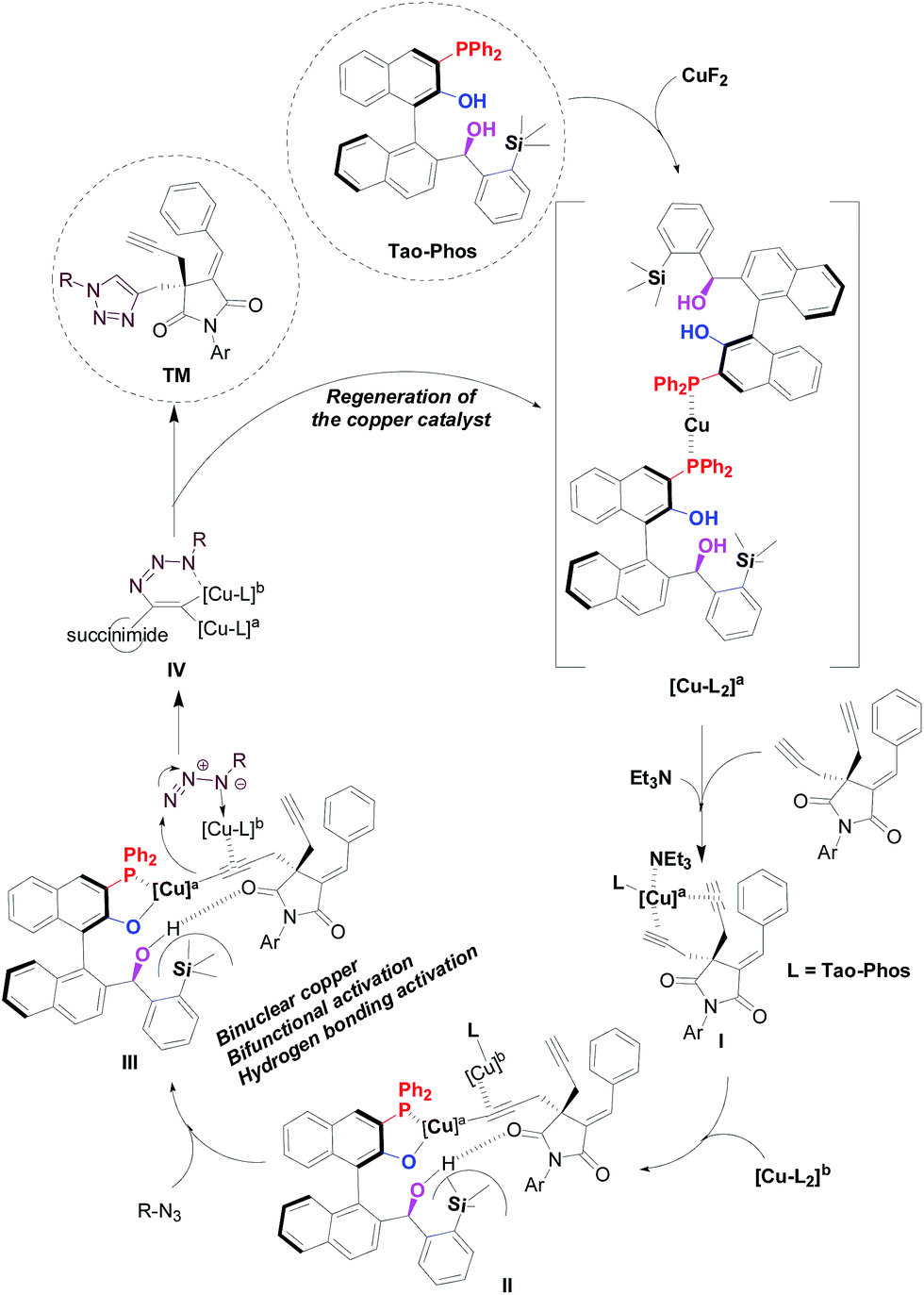 | ||
| Fig. 5 Proposed mechanism for the asymmetric binuclear copper/Tao-Phos promoted Huisgen cycloaddition of bis-alkyne with azide. | ||
As shown in Fig. 5, we suggested that binuclear copper catalysis with the aid of multifunctional Tao-Phos and trimethylamine played crucial role in the enantioselective click chemistry, in which the catalyst–substrate interaction-involved cooperative activation pattern between copper/Tao-Phos catalyst system and two substrates (alkyne and azide respectively) led to the formation of possible reaction assembly for further intramolecular-like cycloaddition with excellent enantioselectivity, in which both the triethylamine and azide acted as a chelating promoter for the formation of the binuclear copper catalyst system. Despite the present binuclear copper model and mechanistic study for the Et3N-promoted copper-catalyzed Huisgen cycloaddition of alkynes with azides is still insufficient for the Tao-Phos-controlled Huisgen cycloaddition at present, the fact that both a bimetallic catalytic pathway and a base are involved in the stabilization of catalyst–substrate interaction, supported it is also a chelation-assisted, and probably binuclear copper(II)-accelerated AAC reaction.24 And consistent with prior experimental results (especially as shown in Table S4,† the effect of fluoride anion or cation on the desymmetrization of bisalkyne supported the importance of fluoride anion, and possible oxidation of copper(I) to high-valent copper(II) or Cu(III) with fluoride cation led to poor reactivity)23,25 and similarly to previous reported mechanism,22,26 each structural component of the in situ formed chiral copper(I)/Tao-Phos catalyst system from CuF2, Tao-Phos, Et3N, and azide, played a pivotal role in this reaction, suggested that a cooperative manner through the magic activation of succinimide-based bisalkyne with a multinuclear copper(I)/copper(II) complex with the aid of base is reasonable. Thus based on the mechanistic investigation of copper/Tao-Phos-promoted Huisgen cycloaddition reaction, binuclear catalysis with two transition-metal centers was expected to be an effective strategy in asymmetric synthesis.27
Experimental
General procedure for the catalytic asymmetric CuAAC reaction (Fig. 4 and Tables 1 and 2)
Method A: under an atmosphere of N2, to an oven-dried Schlenk tube were added Tao-Phos (25 mg, 0.04 mmol) and CuF2 (4 mg, 0.04 mmol), followed by the addition of CH3CN (1.0 mL). After the solution was stirred at 25 °C for 1 hour, NEt3 (7 μL, 0.05 mmol, 0.25 eq.) and benzyl azidoacetate (27.5 μL, 0.22 mmol) were added, and keep stirring for 0.5 hour. The reaction was cooled to 0 °C and the substrate (0.2 mmol) was added in additional CH3CN (1 mL). The resulting mixture was stirred for 72 hours till almost full conversation to product 3 (TLC analysis). When the reaction was complete, it was quenched with saturated aqueous NH4Cl (1 mL) and stirred vigorously for 5 minutes. The aqueous phase was extracted with ethyl acetate (3 × 5 mL). The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (PE/EA, 5/1–3/1) to get the products 3. Characterization data for the products of these reactions are provided in the ESI.† For example, compound 3a was obtained in 61% yield as white solid, 90% ee determined by HPLC analysis (Chiralcel IB-H column, hexane: i-PrOH 70:30, 1.0 mL min−1, 254 nm). Retention time: tminor = 25.92, tmajor = 18.69. 1H NMR (400 MHz, CDCl3) d 7.94 (s, 1H), 7.74 (dd, J = 4.0, 6.4 Hz, 1H), 7.43 (dd, J = 3.6, 6.4 Hz, 1H), 7.36–7.34 (m, 2H), 7.26–7.21 (m, 1H), 7.14 (d, J = 8.4 Hz, 2H), 7.05 (d, J = 8.8 Hz, 2H), 6.98 (s, 2H), 6.89 (d, 8.8 Hz, 2H), 5.39 (dd, J = 14.8, 32.4 Hz, 2H), 4.04 (q, J = 6.8 Hz, 2H), 3.23 (d, J = 14.8 Hz, 1H), 2.93–2.87 (m, 2H), 2.55 (dd, J = 2.4, 16.4 Hz, 1H), 2.30 (s, 3H), 2.08 (q, J = 3.6 Hz, 1H), 1.42 (q, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) d 177.3, 168.6, 159.0, 139.1, 136.0, 134.5, 133.9, 133.0, 132.2, 130.4, 129.7, 129.5, 129.0, 128.6, 127.7, 126.7, 125.0, 124.6, 122.0, 115.0, 78.2, 72.1, 63.7, 54.1, 51.1, 32.4, 27.9, 21.3, 14.8; IR (KBr, cm−1) ?max 3305, 2977, 1709, 1651, 1513, 1396, 1248, 1168, 1050, 745. [α]20D = +20.98 (c = 3.71 CHCl3). HRMS (ESI-TOF): exact mass calcd for C33H29ClN4O3H [M + H]+: 565.2001, found: 565.2001.
Method B: under an atmosphere of N2, to an oven-dried Schlenk tube were added Tao-Phos (25 mg, 0.04 mmol) and CuF2 (4 mg, 0.04 mmol), followed by the addition of CH3CN (1.0 mL). After the solution was stirred at 25 °C for 1 hour, the substrate (0.2 mmol) was added in additional CH3CN (1 mL), and the reaction mixture was kept stirring for 0.5 hour. NEt3 (7 μL, 0.05 mmol, 0.25 eq.) and benzyl azidoacetate (27.5 μL, 0.22 mmol) were added. The reaction was cooled to 0 °C, and the resulting mixture was then stirred for 72 hours till almost full conversation to product 3 (TLC analysis). When the reaction is completed, it was quenched with saturated aqueous NH4Cl (1 mL) and stirred vigorously for 5 minutes. The aqueous phase was extracted with ethyl acetate (3 × 5 mL). The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (PE/EA, 5/1–3/1) to get the products 3.
Conclusions
In summary, it has been shown that the interesting catalytic system, CuF2/Tao-Phos complex, afforded the useful succinimide-derived triazoles bearing quaternary carbon-stereogenic center in good yields with high level of chemoselectivity and enantioselectivity (up to 97% ee). In addition, we have further determined that the Tao-Phos was a privileged ligand in controlling copper(II)-catalyzed desymmetrization of succinimide-derived bisalkynes in comparison to that of other chiral P-ligands in catalytic asymmetric Huisgen [3 + 2] cycloaddition of azides and alkynes (CuACC) that has been just highlighted by Brittain28 and Zhou29 very recently, which is complementary to previously reported enantioselective alkyne–azide cycloaddition. The mechanistic studies based on the effect of reaction parameters on stereoselectivity as well as ESI-MS analysis suggested that the Tao-Phos and related binuclear copper centers play crucial role in this asymmetric click chemistry because of the strong catalyst–substrate interaction with the aid of a possibly in situ formed binuclear or multinuclear copper-based transition state, which provided interesting and important information for the mechanistic model of base-chelating or substrate-promoted copper-catalyzed asymmetric Huisgen alkyne–azide cycloaddition reaction. Remarkably, on the basis of the detailed investigation of reaction parameters, we believed the privileged role of multifunctional Tao-Phos in the copper-catalyzed Huisgen [3 + 2] cycloaddition of alkynes and azides has been revealed in the catalytic asymmetric synthesis of structurally diverse triazoles. This work not only provides a new example accounting for the Cu(II)-mediated asymmetric AAC reactions involving chelating base and azides, but also suggests the significance of binuclear copper species in catalytic asymmetric click reactions where two copper centers-involved enantioselective inductions. Further investigations into the true mechanism of the copper/Tao-Phos catalysis in such desymmetrization via Huisgen cycloaddition reaction are needed to be clarified and reported in the near future.Acknowledgements
This Project was supported by the National Natural Science Founder of China (No. 51303043, 21472031, and 21503060), and Zhejiang Provincial Natural Science Foundation of China (LR14B030001) is appreciated. This work is also supported partially by Science and Technology Department of Zhejiang Province (2015C31138), and Hangzhou Science and Technology Bureau of China (20140432B04).Notes and references
- For representative reviews, see: (a) S. Ahmed, Drug Des. Discovery, 1996, 14, 77–89 CAS; (b) A. R. Katritzky, J. Yao, M. Qi, Y. Chou, D. J. Sikora and S. Davis, Heterocycles, 1998, 48, 2677–2691 CrossRef CAS; (c) C. Le Sann, Nat. Prod. Rep., 2006, 23, 357–367 RSC; (d) A. Gandini, Prog. Polym. Sci., 2013, 38, 1–29 CrossRef CAS; (e) G. Delaittre, N. K. Guimard and C. Barner-Kowollik, Acc. Chem. Res., 2015, 48, 1296–1307 CrossRef CAS PubMed.
- (a) M. K. Hargreaves, J. G. Pritchard and H. R. Dave, Chem. Rev., 1970, 70, 439 CrossRef CAS; (b) M. L. Curtin, R. B. Garland, H. R. Heyman, R. R. Frey, M. R. Michaelidies, J. Li, L. J. Pease, K. B. Glaser, P. A. Marcotte and S. K. Davidsen, Bioorg. Med. Chem. Lett., 2002, 12, 2919–2923 CrossRef CAS PubMed; (c) R. Ballini, G. Bosica, G. Cioci, D. Fiorini and M. Petrini, Tetrahedron, 2003, 59, 3603–3608 CrossRef CAS; (d) J. Pohlmann, T. Lampe, M. Shimada, P. G. Nell, J. Pernerstorfer, N. Svenstrup, N. A. Brunner, G. Schiffer and C. Freiberg, Bioorg. Med. Chem. Lett., 2005, 15, 1189–1192 CrossRef CAS PubMed.
- For recent examples, see: (a) H. P. Cho, D. W. Engers, D. F. Venable, C. M. Niswender, C. W. Lindsley, P. J. Conn, K. A. Emmitte and A. L. Rodriguez, ACS Chem. Neurosci., 2014, 5, 597 CrossRef CAS PubMed; (b) H. Gunosewoyo, A. Midzak, I. N. Gaisina, E. V. Sabath, A. Fedolak, T. Hanania, D. Brunner, V. Papadopoulos and A. P. Kozikowski, J. Med. Chem., 2013, 56, 5115–5129 CrossRef CAS PubMed; (c) E. J. Wright, S. Sosna, S. Bloodworth, J. D. Kilburn and P. N. Bartlett, Chem.–Eur. J., 2014, 20, 5550–5554 CrossRef CAS PubMed; (d) D. W. Manley, A. Mills, C. O'Rourke, A. M. Z. Slawin and J. C. Walton, Chem.–Eur. J., 2014, 20, 5492–5500 CrossRef CAS PubMed; (e) D. J. Nieves, N. S. Azmi, R. Xu, R. Levy, E. A. Yates and D. G. Ferning, Chem. Commun., 2014, 50, 13157–13160 RSC; (f) C. Zhu and S. M. Ma, Adv. Synth. Catal., 2014, 356, 3897–3911 CrossRef CAS; (g) H. Kitagishi, H. Kawasaki and K. Kano, Chem.–Asian J., 2015, 10, 1768–1775 CrossRef CAS PubMed; (h) H. Imoto, K. Kizaki, S. Watase, K. Matsukawa and K. Naka, Chem.–Eur. J., 2015, 21, 12105–12111 CrossRef CAS PubMed; (i) C. H. Zhu, G. Y. Xu, D. Ding, L. Qing and J. T. Sun, Org. Lett., 2015, 17, 4244–4247 CrossRef CAS PubMed.
- For representative reviews, see: P. Chauhan, J. Kaur and S. S. Chimni, Chem.–Asian J., 2013, 8, 328–346 CrossRef CAS PubMed.
- W. Yang, K. Z. Jiang, X. Lu, H. M. Yang, L. Li, Y. Lu and L. W. Xu, Chem.–Asian J., 2013, 8, 1182–1190 CrossRef CAS PubMed.
- (a) G. L. Zhao, Y. Xu, H. Sundén, L. Eriksson, M. Sayah and A. Córdova, Chem. Commun., 2007, 43, 734–735 RSC; (b) F. Yu, X. M. Sun, Z. C. Jin, S. G. Wen, X. M. Liang and J. X. Ye, Chem. Commun., 2010, 46, 4589–4591 RSC; (c) F. Yu, Z. C. Jin, H. C. Huang, T. T. Ye, X. M. Liang and J. X. Ye, Org. Biomol. Chem., 2010, 8, 4767–4774 RSC; (d) J. F. Bai, L. Peng, L. L. Wang, L. X. Wang and X. Y. Xu, Tetrahedron, 2010, 66, 8928–8932 CrossRef CAS; (e) F. Xue, L. Liu, S. L. Zhang, W. H. Duan and W. Wang, Chem.–Eur. J., 2010, 16, 7979–7982 CrossRef CAS PubMed; (f) T. Miura, A. Masuda, M. Ina, K. Nakashima, S. Nishida, N. Tada and A. Itoh, Tetrahedron: Asymmetry, 2011, 22, 1605–1609 CrossRef CAS; (g) T. Tsuyoshi, S. Nishida, A. Masuda, N. Tada and A. Itoh, Tetrahedron Lett., 2011, 52, 4158–4160 CrossRef; (h) Z. W. Ma, Y. X. Liu, W. J. Zhang, Y. Tao, Y. Zhu, J. C. Tao and M. S. Tang, Eur. J. Org. Chem., 2011, 2011, 6747–6754 CrossRef CAS; (i) Z. W. Ma, Y. X. Liu, P. L. Li, H. Ren, Y. Zhu and J. C. Tao, Tetrahedron: Asymmetry, 2011, 22, 1740–1748 CrossRef CAS; (j) T. C. Nugent, A. Sadiq, A. Bibi, T. Heine, L. L. Zeonjuk, N. Vankova and B. S. Bassil, Chem.–Eur. J., 2012, 18, 4088–4098 CrossRef CAS PubMed.
- T. Song, L. Li, W. Zhou, Z. J. Zheng, Y. Deng, Z. Xu and L. W. Xu, Chem.–Eur. J., 2015, 21, 554–558 CrossRef CAS PubMed.
- For selected review, see: (a) M. D. Díaz-de-Villegas, J. A. Gálvez, P. Etayo, R. Badorrey and M. P. López-Ram-de-Víu, Chem. Soc. Rev., 2011, 40, 5564–5587 RSC; (b) E. Garcia-Urdiales, I. Alfonso and V. Gotor, Chem. Rev., 2005, 105, 313–354 (Chem. Rev., 2011, 111, PR110–PR180) CrossRef CAS PubMed , update.
- (a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS; (b) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS; (c) C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef.
- For recent reviews, see: (a) W. Xi, T. F. Scott, C. J. Kloxin and C. N. Bowman, Adv. Funct. Mater., 2014, 24, 2572–2590 CrossRef CAS; (b) A. Lauria, R. Delisi, F. Mingoia, A. Terenzi, A. Martorana, G. Barone and A. M. Almerico, Eur. J. Org. Chem., 2014, 2014, 3289–3306 CrossRef CAS; (c) P. C. Trippier, ChemMedChem, 2013, 8, 190–203 CrossRef CAS PubMed; (d) K. Kempe, A. Krieg, C. R. Becer and U. S. Schubert, Chem. Soc. Rev., 2012, 41, 176–191 RSC; (e) C. Chu and R. Liu, Chem. Soc. Rev., 2011, 40, 2177–2188 RSC; For recent highlight on the synthesis of triazoles, see: J. P. Wan, D. Q. Hu, Y. Y. Liu and S. R. Sheng, ChemCatChem, 2015, 7, 901–903 CrossRef CAS.
- For recent examples, see: (a) I. Glassford, C. N. Teijaro, S. S. Daher, A. Weil, M. C. Small, S. K. Redhu, D. J. Colussi, M. A. Jacobsen, W. E. Childers, B. Buttaro, A. W. Nicholson, A. D. MacKerell, B. S. Cooperman and R. B. Andrade, J. Am. Chem. Soc., 2016, 138, 3136–3144 CrossRef CAS PubMed; (b) S. H. Etschel, L. Portilla, J. Kirschner, M. Drost, F. Tan, H. Marbach, R. R. Tykwinski and M. Halik, Angew. Chem., Int. Ed., 2015, 54, 9235–9238 CrossRef CAS PubMed; (c) R. Wirth, J. D. White, A. D. Moghaddam, A. L. Ginzburg, L. N. Zakharov, M. M. Haley and V. J. DeRose, J. Am. Chem. Soc., 2015, 137, 15169–15175 CrossRef CAS PubMed; (d) Y. Shi, R. W. Graff, X. Cao, X. Wang and H. Gao, Angew. Chem., Int. Ed., 2015, 54, 7631–7635 CrossRef CAS PubMed; (e) P. Michael and W. H. Binder, Angew. Chem., Int. Ed., 2015, 54, 13918–13922 CrossRef CAS PubMed; (f) A. Makarem, R. Berg, F. Rominger and B. F. Straub, Angew. Chem., Int. Ed., 2015, 54, 7431–7435 CrossRef CAS PubMed; (g) A. Macdonell, N. A. B. Johnson, A. J. Surman and L. Cronin, J. Am. Chem. Soc., 2015, 137, 5662–5665 CrossRef CAS PubMed; (h) S. De, S. Pramanik and M. Schmittel, Angew. Chem., Int. Ed., 2014, 53, 14255–14259 CrossRef CAS PubMed; (i) V. Bevilacqua, M. King, M. Chaumonter, M. Nothisen, S. Gabillet, D. Buisson, C. Puente, A. Wagner and F. Taran, Angew. Chem., Int. Ed., 2014, 53, 5872–5876 CrossRef CAS PubMed; (j) C. Deraedt, A. Rapakousiou, Y. Wang, L. Salmon, M. Bousquet and D. Astruc, Angew. Chem., Int. Ed., 2014, 53, 8445–8449 CrossRef CAS PubMed; (k) C. Deraedt, N. Pinaud and D. Astruc, J. Am. Chem. Soc., 2014, 136, 12092–12098 CrossRef CAS PubMed; (l) N. S. Teske, J. Voigt and V. P. Shastri, J. Am. Chem. Soc., 2014, 136, 10527–10533 CrossRef CAS PubMed; (m) S. Yoshida, Y. Hatakeyama, K. Johmoto, H. Uekusa and T. Hosoya, J. Am. Chem. Soc., 2014, 136, 13590–13593 CrossRef CAS PubMed; (n) J. M. Ren, K. Satoh, T. K. Goh, A. Blencowe, K. Nagai, K. Ishitake, A. J. Christofferson, G. Yiapanis, I. Yarovsky, M. Kamigaito and G. G. Qiao, Angew. Chem., Int. Ed., 2014, 53, 459–464 CrossRef CAS.
- (a) J. C. Meng, V. V. Fokin and M. G. Finn, Tetrahedron Lett., 2005, 46, 4543–4546 CrossRef CAS; (b) F. Zhou, C. Tan, J. Tang, Y. Y. Zhang, W. M. Gao, H. H. Wu, Y. H. Yu and J. Zhou, J. Am. Chem. Soc., 2013, 135, 10994–10997 CrossRef CAS PubMed; (c) G. R. Stephenson, J. P. Buttress, D. Deschamps, M. Lancelot, J. P. Martin, A. I. G. Sheldon, C. Alayrac, A. C. Gaumont and P. C. B. Page, Synlett, 2013, 24, 2723–2729 CrossRef CAS; (d) T. Osako and Y. Uozumi, Org. Lett., 2014, 16, 5866–5869 CrossRef CAS PubMed; (e) T. Osako and Y. Uozumi, Synlett, 2015, 26, 1475–1479 CrossRef CAS; (f) W. D. Brittain, B. R. Buckley and J. S. Fossey, Chem. Commun., 2015, 51, 17217–17220 RSC.
- L. Hong, W. Sun, D. Yang, G. Li and R. Wang, Chem. Rev., 2016, 116, 4006–4123 CrossRef CAS PubMed.
- (a) X. F. Bai, L. W. Xu, L. S. Zheng, J. X. Jiang, G. Q. Lai and J. Y. Shang, Chem.–Eur. J., 2012, 18, 8174–8179 CrossRef CAS PubMed. And the hypothesis of springboard chemistry that high-reactive substrate as a “tractor” or “reactive springboard” to drive transformation of low-reactive substrate was first discussed as the concept of HDL catalysis in our previous work, see: (b) H. Wang, K. F. Yang, L. Li, Y. Bai, Z. J. Zheng, W. Q. Zhang, Z. W. Gao and L. W. Xu, ChemCatChem, 2014, 6, 580–591 CrossRef CAS.
- Y. Schramm, F. Barrios-Landeros and A. Pfaltz, Chem. Sci., 2013, 4, 2760–2766 RSC.
- (a) P. Schnider, G. Koch, R. Prétôt, G. Wang, F. M. Bohnen, C. Krüger and A. Pfaltz, Chem.–Eur. J., 1997, 3, 887–892 CrossRef CAS; (b) A. Baeza and A. Pfaltz, Chem.–Eur. J., 2010, 16, 4003–4009 CrossRef CAS PubMed.
- It was found the chiral sugar-based Huisgen cycloaddition could be used to the preparation of atropisomeric bistriazoles by amine-functional polysiloxanes (AFPs)-controlled Huisgen-oxidative coupling reaction, see: (a) Z. J. Zheng, F. Ye, L. S. Zheng, K. F. Yang, G. Q. Lai and L. W. Xu, Chem.–Eur. J., 2012, 18, 14094–14099 CrossRef CAS PubMed ; And for the BINOL-linked chiral triazoles, see:; (b) C. Y. Wang, J. F. Zou, Z. J. Zheng, W. S. Huang, L. Li and L. W. Xu, RSC Adv., 2014, 4, 54256–54262 RSC.
- (a) C. A. Coleman and C. J. Murray, J. Am. Chem. Soc., 1991, 113, 1677–1684 CrossRef CAS; (b) J. Atzrodt, V. Derdau, T. Fey and J. Zimmermann, Angew. Chem., Int. Ed., 2007, 46, 7744–7765 CrossRef CAS PubMed.
- (a) T. Satyanarayana, S. Abraham and H. B. Kagan, Angew. Chem., Int. Ed., 2009, 48, 456–494 CrossRef CAS PubMed; (b) M. Tsukamoto, K. Gopalaiah and H. B. Kagan, J. Phys. Chem. B, 2008, 112, 15361 CrossRef CAS PubMed. For the early example, see: (c) C. Puchot, O. Samuel, E. Duñach, S. Zhao, C. Agami and H. B. Kagan, J. Am. Chem. Soc., 1986, 108, 2353–2357 CrossRef CAS PubMed; (d) D. Guillaneux, S. H. Zhao, O. Samuel, D. Rainford and H. B. Kagan, J. Am. Chem. Soc., 1994, 116, 9430–9439 CrossRef CAS. For the representative review, see: (e) C. Girard and H. B. Kagan, Angew. Chem., Int. Ed., 1998, 37, 2922–2959 CrossRef.
- C. Iacobucci, S. Reale, J. F. Gal and F. D. Angelis, Angew. Chem., Int. Ed., 2015, 54, 3065–3068 CrossRef CAS PubMed.
- Electrospray ionization mass spectrometry (ESI-MS) has become a powerful tool for the mechanistic studies in the past decades, see: (a) F. M. Nachtigall and M. N. Eberlin, Organic Reaction Studies by ESI-MS, in Reactive Intermediates: MS Investigations in Solution, ed. L. S. Santos, Wiley, Weinheim, 2010 Search PubMed; (b) D. Schröder, Acc. Chem. Res., 2012, 45, 1521–1532 CrossRef PubMed; (c) L. P. E. Yunker, R. L. Stoddard and J. S. McIndoe, J. Mass Spectrom., 2014, 49, 1–8 CrossRef CAS PubMed; (d) C. Hinderling, C. Aldhart and P. Chen, Angew. Chem., Int. Ed., 1998, 37, 2685–2689 CrossRef CAS; (e) C. A. Marquez, F. Fabretti and J. O. Metzger, Angew. Chem., Int. Ed., 2007, 119, 7040–7042 CrossRef; (f) F. Coelho and M. N. Eberlin, Angew. Chem., Int. Ed., 2011, 50, 5261–5263 CrossRef CAS PubMed; (g) F. Ye, Z. J. Zheng, W. H. Deng, L. S. Zheng, Y. Deng, C. G. Xia and L. W. Xu, Chem.–Asian J., 2013, 8, 2242–2253 CrossRef CAS PubMed; (h) W. Yang, K. Z. Jiang, X. Lu, H. M. Yang, L. Li, Y. Lu and L. W. Xu, Chem.–Asian J., 2013, 8, 1182–1190 CrossRef CAS PubMed; (i) T. Song, L. S. Zheng, F. Ye, W. H. Deng, Y. L. Wei, K. Z. Jiang and L. W. Xu, Adv. Synth. Catal., 2014, 356, 1708–1718 CrossRef CAS; (j) C. Dong, T. Song, X. F. Bai, Y. M. Cui, Z. Xu and L. W. Xu, Catal. Sci. Technol., 2015, 5, 4755–4759 RSC.
- B. T. Worrell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457–460 CrossRef CAS PubMed.
- (a) V. O. Rodionov, S. I. Presolski, D. D. Díaz, V. V. Fokin and M. G. Finn, Angew. Chem., Int. Ed., 2005, 44, 2210–2215 CrossRef CAS PubMed; (b) V. D. Bock, H. Hiemstra and J. H. van Maarseveen, Eur. J. Org. Chem., 2005, 2005, 51–68 CrossRef; (c) V. O. Rodionov, S. I. Presolski, D. D. Díaz, V. V. Fokin and M. G. Finn, J. Am. Chem. Soc., 2007, 129, 12705–12712 CrossRef CAS PubMed; (d) B. F. Straub, Chem. Commun., 2007, 43, 3668 Search PubMed; (e) M. Ahlquist and V. V. Fokin, Organometallics, 2007, 26, 4389–4391 CrossRef CAS; (f) B. R. Buckley, S. E. Dann and H. Heaney, Chem.–Eur. J., 2010, 16, 6278–6284 CrossRef CAS PubMed; (g) H. B. Chen, N. Abeyrathna and Y. Liao, Tetrahedron Lett., 2014, 55, 6575–7576 CrossRef CAS; (h) C. Deraedt, N. Pinaud and D. Astruc, J. Am. Chem. Soc., 2014, 136, 12092–12098 CrossRef CAS PubMed; (i) E. Haldón, M. Besora, I. Cano, X. C. Cambeiro, M. A. Pericás, F. Maseras, M. C. Nicasio and P. J. Pérez, Chem.–Eur. J., 2014, 20, 3463–3474 CrossRef PubMed; (j) A. Gorbunov, D. Cheshkov, V. Kovalev and I. Vatsouro, Chem.–Eur. J., 2015, 21, 9528–9534 CrossRef CAS PubMed.
- G. C. Kuang, P. M. Guha, W. S. Brotherton, J. T. Simmons, L. A. Stankee, B. T. Nguyen, R. J. Clark and L. Zhu, J. Am. Chem. Soc., 2011, 133, 13984–14001 CrossRef CAS PubMed.
- The confirm of Cu(I) or Cu(II) complex was difficult at present for this reaction. However, previous investigations on the copper-catalyzed fluorination in the presence of fluoride ion suggested Cu(I)-based species was probably the true catalyst in many reactions. See: (a) H. Dang, M. Mailig and G. Lalic, Angew. Chem., Int. Ed., 2014, 53, 6473–7476 CrossRef CAS PubMed; (b) B. D. Zlatopolskiy, J. Zischler, P. Krapf, F. Zarrad, E. A. Urusova, E. Kordys, H. Endepols and B. Neumaier, Chem.–Eur. J., 2015, 21, 5972–5979 CrossRef CAS PubMed; (c) N. Ichiishi, A. J. Canty, B. Yates and M. S. Sanford, Organometallics, 2014, 33, 5525–5534 CrossRef CAS PubMed; (d) M. Tredwell, S. M. Preshlock, N. J. Taylor, S. Gruber, M. Huiban, J. Passchier, J. Mercier, C. Génicot and V. Gouverneur, Angew. Chem., Int. Ed., 2014, 53, 7751–7755 CrossRef CAS PubMed; (e) Y. Liu, C. Chen, H. Li, K. W. Huang, J. Tan and Z. Weng, Organometallics, 2013, 32, 6587–6592 CrossRef CAS; (f) X. Mu, H. Zhang, P. Chen and G. Liu, Chem. Sci., 2014, 5, 275–280 RSC.
- (a) C. Wang, D. Ikhlef, S. Kahlal, J. Y. Saillard and D. Astruc, Coord. Chem. Rev., 2016, 316, 1–20 CrossRef CAS; (b) R. Berg and B. F. Straub, Beilstein J. Org. Chem., 2013, 9, 2715–2750 CrossRef PubMed.
- For recent examples, see: binuclear gold catalysis, (a) S. Klimczyk, A. Misale, X. Huang and N. Maulide, Angew. Chem., Int. Ed., 2015, 54, 10365–10369 CrossRef CAS PubMed; binuclear copper catalysis, (b) F. von Rekoeski, C. Koch and R. M. Gschwind, J. Am. Chem. Soc., 2014, 136, 11389–11395 CrossRef PubMed; binuclear silver catalysis, (c) X. F. Bai, T. Song, Z. Xu, C. G. Xia, W. S. Huang and L. W. Xu, Angew. Chem., Int. Ed., 2015, 54, 5255–5259 CrossRef CAS PubMed; (d) X. F. Bai, Z. Xu, C. G. Xia, Z. J. Zheng and L. W. Xu, ACS Catal., 2015, 5, 6016–6020 CrossRef CAS.
- As highlighted by Brittain and co-workers very recently, the area of asymmetric CuAAC is still in its infancy in comparison to that of well-established metal-mediated asymmetric transformations: W. D. G. Brittain, B. R. Buckley and J. S. Fossey, ACS Catal., 2016, 6, 3629–3636 CrossRef CAS.
- X. P. Zeng, Z. Y. Cao, Y. H. Wang, F. Zhou and J. Zhou, Chem. Rev., 2016 DOI:10.1021/acs.chemrev.6b00094 , ASAP.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra13687g |
| ‡ M. Y. Chen and T. Song contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |