
Continuous photoflow synthesis of precision polymers
T.
Junkers
*ab and
B.
Wenn
a
aPolymer Reaction Design Group, Institute of Materials Research (IMO), Hasselt University, Martelarenlaan 42, BE-3500 Hasselt, Belgium. E-mail: tanja.junkers@uhasselt.be
bIMEC Associated Lab IMOMEC, Wetenschapspark 1, BE-3590 Diepenbeek, Belgium
First published on 17th November 2015
Abstract
Precision polymer design in continuous photoflow reactors is a young, yet rapidly growing research field. By switching from batch to flow processing, polymerizations can be carried out with unmatched efficiency under mild reaction conditions, while concommitantly providing conditions for simple scale up of reactions. Specifically the reversible deactivation radical polymerization (RDRP) techniques have gained significant interest in this respect within the past one to two years. The different photoRDRP methods are herein compared and the underlying principles of the advantage of carrying polymerization out under photoflow conditions are elucidated. Further, the yet unexplored potential of these techniques is identified and discussed towards future development.
Photoflow chemistry
Controlled polymerization techniques, starting from anionic polymerization to the plethora of controlled radical polymerization techniques (referred to as reversible deactivation radical polymerization, RDRP) are without doubt the gold standard of contemporary polymer synthesis towards advanced materials. The possibilities in macromolecular design are virtually endless – especially in combination with modular click chemistry1 approaches – and almost any macromolecular architecture can nowadays be targeted in one way or another. While such tailor-made materials open the window to a realm of materials with unprecedented biological, physical, thermal and mechanical properties, this raises as well the need for efficient pathways to synthesize these compounds in significant amounts. In research, highly precise polymer materials are often only obtained on the milligram scale, a hurdle that must be overcome in order to give access to material testing and ultimately to application.Flow techniques take hereby a prominent role and in recent years much focus was spend not only on bulk polymer and polymer particle synthesis,2 but also to precision polymerization techniques.3 As could be shown, small diameter flow reactors allow not only for simple scale up of such polymerizations, but also allow to synthesize materials with increased precision and hence advanced properties due to the more stable reaction conditions and improved isothermicity of the reactions.4 To date, practically all relevant polymerization techniques have been translated to micro- or mesoflow processing, including anionic and cationic polymerization,5,6 reversible addition fragmentation radical chain transfer polymerization (RAFT),7 atom transfer radical polymerization,8,9 single-electron transfer living radical polymerization (SET-LRP),10 nitroxide-mediated polymerization (NMP)11 or classical azide–alkyne cycloadditions12 to name the most relevant synthesis techniques.
A development that occurred at the same time in the field of polymer design is a renaissance of photo-induced reactions. Within only a few years, photo-induced reaction routes for all above mentioned synthesis techniques have been developed and optimized. Research activities in the field are on a steep rise and constantly new variations of photoactivated RDRP methods are published. Especially the ability to choose between different activator/sensitizer systems gives room for broad and efficient protocol optimizations.13–18 Photopolymerizations feature less side product formation and give access to fast, yet simple reaction protocols. As advantageous as these reactions are, they also feature a significant disadvantage; inherently, photo-induced reactions are difficult to scale up since increasing reactor volumes lead to intensity gradients in the reactors and hence loss of reaction efficiency. Only few options exist to overcome this problem. Light gradients occur according to Beer–Lambert's law, which correlates decreasing light intensity with increasing optical path lengths. Often, this effect is underestimated, but in fact even with moderate extinction coefficients, no significant light penetration is achieved in a batch reactor above the length of tens of millimeters. A visualization of the light gradient effect is shown in Fig. 1, which compares the light intensity profile in a batch reactor with a typical flow reactor (in which a small diameter PFA tubing is wrapped around a light source).19–22 Both reactors are filled with a solution of poly(3-hexylthiophene), a fluorescent polymer. As can easily be seen, all light is absorbed in the batch reactor in the top layers, even though a 100 W UV lamp was used for irradiation. Strong light gradients exist and any chemical reaction will only occur at the reactor surface. In contrast, the flow reactor – due to its much smaller average diameter, is fully illuminated and practically at no position intensity gradients are observed.
 | ||
| Fig. 1 Light intensity profile in a batch reactor (left) and a tubular flow reactor (right). | ||
The ‘Beer–Lambert penalty’ can in batch not be fully overcome. Flow reactors deliver a very convenient, and at the same time very economical way to scale photoreactions up. Due to the much better light efficiency of photoflow reactors, reactions are also significantly speed up, in some cases from days of reaction times to few minutes.19 Also, formation of degradation products can be avoided as total illumination times are shorter, and lower light intensities can be used. A good example for such scale up from the polymer world is the [2 + 2] cycloaddition reaction between polymers bearing maleimide endgroups and functionalized alkenes. Such reaction proceeds on the timescale of tens of hours in conventional batch reactors, but is efficiently carried out within 1 min under photoflow processing.23,24 While batch reactions are typically performed on milligrams of materials, even in small mesoscale flow reactors up to hundreds of grams can be synthesized with ease. Also, less side products are found in the crude, and the reaction becomes significantly more efficient, as the large excess of ene component required in batch is reduced to almost equimolar ratios in flow.
Photoflow reactors are thus the ideal solution for the above sketched problem of upscaling photoRDRPs. Further, they still retain the classical advantages of flow reactors for precision polymer synthesis (stable reaction conditions, scalability, reliability and reproducibility) and combine them with the advantages of photo-induced chemical transformations such as mild reaction conditions and energy saving. In other words, the combination of flow technology with photo-induced polymer reactions is a perfect couple. The very high potential of the combination has already been exploited in first proof-of-concept studies. This minireview summarizes these advances to highlight the achievable efficiency gain that is reached, and gives an outlook to future potentials and endeavours in this rapidly developing field.
PhotoRDRP flow methods
Controlled polymerization techniques generally give access to polymer materials with defined length, endgroup functionality and narrow dispersity. Polymers are ‘reactivatable’, and hence allow for block copolymer formation. Combined with advanced initiator and control agent design, macromolecular architectures ranging from rather simple linear structures to brush or star-shaped complex dendritic materials can be produced (Fig. 2). By adjusting the solubility of the various blocks incorporated in the macromolecules, so called ‘smart materials’ can be obtained, which are able to react to outer stimuli such as pH, temperature, light or an electrical current. Photo-induced reaction modes exist for practically all reversible deactivation radical polymerization methodologies, but are to date not yet exploited to full potential. These RDRP protocols are often very robust and can be carried out under less stringent reaction conditions compared to other living polymerizations, e.g. anionic polymerization. Generally, two different types of RDRP exist. Control over the polymerization reactions can be exerted via complex degenerative chain transfer equilibria in which chain propagation probability is evenly distributed over all present polymer chains, or via reversible termination reactions, which work via a reduction of free-radical concentrations during polymerization. Both types are discussed in the following.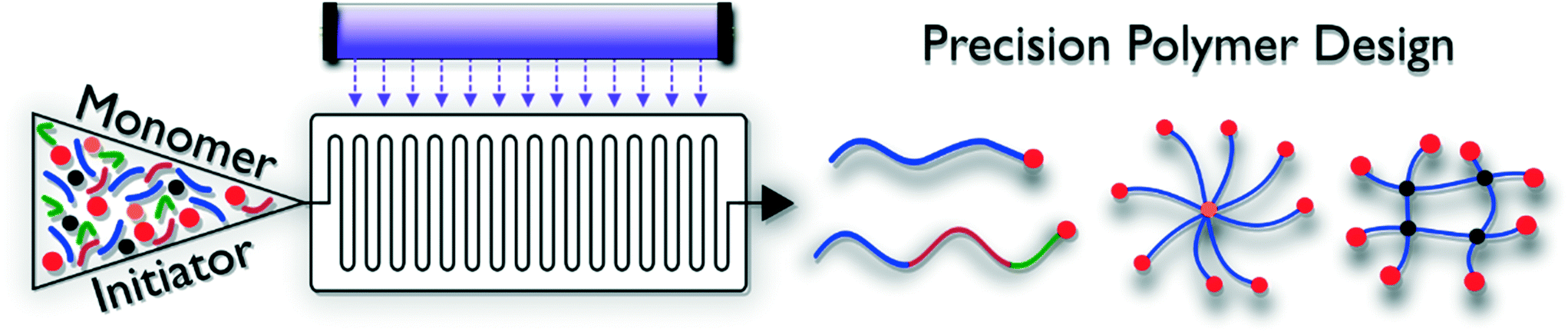 | ||
| Fig. 2 General scheme displaying precision polymer synthesis in photoflow reactors. | ||
Photo-induced reversible termination methods
In reversible termination polymerization methods the propagating radicals are terminated reversibly in an efficient control equilibrium. The majority of chains are at any instance in the dormant state and radical concentration are largely reduced, avoiding radical termination and hence inducing living character. The most known techniques are transition metal-mediated polymerizations (ATRP and SET-LRP) and NMP (for mechanisms the reader is referred to the cited literature). Metal-mediated photoinitiated polymerizations employing cobalt, copper and iridium for different monomers are reported in literature. For all systems a significant rate increase from batch to flow reactors is observed (Table 1). So far, the largest increase in polymerization rate was observed for Co-mediated polymerization of vinyl acetate from 30 to 1 hour, nicely demonstrating the flow advantage. Further, batch Co-mediated reactions are unstable and yield significant side products. The constant reaction conditions in flow improved the yield quality and no side products were found.25 Also Ir-catalysed systems show faster reactions in photoflow compared to batch reactions. However, the increase is not as dramatic as with Co-catalysts. The third reported metal, copper (by far the most widespread system), displays similar advantages with increasing reaction rates (Table 1). A direct comparison of Ir- and Cu-mediated polymerizations of methyl methacrylate show that copper enables much faster polymerizations, even though the underpinning reaction mechanism is relatively similar. For both metals, however, reaction rates are increased from batch to flow by a factor of roughly 6. Copper leads in comparison to all other metal catalysed systems to the fastest polymerization, allowing for good yields with concomitant high product quality.| # | Method | Reactor | Monomer | Reaction time [min] | Conv. [%] | M n [g mol−1] | Đ (Mw/Mn) |
|---|---|---|---|---|---|---|---|
| 1 (ref. 25) | Co-mediated | Batch | Vinyl acetate | 1800 | 26 | 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 500 |
1.33 |
| 2 (ref. 25) | Co-mediated | Flow | Vinyl acetate | 60 | 30 | 14200 |
1.28 |
| 3 (ref. 13) | Cu-mediated | Batch | Methyl acrylate | 90 | 95 | 4500 | 1.05 |
| 4 (ref. 26) | Cu-mediated | Flow | Methyl acrylate | 20 | 79 | 3100 | 1.10 |
| 5 (ref. 27) | Cu-mediated | Batch | Methyl methacrylate | 420 | 88 | 2900 | 1.23 |
| 6 (ref. 27) | Cu-mediated | Flow | Methyl methacrylate | 60 | 48 | 3100 | 1.24 |
| 7 (ref. 28) | Ir-mediated | Batch | Methyl methacrylate | 1260 | 80 | 6300 | 1.19 |
| 8 (ref. 29) | Ir-mediated | Flow | Methyl methacrylate | 220 | 42 | 5400 | 1.21 |
| 9 (ref. 30) | PhotoRAFT | Batch | n-Butyl acrylate | 3000 | 46 | 20700 |
1.12 |
| 10 (ref. 31) | PhotoRAFT | Flow | tert-Butyl acrylate | 60 | 81 | 29800 |
1.21 |
| 13 (ref. 32) | NMP2 | Batch | Methyl methacrylate | 40 | 58 | 20100 |
1.36 |
For methyl acrylate, 79% conversion is reached within 20 minutes reaction time in a photoflow reactor with good yield quality. Thus, all three systems display large efficiency gains. The absolute rate obviously also depends on the chosen light source. Yet, as in the above discussed reports similar lamps were employed, the conclusion can be made that copper is the best photo-mediator, at least when polymerizing (meth)acrylate monomers. Still, it needs to be noted that not only the rate increase is of significance. As mentioned above, flow processing gives access to significant amounts of products, while batch processing usually does not yield much more than products on the lower gram scale. The third reversible termination method, NMP, has not been carried out in a photoflow reactor yet. Nevertheless, NMP2 (nitroxide-mediated photopolymerization) exhibits by far the fastest polymerization rate in batch. It is hence a matter of priority to study also this method under photoflow conditions.
Photo-induced degenerative transfer methods
The best known degenerative transfer RDRP is RAFT polymerization. RAFT is very versatile and features the advantage that it is driven by conventional radical initiators, and as such is inherently photoactivatable under any condition (unlike reversible termination, where specific reaction conditions need to be established). RAFT is controlled by di- or trithiocarbonyl endgroups. Until today, research on RAFT was mainly focused on thermal initiated reactions in batch33 as well as in flow reactors.34 However, photoinitiated RAFT is long known.30,35,36 Consequently, several methods exist to drive RAFT polymerization by use of classical initiators or via use of photoelectron transfer reactions (PET-RAFT).37 In here, however, we refer to photoRAFT as technique in which the RAFT agent itself absorbs UV light and triggers polymerization, thus in absence of any photoinitiator system. Recently, Chen and Johnson translated such iniferter reaction38 from batch to a flow process.31 Also here, a dramatic increase in reaction rate for acrylate polymerization (see Table 1) is observed, without loss of control over the reaction. Compared to the metal-mediated methods the photoRAFT process so far allowed for synthesis of the largest molecular weights. Molar masses up to 100000 g mol−1 are obtained,31 for Cu and Ir only lower masses up to 10000 g mol−1 are reported,26,29 hinting at some limitations of the reversible termination type reactions, even though further research is probably required to confirm this difference. Overall, photoRAFT is complex, and also here a distinct need is identified for further detailed studies. PET-RAFT, or photoinitiator-driven RAFT may be as favourable, yet, no data is available for such systems (even in batch only limited information is yet available) and their potential remains currently unexplored.
Conclusion and future outlook
PhotoRDRP has only recently received significant attention, even in classical batch chemistry. The interest in these methods is rapidly growing, owing to the high efficiencies that can be achieved with ease. As described above, the reaction efficiencies can be further significantly increased when switching to photoflow processes. Photoflow offers additionally the engineering advantage that products can be synthesized in significant amounts without the requirement of expensive equipment. All above described reactions can be carried out in simple tubular reactors consisting only of a light source, translucent tubing and a HPLC pump. Reaction mixtures can be kept in the dark before injection, thus mixers are often not even required.With the eminent advantages of photoflow it may be speculated that most laboratories will in future switch (or extend) to flow processing. This not only due to the reasons above, but also because flow offers general benefits to precision polymer design. First studies are already carried out in which complex macromolecular architectures such as star, brush or cyclic polymers are also targeted. Further, flow processes can be conveniently combined with (dark) thermal reactions, thus allowing for sequential modification. Also λ-orthogonal reactions may in future come in the focus of photoflow material design, expanding the tool box from RDRP also to the realm of photoclick reactions.39
The combination of photoRDRP, photoclick and thermal polymer modification reactions (in conjunction with in-line purification) will give access to highly complex material synthesis virtually in one-step procedures. The polymer chemistry community – outside classical polymer reaction engineering – is only at the beginning of implementing flow techniques to the synthesis portfolio. Photoflow, itself even more so an infant research topic in the polymer community, will give further boost to flow adoption due to its striking benefits. With certainty, the above described data are only the beginning of a rapid development, and the full potential of photoflow precision polymer material design and synthesis will soon fully unfold.
Acknowledgements
The authors are grateful for funding from the Fonds Wetenschappelijk Onderzoek (FWO).References
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- M. T. Gokmen, B. Dereli, B. G. De Geest and F. E. DuPrez, Part. Part. Syst. Charact., 2013, 30, 438–444 CrossRef CAS.
- C. Tonhauser, A. Natalello, H. Löwe and H. Frey, Macromolecules, 2012, 45, 9551–9570 CrossRef CAS.
- P. Derboven, P. H. M. Van Steenberge, J. Vandenbergh, M.-F. Reyniers, T. Junkers, D. R. D'Hooge and G. B. Marin, Macromol. Rapid Commun., 2015 DOI:10.1002/marc.201500357.
- A. Nagaki, Y. Takahashi, K. Akahori and J.-I. Yoshida, Macromol. React. Eng., 2012, 6, 467–472 CrossRef CAS.
- E. Baeten, B. Verbraeken, R. Hoogenboom and T. Junkers, Chem. Commun., 2015, 51, 11701–11704 RSC.
- J. Vandenbergh, T. de Moraes Ogawa and T. Junkers, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2366–2374 CrossRef CAS.
- T. Noda, A. J. Grice, M. E. Levere and D. M. Haddleton, Eur. Polym. J., 2007, 43, 2321–2330 CrossRef CAS.
- N. Chan, M. F. Cunningham and R. A. Hutchinson, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3081–3096 CrossRef CAS.
- J. A. Burns, C. Houben, A. Anastasaki, C. Waldron, A. A. Lapkin and D. M. Haddleton, Polym. Chem., 2013, 4, 4809–4813 RSC.
- T. Fukuyama, Y. Kajihara, I. Ryu and A. Studer, Synthesis, 2012, 44, 2555–2559 CrossRef CAS.
- J. Vandenbergh, T. Tura, E. Baeten and T. Junkers, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1263–1274 CrossRef CAS.
- A. Anastasaki, V. Nikolaou, Q. Zhang, J. Burns, S. R. Samanta, C. Waldron, A. J. Haddleton, R. McHale, D. Fox, V. Percec, P. Wilson and D. M. Haddleton, J. Am. Chem. Soc., 2014, 136, 1141–1149 CrossRef CAS PubMed.
- D. Konkolewicz, K. Schröder, J. Buback, S. Bernhard and K. Matyjaszewski, ACS Macro Lett., 2012, 1, 1219–1223 CrossRef CAS.
- E. Yoshida, Colloid Polym. Sci., 2010, 288, 73–78 CAS.
- J. F. Quinn, L. Barner, C. Barner-Kowollik, E. Rizzardo and T. P. Davis, Macromolecules, 2002, 35, 7620–7627 CrossRef CAS.
- S. H. Qin and K. Y. Qiu, Eur. Polym. J., 2001, 37, 711–717 CrossRef CAS.
- M. A. Tasdelen, M. Uygun and Y. Yagci, Macromol. Rapid Commun., 2011, 32, 58–62 CrossRef CAS PubMed.
- L. D. Elliott, J. P. Knowles, P. J. Koovits, K. G. Maskill, M. J. Ralph, G. Lejeune, L. J. Edwards, R. I. Robinson, I. R. Clemens, B. Cox, D. D. Pascoe, G. Koch, M. Eberle, M. B. Berry and K. I. Booker-Milburn, Chem. – Eur. J., 2014, 20, 15226–15232 CrossRef CAS PubMed.
- J. P. Knowles, L. D. Elliott and K. I. Booker-Milburn, Beilstein J. Org. Chem., 2012, 8, 2025–2052 CrossRef CAS PubMed.
- B. D. A. Hook, W. Dohle, P. R. Hirst, M. Pickworth, M. B. Berry and K. I. Booker-Milburn, J. Org. Chem., 2005, 70, 7558–7564 CrossRef CAS PubMed.
- K. Gilmore and P. H. Seeberger, Chem. Rec., 2014, 14, 410–418 CrossRef CAS PubMed.
- M. Conradi and T. Junkers, Macromolecules, 2014, 47, 5578–5585 CrossRef CAS.
- M. Conradi and T. Junkers, J. Photochem. Photobiol., A, 2013, 259, 41–46 CrossRef CAS.
- A. Kermagoret, B. Wenn, A. Debuigne, C. Jerome, T. Junkers and C. Detrembleur, Polym. Chem., 2015, 6, 3847–3857 RSC.
- B. Wenn, M. Conradi, A. D. Carreiras, D. M. Haddleton and T. Junkers, Polym. Chem., 2014, 5, 3053–3060 RSC.
- Y.-M. Chuang, B. Wenn, S. Gielen, A. Ethirajan and T. Junkers, Polym. Chem., 2015, 6, 6488–6497 RSC.
- B. P. Fors and C. J. Hawker, Angew. Chem., Int. Ed., 2012, 51, 8850–8853 CrossRef CAS PubMed.
- A. Melker, B. P. Fors, C. J. Hawker and J. E. Poelma, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2693–2698 CrossRef CAS.
- Y.-Z. You, C.-Y. Hong, R.-K. Bai, C.-Y. Pan and J. Wang, Macromol. Chem. Phys., 2002, 203, 477–483 CrossRef CAS.
- M. Chen and J. A. Johnson, Chem. Commun., 2015, 51, 6742–6745 RSC.
- J. Su, X. Liu, J. Hu, Q. You, Y. Cui and Y. Chen, Polym. Int., 2015, 64, 867–874 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- C. H. Hornung, C. Guerrero-Sanchez, M. Brasholz, S. Saubern, J. Chiefari, G. Moad, E. Rizzardo and S. H. Thang, Org. Process Res. Dev., 2011, 15, 593–601 CrossRef CAS.
- T. Junkers, A. Theis, M. Buback, T. P. Davis, M. H. Stenzel, P. Vana and C. Barner-Kowollik, Macromolecules, 2005, 38, 9497–9508 CrossRef CAS.
- S. Muthukrishnan, E. H. Pan, M. H. Stenzel, C. Barner-Kowollik, T. P. Davis, D. Lewis and L. Barner, Macromolecules, 2007, 40, 2978–2980 CrossRef CAS.
- S. Shanmugam, J. Xu and C. Boyer, Macromolecules, 2014, 47, 4930–4942 CrossRef CAS.
- T. Otsu and M. Yoshida, Makromol. Chem., Rapid Commun., 1982, 3, 127–132 CrossRef CAS.
- A. S. Goldmann, M. Glassner, A. J. Inglis and C. Barner-Kowollik, Macromol. Rapid Commun., 2013, 34, 810–849 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |