
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rhodium-catalyzed oxidative amidation of allylic alcohols and aldehydes: effective conversion of amines and anilines into amides†
Zhao
Wu
 and
Kami L.
Hull
*
and
Kami L.
Hull
*
University of Illinois, Urbana-Champaign, Department of Chemistry, 600 S. Mathews, Urbana, IL 61820, USA. E-mail: kamihull@illinois.edu
First published on 27th October 2015
Abstract
The rhodium-catalyzed oxidative amidation of allylic alcohols and aldehydes is reported. In situ generated [(BINAP)Rh]BF4 catalyzes the one-pot isomerization/oxidative amidation of allylic alcohols or direct amidation of aldehydes using acetone or styrene as the hydrogen acceptor. The conditions are general, affording good to excellent yields with a wide array of amine and aniline nucleophiles, and chemoselective, other alcohols do not participate in the oxidation reaction. Utilization of biphasic conditions is critical, as they promote an equilibrium between the imine/enamine byproducts and the hemiaminal, which can undergo oxidation to the amide.
Introduction
Amides are a common functionality found throughout natural products, pharmaceuticals, agrochemicals, and organic materials.1 Approaches have been developed for the coupling of carboxylic acids and amines to generate amides in high yields.2 While these reactions are versatile and widely employed, there are significant drawbacks associated with them: they generate super-stoichiometric quantities of high molecular weight byproducts and are not functional group tolerant.3 The transition metal-catalyzed oxidative amidation of alcohols4 and aldehydes5 is a promising alternative to traditional coupling methods,6,7 as it allows for the generation of amides along with molecular hydrogen as the only by-product (Scheme 1). For example, Misltein and Madsen reported an acceptorless oxidative amidation of alcohols using Ru catalysis with pincer4c and NHC4e ligands, respectively. Although no hydrogen acceptor is needed, substrate scopes are limited to unhindered alcohols and amines. Secondary cyclic amines require a less-hindered pincer ligand8b and acyclic amines are still challenging.8 Moreover, elevated temperatures (>110 °C) and refluxing solvents are needed to favor the evolution of H2 gas4c–e,i–l or transfer hydrogenation to ketones.4f Direct amidation of aryl aldehydes with external oxidants have been reported by Rh5b and Cu5c catalysis. However, yields are usually low when enolizable aldehydes are used due to the formation of amine byproducts.5b Recently, Dong reported a Ni-catalyzed C–H activation of aldehydes followed by coupling with alcohols or amines to form esters and amides respectively.5i A variety of aldehydes and amines are shown to be reactive, but an additional equivalent aldehyde or trifluoroacetophenone is required as the hydrogen acceptor. Herein, we report an easily accessible, chemoselective, and general catalytic method, which selectively couples allylic alcohols or aldehydes with aliphatic and aryl amines to form amides.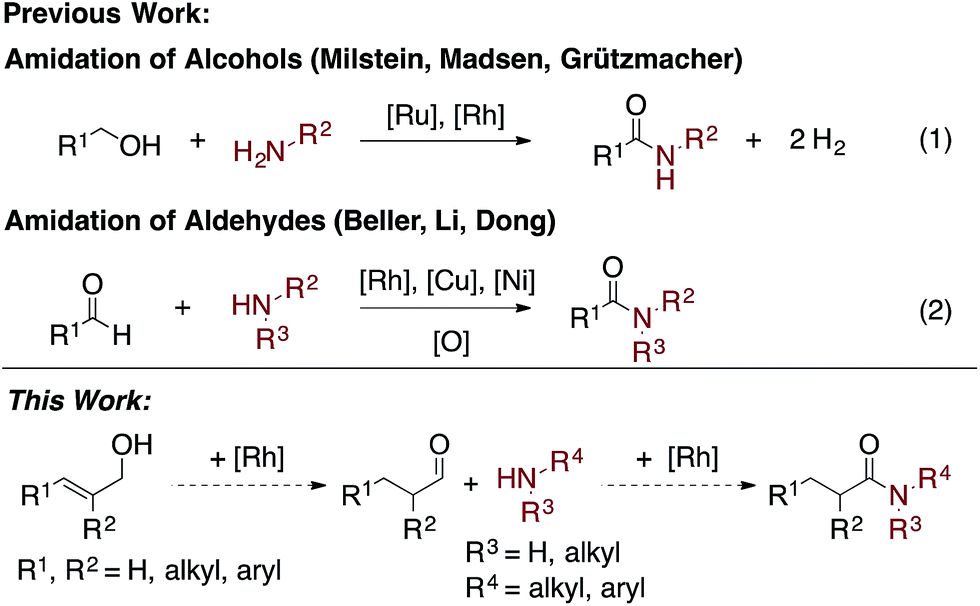 | ||
| Scheme 1 Transition metal-catalyzed oxidative amidation of alcohols and aldehydes. | ||
Results and discussion
Research hypothesis
In seeking to develop general conditions for amide bond formation, we proposed that allylic alcohols could serve as an aldehyde precursor.9,10 Such a system would be advantageous as allylic alcohols have been previously shown to isomerize to an aldehyde in the presence of a [Rh]-catalyst.10 Then, the in situ generated aldehydes could undergo a Rh-catalyzed oxidative amidation with an amine to afford the amide and a Rh–H. Subsequent H2 formation or transfer hydrogenation would regenerate the active catalyst species.4g,5bOptimization studies
Initial efforts focused on developing a Rh-catalyzed oxidative amidation of cinnamyl alcohol (1a) and N-methylpiperazine (2a). When they are combined with a cationic rhodium catalyst, the enamine (E)-1-methyl-4-(3-phenylprop-1-en-1-yl)piperazine (4a) predominates,12 suggesting that isomerization/condensation is significantly faster than the subsequent oxidation of the hemiaminal (vide infra). Moreover, reduced starting material 1h was also observed, indicating that the cinnamyl alcohol was acting as the hydrogen acceptor.12As such, two main challenges in optimization of the desired oxidative amidation reaction were to identify: (1) conditions that promote amide over enamine formation and (2) an oxidant that is selectively reduced over the allylic alcohol.
As summarized in Table 1, and further elaborated in Tables S1–S8,† a variety of reaction conditions were explored: varying solvent systems, bases, and oxidants. As enamine byproducts do not undergo the oxidative amidation, it was postulated that adding water to the reaction might promote reformation of the hemiaminal; indeed, the addition of an equal volume of H2O to non-polar solvents like benzene and toluene significantly suppressed the formation of the enamine 4a (Table S3†) and favored the formation of the amide 3a (Table 1, entries 1–4; Table S2†). Bases were next investigated: addition of CsOAc gave 56% yield of 3a; stronger bases lead to lower yields of the desired amide, with KOH and Et3N affording 3a in 24% and 41% yields with 1h as the major byproduct (Table 1, entry 5–8; Table S4†). Next, hydrogen acceptors were investigated; styrene proved to be superior, over acetone, affording 3a in 80% yield and 1h in 9% yield (Table 1, entry 11). Cyclohexanone, norbornene and MMA lead to lower yields of 3a (Table 1, entries 9–12; Table S4†). Finally, increasing the equivalents of styrene and CsOAc slightly improved yields and shortened the reaction time to 4 hours (Table 1, entries 13–16; Tables S5 and S7†).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Base | Oxidant | % yield 3a | % yield 1h |
| a Unless otherwise noted, reaction conditions are: allyl alcohol (0.25 mmol, 1.0 equiv.), amine (3.0 equiv.), base (1.5 equiv.), oxidant (3.0 equiv.), solvent (0.2 mL, 1.2 M), DI H2O (0.2 mL). Yields are determined by GC analysis and comparison to an internal standard. b MMA = methyl methacrylate. c 1.0 equiv. d 5.0 equiv. e 2.0 equiv. f 2.5 equiv. | |||||
| 1 | DME | None | Acetone | 9 | 0 |
| 2 | DME/H2O | None | Acetone | 14 | 0 |
| 3 | C6H6/H2O | None | Acetone | 42 | 20 |
| 4 | Tol/H2O | None | Acetone | 38 | 19 |
| 5 | C6H6/H2O | KOH | Acetone | 24 | 19 |
| 6 | C6H6/H2O | NEt3 | Acetone | 41 | 19 |
| 7 | C6H6/H2O | K2CO3 | Acetone | 51 | 24 |
| 8 | C6H6/H2O | CsOAc | Acetone | 56 | 22 |
| 9 | C6H6/H2O | CsOAc | Cyclohexanone | 63 | 22 |
| 10 | C6H6/H2O | CsOAc | Norbornene | 62 | 23 |
| 11 | C6H6/H2O | CsOAc | Styrene | 80 | 9 |
| 12 | C6H6/H2O | CsOAc | MMAb | 65 | 12 |
| 13 | C6H6/H2O | CsOAc | Styrenec | 68 | 20 |
| 14 | C6H6/H2O | CsOAc | Styrened | 91 | 5 |
| 15 | C6H6/H2O | CsOAce | Styrene | 84 | 10 |
| 16 | C6H6/H2O | CsOAcf | Styrene | 90 | 10 |

Interestingly, as seen in Table 2, under the optimized conditions benzyl amine gave only 11% yield of amide 3h and 89% yield of imine 4h (Table 2, entry 1). Fortunately, by changing the base and hydrogen acceptor to KOH and acetone, respectively, the desired amide 3h is formed in 82% yield along with only 6% yield of 1h (Table 2, entry 6).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Oxidant | % yield 3h | % yield 4h | % yield 1h |
| a Unless otherwise noted, reaction conditions are: allyl alcohol (0.25 mmol, 1.0 equiv.), amine (3.0 equiv.), base (1.5 equiv.), oxidant (3.0 equiv.), solvent (0.2 mL, 1.2 M), DI H2O (0.2 mL). Yields are determined by GC analysis and comparison to an internal standard. b 5.0 equiv. c No H2O added. | |||||
| 1 | CsOAc | Styrene | 11 | 89 | 0 |
| 2 | Cs2CO3 | Styrene | 50 | 30 | 20 |
| 3 | KOH | Styrene | 51 | 19 | 30 |
| 4 | KOH | Norbornene | 56 | 10 | 26 |
| 5 | KOH | NMO | 48 | 6 | 37 |
| 6 | KOH | Acetone | 82 | 0 | 6 |
| 7 | Cs2CO3 | Acetone | 60 | 11 | 18 |
| 8 | KOH | Acetoneb | 84 | 0 | 4 |
| 9c | KOH | Acetone | 15 | 3 | 0 |

Substrate scope
The optimization experiments suggest that the allylic alcohol rapidly converts into the aldehyde under the reaction conditions, which could then go on to form the amide. This suggested that under the optimized conditions, aldehydes should be effective substrates for the oxidative coupling reaction. Indeed, when 3-phenylpropanal (5a) is subjected to the optimized conditions amide 3a is formed in a 76% yield, which is nearly identical to the 81% yield from the cinnamyl alcohol (Table 3). The scope of amines was then explored with both 1a and 5a (Table 3). Secondary cyclic amines, including 1-methylpiperazine, morpholine, piperidine, pyrrolidine, tetrahydroisoquinoline (2a–2e) and more sterically hindered acyclic amines, such as dimethyl amine (2f) and N-benzyl methyl amine (2g) are incorporated in very good, and nearly equivalent, yields from either the 1a or 5a. cis-Cinnamyl alcohol also undergoes the amidation reaction with morpholine to afford 3b, although at a significantly reduced rate and in moderately reduced yields.12 Likewise, primary amines afforded 3h–3l in good to excellent yields.| a Alcohol or aldehyde (0.25 mmol, 1.0 equiv.), amine (3.0 equiv.), [(COD)2Rh]BF4 (3.0 mol%), BINAP (3.0 mol%), oxidant (3.0–5.0 equiv.): styrene (2° amine and aniline) or acetone (1° amine), base (1.5–2.5 equiv.), benzene (0.2 mL, 1.2 M), DI H2O (0.2 mL).12 b 82% yield was observed from (Z)-cinnamyl alcohol.12 |
|---|

|
The reaction is sensitive to steric hindrance of the amine, as cyclohexylamine affords 54%/48% yield of amide 3j while n-butylamine affords amide 3i in 72%/63% yield. Unlike other oxidative amidation processes,4a,d–l electron rich and electron poor aniline derivatives (2m–2r) were all effective nucleophiles in the coupling reaction without requiring higher temperatures or specialized reaction conditions.11 It is important to note, that for all substrates, the allylic alcohol and the aldehyde gave similar yields.
The allylic alcohols that undergo the oxidative amidation reaction were explored, as seen in Table 4. Functional groups such as ketones, esters, ethers, aryl bromides, and chlorides are tolerant under the standard conditions based on a chemical robustness screen.12 Products bearing active aryl bromides could be synthesized in good yields (3ba, 3ha), allowing for facile subsequent coupling reactions to increase molecule complexity. 1,1-Di, 1,2-di-, tri-, and tetra-substituted allylic alcohols give the corresponding α- or/and β-branched secondary (6i–6p) and tertiary (6a–6h) amides in moderate to good yields with primary and secondary amines, respectively. Notably, allylic alcohols known to be slow to isomerize, such as cyclohex-1-en-1-ylmethanol (1g) afforded moderate yields (54%) of the oxidative amidation product 6f after 24 hours.10c Notably, geraniol was coupled to afford 6g and 6o in 64% and 65% yield, respectively; neither reduction nor isomerization of the distal alkene was observed.

Tetra-substituted allylic alcohols react, affording 6h and 6p in 41% and 31% yield, respectively, along with unreacted starting material. Unfortunately, the diastereoselectivity of these reactions was poor, affording the amides in only 1.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:1 dr.
:1 and 1:1 dr.
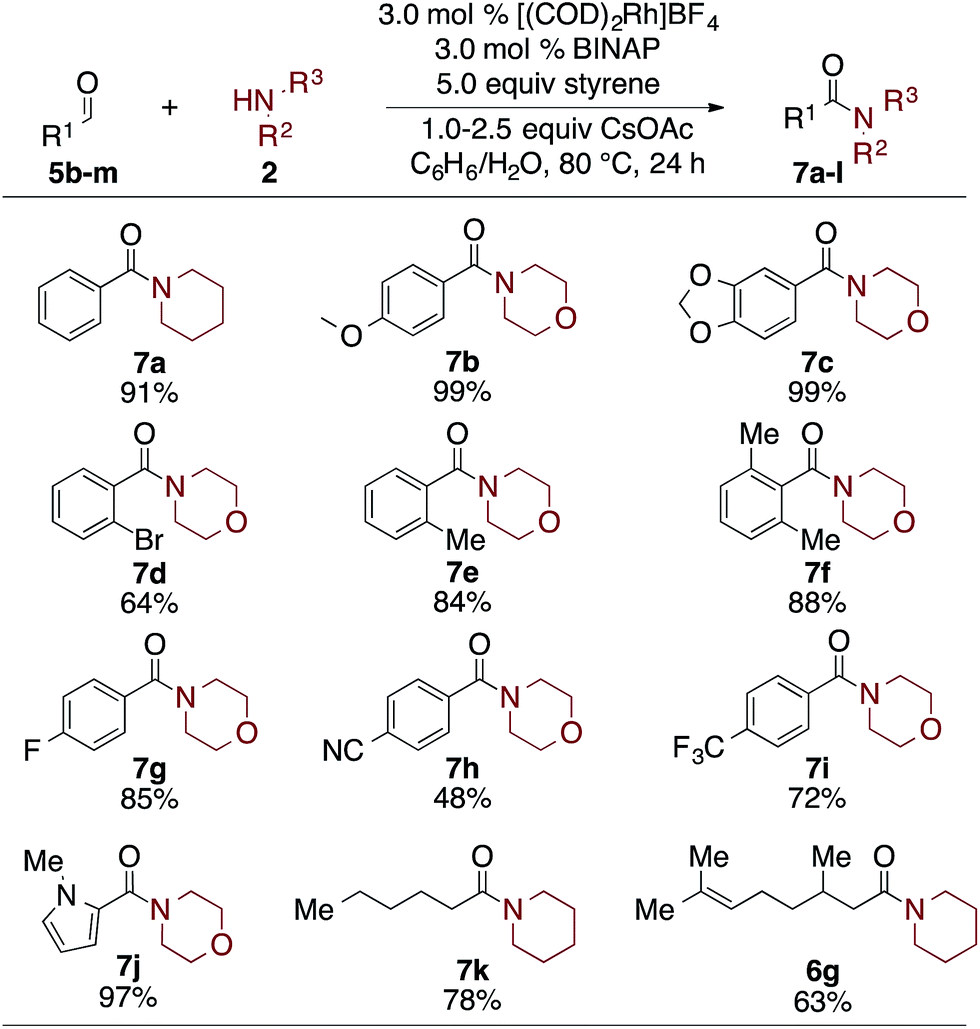
Finally, as seen in Table 5, the scope of aldehydes that undergo the oxidative amidation reaction was investigated. The reactions are tolerant of a variety of functionalities, including ethers (7b), acetals (7c), aryl bromides (7d), aryl fluorides (7g), trifluromethyls (7i), nitriles (7h), and heteroaromatics (7j). Benzaldehyde derivatives with electron donating groups, such as p-MeO, afford the desire amide in excellent yield; electron poor benzaldehydes, such as p-CN or p-F3C, undergo the oxidative amidation to afford 7h and 7i, albeit in reduced yields. Steric hindrance of the aldehyde did not affect its reactivity, as 2,6-dimethylbenzaldehyde afforded amide 7f in 88% yield. Aliphatic aldehydes, which have proven challenging for other oxidative amidation reactions,5a–h also afford the desired amides in good to very good yields (7k and 6g).
Competition experiments
The synthetic utility of this oxidative amidation reaction would be significantly increased if the reaction proved to be chemoselective for allylic alcohols and aldehydes over other oxidizable functionalities, i.e., simple primary alcohols. To explore the chemoselectivity of the reaction conditions, a series of competition studies were carried out (Scheme 2). | ||
| Scheme 2 Competition experiments. a Standard conditions are: alcohol/aldehyde 1 (1.0 equiv.), alcohol/aldehyde 2 (1.0 equiv.), 2a (3.0 equiv.), [(COD)2Rh]BF4 (3.0 mol%), BINAP (3.0 mol%), styrene (3.0 equiv.), CsOAc (2.5 equiv.), benzene (0.2 mL, 1.2 M), DI H2O (0.2 mL), 80 °C, 4 hours. b Benzyl 3-phenylpropanoate was formed in 7% yield. c Hexyl 3-phenyl-propanoate was formed in 8% yield. | ||
In competition experiment 1, cinnamyl alcohol (1a) competes against hexanal (5k) to compare the relative rates of the two coupling partners. Unsurprisingly, given the rapid rate of 1,3-hydride shift,10c the reaction is unselective, affording a 33% yield of 3a and a 25% yield of 8 after four hours. This lack of selectivity supports the rapid Rh-catalyzed isomerization of the cinnamyl alcohol to the corresponding aldehyde; the oxidative amidation of the two aliphatic aldehydes then occurs at similar rates.
Next, in competition experiment 2, the comparative reactivity of two different allylic alcohols was explored. Under the standard reaction conditions, cinnamyl alcohol (1a) reacts selectively over cyclohex-1-en-1-ylmethanol (1g). This excellent chemoselectivity is consistent with the known rates of the Rh-catalyzed isomerization of allylic alcohols.10c Importantly, 1g was observed, unisomerized, at the end of the reaction.
Competition experiments 3–5 investigate the chemo-selectivity of the oxidative amidation reaction with respect to benzylic alcohols, homoallylic alcohols, and aliphatic alcohols. When equimolar amounts of cinnamyl alcohol (1a) and benzyl alcohol were treated with 3.0 equivalents of 2a under the standard reaction conditions, product 3a was formed in 93% yield while 10 was not observed. This indicates that our conditions are highly selective for allylic alcohols over easily oxidizable benzylic alcohols.4h,m Moreover, in competition experiment 4, less than 5% amide product 11 was observed when cinnamyl alcohol and 3-buten-1-ol are subjected to the reaction conditions, which afforded an 82% yield of 3a. Notably, 3-buten-1-ol affords <5% yield of 11 under the standard reaction conditions, in the absence of cinnamyl alcohol.12 Finally, competition experiment 5 demonstrates that cinnamyl alcohol (1a) reacts selectively over hexan-1-ol (12), to afford an 88% yield of 3a; 8 was not observed. These experiments exhibit the excellent chemoselectivity of this Rh-catalyzed oxidative amidation reaction for coupling allylic alcohols and aldehydes selectively.
Mechanistic investigations
Next, the requirement of excess amine and super-stoichiometric base was investigated. When a equimolar amounts of amine and allyl alcohol/aldehyde are added to the reaction the desired amide is formed in only 20% yield along with 13% enamine along with both unreacted aldehyde and aldol condensation products (Table S6,† entry 1). However, in the presence of excess amine excellent yields of the amide are observed. It is proposed that the excess amine forces the equilibrium, between aldehyde and enamine/imine, to the enamine/imine thereby decreasing the concentration of aldehyde and preventing the undesired aldol condensation from occurring. The base additive, CsOAc or KOH, could serve two possible roles, either acting as a proton shuttle or as a ligand on the catalyst. The pH of the water layer in the oxidative amidation reactions is 9.2; when the CsOAc solution is replaced with various buffers (pH = 5.2, 7.5, or 9.0) the reaction are equally efficient as to those in the absence of base (Table S9†). This suggests that the base is acting as a ligand to the catalyst and that the requirement of excess base is due to the solubility differential of the anion in water versus benzene.To gain further mechanistic insight into this reaction, we conducted an isotope incorporation experiment, replacing H2O with D2O. After the reaction had gone to completion, 83% deuterium was incorporated at the α-position of the product (Scheme 3). Importantly, in the absence of catalyst and N-methylpiperazine, no deuterium incorporation is observed into amide 3a or 3-phenylpropanal. This suggests that water reacts with the enamine/imine to reform the reactive hemiaminal,13 which is different from many Ru-catalyzed dehydrogenative coupling of alcohols with amines, where in situ formed aldehydes stay bounded to metal center to avoid the imine formation.4c–f,i–k Comparison of the initial rate of the reaction in H2O and D2O revealed a kH/kD = 2.2, indicating the enamine–hemiaminal equilibrium occurs at or before the turnover-limiting step. Alternatively, the primary k.i.e. could be attributed to cleavage of N–H(D) bond in hemiaminal formation, due to the exchange of amine proton with D2O.
 | ||
| Scheme 3 Deuterium studies. | ||
The proposed dual catalytic cycles for this oxidative amidation reaction are shown in Scheme 4. First, a Rh-mediated 1,3-hydride shift10 occurs to form an aldehyde from the allylic alcohol. The aldehyde then condenses with the amine to generate the enamine/imine, which is in equilibrium with the hemiaminal. The oxidation of the hemiaminal could occur through either a Rh(I)/Rh(III) or a redox neutral Rh(I) catalytic cycle. In the first, oxidative addition of the hemiaminal OH into the Rh(I) generates a Rh(III) (H)OCHNR2 (III). Subsequent β-hydride elimination generates the amide and a Rh(III)–(H)2 (IV) complex which is reduced to Rh(I) through hydrogenation of the styrene or acetone. Alternatively, in a redox neutral catalytic cycle, ligand exchange of the [Rh] (I) with the hemiaminal affords the Rh–alkoxide (III) then undergoes β-hydride elimination to generate the amide and the Rh–H (IV). Insertion of the hydride into styrene/acetone followed by protolytic cleavage or ligand exchange affords the ethyl benzene/isopropanol, respectively, and either [Rh] complex I or III. Currently, we can not distinguish between the two possible mechanistic pathways and further mechanistic investigations are ongoing.14
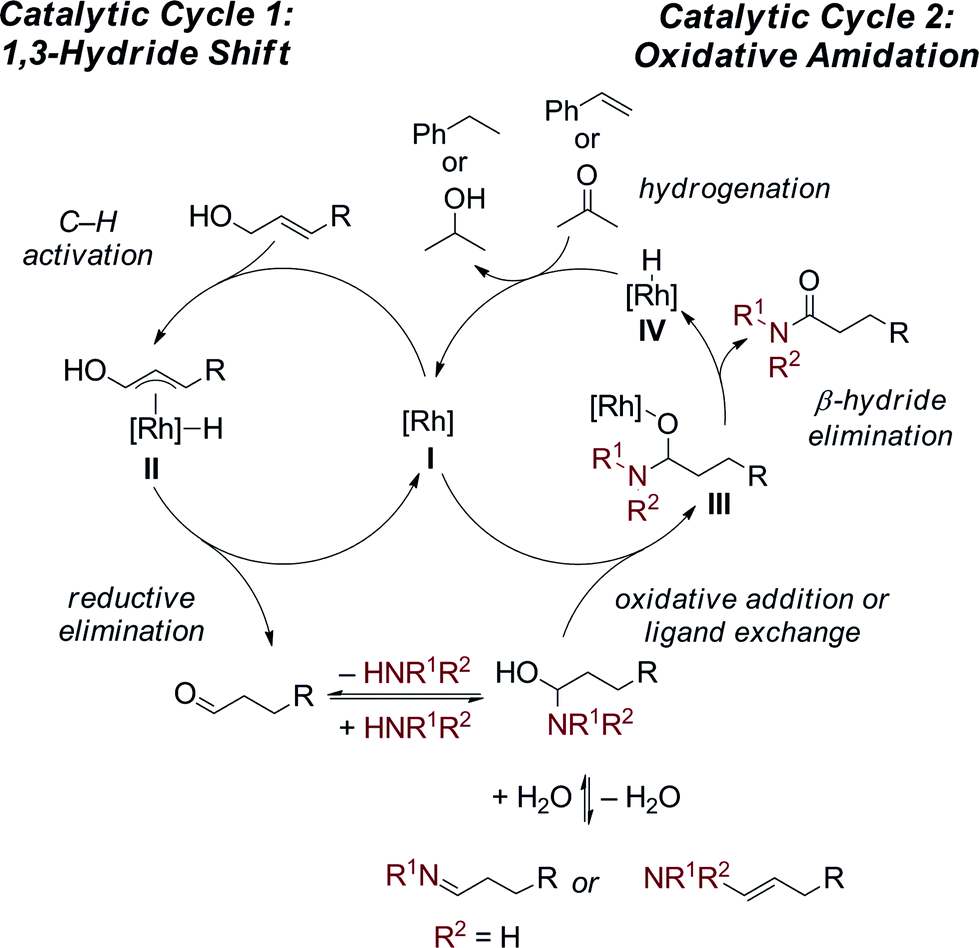 | ||
| Scheme 4 Proposed dual catalytic cycle. | ||
Conclusions
In conclusion, conditions have been developed for the chemoselective oxidative amidation of allylic alcohols or aldehydes, using styrene or acetone as hydrogen acceptors. This methodology presents a general protocol for the synthesis of amides, which is effective for both primary and secondary alkyl/aryl amines. Current efforts are focusing on expanding the scope of nucleophiles and developing asymmetric conditions15 for the transformation.Acknowledgements
The authors would like to thank the University of Illinois, Urbana-Champaign for their generous support.Notes and references
- (a) J. Zabicky, The Chemistry of Amides, Wiley-VCH, New York, 1970 Search PubMed; (b) A. Greenberg and C. M. Breneman, The Amide Linkage: Structural Significance in Chemistry, Biochemistry and Material Science, Wiley-VCH, New York, 2000 Search PubMed; (c) J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337 RSC.
- For a review: E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606 RSC.
- (a) I. Coin, M. Beyermann and M. Bienert, Nat. Protoc., 2007, 2, 3247 CrossRef CAS PubMed; (b) D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer Jr, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411 RSC.
- From alcohols: (a) T. Naota and S. I. Murahashi, Synlett, 1991, 10, 693 CrossRef; (b) K. Fujita, Y. Takahashi, M. Owaki, K. Yamamoto and R. Yamaguchi, Org. Lett., 2004, 6, 2785 CrossRef CAS PubMed; (c) C. Gunanathan, Y. Ben-David and D. Milstein, Science, 2007, 317, 790 CrossRef CAS PubMed; (d) N. A. Owston, A. J. Parker and J. M. J. Williams, Org. Lett., 2007, 9, 73 CrossRef CAS PubMed; (e) L. U. Nordstrom, H. Vogt and R. Madsen, J. Am. Chem. Soc., 2008, 130, 17672 CrossRef CAS PubMed; (f) A. J. A. Watson, A. C. Maxwell and J. M. J. Williams, Org. Lett., 2009, 11, 2667 CrossRef CAS PubMed; (g) T. Zweifel, J.-V. Naubron and H. Grützmacher, Angew. Chem., Int. Ed., 2009, 48, 559 CrossRef CAS PubMed; (h) K.-I. Shimizu, K. Ohshima and A. Satsuma, Chem.–Eur. J., 2009, 15, 9977 CrossRef CAS PubMed; (i) S. C. Ghosh, S. Muthaiah, Y. Zhang, X. Xu and S. H. Hong, Adv. Synth. Catal., 2009, 351, 2643 CrossRef CAS; (j) Y. Zhang, C. Chen, S. C. Ghosh, Y. Li and S. H. Hong, Organometallics, 2010, 29, 1374 CrossRef CAS; (k) S. Muthaiah, S. C. Ghosh, J.-E. Jee, C. Chen, J. Zhang and S. H. Hong, J. Org. Chem., 2010, 75, 3002 CrossRef CAS PubMed; (l) J. H. Dam, G. Osztrovszky, L. U. Nordstrom and R. Madsen, Chem.–Eur. J., 2010, 16, 6820 CrossRef CAS PubMed; (m) Y. Wang, D. Zhu, L. Tang, S. Wang and Z. Wang, Angew. Chem., Int. Ed., 2011, 50, 8917 CrossRef CAS PubMed.
- From aldehydes: (a) Y. Tamaru, Y. Yamada and Z.-I. Yoshida, Synthesis, 1983, 474 CrossRef CAS; (b) A. Tillack, I. Rudloff and M. Beller, Eur. J. Org. Chem., 2001, 523 CrossRef CAS; (c) W.-J. Yoo and C.-J. Li, J. Am. Chem. Soc., 2006, 128, 13064 CrossRef CAS PubMed; (d) J. W. W. Chang and P. W. H. Chan, Angew. Chem., Int. Ed., 2008, 47, 1138 CrossRef CAS PubMed; (e) S. Y. Seo and T. J. Marks, Org. Lett., 2008, 10, 317 CrossRef CAS PubMed; (f) ref. 4i ; (g) S. C. Ghosh, J. S. Y. Ngiam, A. M. Seayad, D. T. Tuan, C. L. L. Chai and A. Chen, J. Org. Chem., 2012, 77, 8007 CrossRef CAS PubMed; (h) Y. Li, F. Jia and Z. Li, Chem.–Eur. J., 2013, 19, 82 CrossRef CAS PubMed; (i) A. M. Whittaker and V. M. Dong, Angew. Chem., Int. Ed., 2015, 54, 1312 CrossRef CAS PubMed.
- For recent reviews, see: (a) G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681 CrossRef CAS PubMed; (b) V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471 CrossRef CAS PubMed; (c) C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405 RSC; (d) C. Singh, V. Kumar, U. Sharma, N. Kumar and B. Singh, Curr. Org. Synth., 2013, 10, 241 CrossRef CAS; (e) P. Crochet and V. Cadierno, Top. Organomet. Chem., 2014, 48, 81 CrossRef.
- For related NHC catalyzed amidation see: (a) H. U. Vora and T. Rovis, J. Am. Chem. Soc., 2007, 129, 13796 CrossRef CAS PubMed; (b) J. W. Bode and S. S. Sohn, J. Am. Chem. Soc., 2007, 129, 13798 CrossRef CAS PubMed; (c) S. D. Sarkar and A. Studer, Org. Lett., 2010, 12, 1992 CrossRef PubMed.
- Formation of tertiary amides: (a) C. Chen, Y. Zhang and S. H. Hong, J. Org. Chem., 2011, 76, 10005 CrossRef CAS PubMed; (b) D. Srimani, E. Balaraman, P. Hu, Y. Ben-David and D. Milstein, Adv. Synth. Catal., 2013, 355, 2525 CrossRef CAS.
- Transition metal-catalyzed functionalization of allylic alcohol: (a) K. Hirai, Y. Takahashi and I. Ojima, Tetrahedron Lett., 1982, 23, 2491 CrossRef CAS; (b) Y. Watanabe, Y. Tsuji, Y. Ohsugi and J. Shida, Bull. Chem. Soc. Jpn., 1983, 56, 2452 CrossRef CAS.
- Rh-catalyzed allylic alcohol isomerization: (a) K. Tanaka and G. C. Fu, J. Org. Chem., 2001, 66, 8177 CrossRef CAS PubMed; (b) K. Tanaka, S. Qiao, M. Tobisu, M. M.-C. Lo and G. C. Fu, J. Am. Chem. Soc., 2000, 122, 9870 CrossRef CAS; (c) S. H. Bergens and B. Bosnich, J. Am. Chem. Soc., 1991, 113, 958 CrossRef CAS; (d) K. Tani, Pure Appl. Chem., 1985, 57, 1845 CrossRef CAS.
- Anilines have been demonstrated to undergo oxidative amidation with heterogeneous gold catalysts, see ref. 4m.
- See ESI†.
- Amide formation using H2O as the oxygen atom source: (a) J. R. Khusnutdinova, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2014, 136, 2998 CrossRef CAS PubMed; (b) U. Gellrich, J. R. Khusnutdinova, G. M. Leitus and D. Milstein, J. Am. Chem. Soc., 2015, 137, 4851 CrossRef CAS PubMed.
- When the reaction is monitored by both 1H and 31P NMR no Rh–H are observed. This suggests that either oxidative addition or β-hydride elimination is between the turnover limiting step and the catalyst resting state if the reaction is occurring through the Rh(I)/(III) or Rh(I) catalytic cycles, respectively.
- Under standard condition, reaction of prochiral (E)-3-phenylbut-2-en-1-ol with morpholine affords 1-morpholino-3-phenylbutan-1-one in 86% yield and 3:1 er by using R-BINAP as chiral ligand. For comparison, [((R)-BINAP)Rh]ClO4 catalyzes the asymmetric isomerization of (E)-3-phenylbut-2-en-1-ol to afford the β-chiral aldehyde in 70% yield and 3.3:1 er. See ref. 10d.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc03103f |
| This journal is © The Royal Society of Chemistry 2016 |