
Graphene-directed two-dimensional porous carbon frameworks for high-performance lithium–sulfur battery cathodes†
Jieqiong
Shan‡
a,
Yuxin
Liu‡
a,
Yuezeng
Su
b,
Ping
Liu
*c,
Xiaodong
Zhuang
c,
Dongqing
Wu
*c,
Fan
Zhang
c and
Xinliang
Feng
d
aSchool of Aeronautics and Astronautics, Shanghai Jiao Tong University, 800 Dongchuan RD, Shanghai 200240, P. R. China
bSchool of Electronic Information and Electrical Engineering, Shanghai Jiao Tong University, 800 Dongchuan RD, Shanghai 200240, P. R. China
cSchool of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, 800 Dongchuan RD, Shanghai 200240, P. R. China. E-mail: liupingsjj@sjtu.edu.cn; wudongqing@sjtu.edu.cn
dDepartment of Chemistry and Food Chemistry (cfaed), Technische Universität Dresden, Mommsenstrasse 4, 01062, Germany
First published on 17th November 2015
Abstract
Graphene-directed two-dimensional (2D) nitrogen-doped porous carbon frameworks (GPF) as the hosts for sulfur were constructed via the ionothermal polymerization of 1,4-dicyanobenzene directed by the polyacrylonitrile functionalized graphene nanosheets. As cathodes for lithium–sulfur (Li–S) batteries, the prepared GPF/sulfur nanocomposites exhibited a high capacity up to 962 mA h g−1 after 120 cycles at 2 A g−1. A high reversible capacity of 591 mA h g−1 was still retained even at an extremely large current density of 20 A g−1. Such impressive electrochemical performance of GPF should benefit from the 2D hierarchical porous architecture with an extremely high specific surface area, which could facilitate the efficient entrapment of sulfur and polysulfides and afford rapid charge transfer, fast electronic conduction as well as intimate contact between active materials and the electrolyte during cycling.
Introduction
Motivated by the ever-growing requirement for advanced batteries, lithium–sulfur (Li–S) batteries with high theoretical energy density, cost effectiveness and environmental benignity have been lately regarded as a promising successor to lithium-ion batteries (LIBs).1–5 Although the concept of Li–S batteries emerged in the 1960s, their large-scale commercial applications have long been precluded by the following factors: (i) fast capacity fade and unsatisfactory cycle life due to the diffusion of polysulfide (Li2Sn) species in the electrolyte; (ii) the low utilization rate of active materials for the insulating nature of both sulfur and lithium sulfides (Li2S and Li2S2).2,5To address the above-mentioned problems, numerous novel sulfur cathodes including nanoporous carbon–sulfur composites,6–8 graphene–sulfur composites,9–12 one dimensional (1D) carbon–sulfur composites,13–16 polymer–sulfur composites,17,18 and inorganic–sulfur composites (metal oxides,19,20 metal–organic frameworks,21 and mesoporous molecular sieves22) have been extensively explored. Among these materials, nanostructured porous carbons are confirmed to be the most promising hosts for sulfur in Li–S batteries. Given that nanostructured porous carbons can improve the utilization of an active material by keeping sulfur particles nanometer-sized and electrically connected in their well-defined porous structure with intrinsic high electronic conductivity;23 meanwhile, they are able to adsorb sulfur and polysulfides on their large internal surfaces and mitigate polysulfide diffusion.24 Moreover, compared to zero-dimensional (0D) or one-dimensional (1D) counterparts, two-dimensional (2D) porous carbon materials provide large surface-to-volume ratios for better contact between active materials and the electrolyte, continuous pathways for electron conduction, short distance for charge transfer, and enormous potential in tuning the pore and channel configuration with a large specific surface area.25,26 The rational design and construction of 2D porous carbons for energy-storage devices have attracted extensive interest and aroused favorable results.27–30 However, developing effective templates to control the formation of 2D porous carbon frameworks remains a challenge for researchers. In this respect, functionalized graphene nanosheets such as graphene oxide (GO), reduced graphene oxide (RGO) and other graphene derivatives containing diversified functional groups are believed to be qualified templates for the construction of 2D porous carbon materials since they can direct the synthesis of porous polymers in a 2D manner, which can be further converted to porous carbons via appropriate thermal treatments.23,31,32
Recent research studies of graphene-based sulfur cathodes mostly focused on graphene-coated sulfur nanoparticle composites33 and sandwich-like graphene (oxide)–sulfur composites.34,35 Nevertheless, further applications might be restricted due to the hindering of lithium ion transportation by the graphene lattice. Moreover, the conductivity of graphene oxide greatly depends on its oxidation degree.5 Therefore, it is constructive to fabricate graphene-based 2D porous carbons as the sulfur host, which will combine the merits of 2D materials with the advantages of hierarchical porous carbons. The strategy aims at realizing efficient entrapment of sulfur and polysulfides as well as facilitating electron and charge transportation to improve the cell performance.
Herein, we present the graphene-directed 2D nitrogen-doped porous carbon frameworks (denoted as GPF-n, n = 1, 2, and 3) as sulfur hosts in Li–S batteries. The 2D porous structure with a large specific surface area of up to 1683 m2 g−1 benefits the intimate contact between active materials and the electrolyte and provides short pathways for charge transfer.23,36 The hierarchically micro-/meso-porous carbon doped by nitrogen facilitates the chemical adsorption of polysulfide by enhancement of the surface electronegativity.37 As the cathode material in Li–S batteries, the nanocomposite of GPF-3 and sulfur (GPF-S-3) manifested an initial specific capacity of 1461 mA h g−1 and a stabilized capacity of 962 mA h g−1 after 120 discharge/charge cycles at a large current density of 2 A g−1. It also exhibited an excellent rate performance by retaining a high reversible capacity of 591 mA h g−1 even at an extremely large current density of 20 A g−1.
Experimental section
Materials
Flake graphite was purchased from Aldrich. Acrylonitrile was of industrial polymerization grade and purchased from Xiya Reagent Co. (Chengdu, China) and used without further purification. Organic solvents were purified, dried, and distilled under dry nitrogen. All the other chemicals were purchased from Aladdin Reagent (Shanghai) and used without further purification.Preparation of GPF-n and PF
Graphene oxide (GO) was synthesized from natural graphite flakes by a modified Hummers method and then reduced by hydrazine hydrate to obtain RGO. The preparation of polyacrylonitrile functionalized graphene nanosheet (RGO-PAN) templates was based on our previous report.31 GPF-n was synthesized through a classic ionothermal polymerization in molten ZnCl2.32,38 The mixtures of as-prepared RGO-PAN (30 mg), 1,4-dicyanobenzene (pDCB, 300 mg) and ZnCl2 (3.2 g) in quartz ampoules were heated to 400 °C for 40 h, 600 °C for 20 h, or 400 °C for 20 h and then 600 °C for 20 h. The resulting products were denoted as GPF-1, GPF-2 and GPF-3, respectively. A nitrogen-doped porous carbon framework (PF) was prepared as a control sample by heating to 400 °C for 40 h without using RGO-PAN.Preparation of GPF-S-n and PF-S nanocomposites
Based on a facile melt-diffusion method, GPF-n (n = 1, 2, and 3) and PF were mixed and ground separately with a certain amount of sublimed sulfur and loaded into a closed container before being heated in a muffle furnace at 155 °C for 12 h (denoted as GPF-S-n and PF-S). The theoretical maximal amounts of sulfur that GPF-n and PF can load were calculated according to the results from N2 adsorption/desorption isothermal analysis and by the following method: the loading amount of sulfur = weight of the sample × its total pore volume × density of lithium sulfide (1.66 g cm−3) × the weight ratio of sulfur in lithium sulfide (69.78%). Next, GPF-1, GPF-2, GPF-3, and PF were hybridized with sulfur to obtain GPF-S-1, GPF-S-2, GPF-S-3 and PF-S with sulfur proportions of 50%, 75%, 65% and 43%, respectively.Characterization
The morphology and elemental mapping information of the samples were characterized by scanning electron microscopy (SEM, Sirion 200, 25 kV) and transmission electron microscopy (TEM, JEOL JEM-2010, 200 kV). X-ray diffraction (XRD) measurements were executed on a D/max-2200/PC (Rigaku Corporation, Japan) using Cu (40 kV, 30 mA) radiation. Raman spectra were recorded on a SENTERRA with a 532 nm excitation of an Ar-ion laser with a power of about 5 mW. N2 adsorption was measured with a Micromeritics ASAP 2010 analyzer at 77 K. The Brunauer–Emmett–Teller (BET) method and density functional theory (DFT) pore model were utilized to calculate the specific surface area and pore size distribution. Thermogravimetric analysis (TGA) of the samples was performed with a Q5000IR (TA Instruments, USA) thermogravimetric analyzer at a heating rate of 20 °C min−1 under nitrogen. X-ray photoelectron spectroscopy (XPS) measurements were carried out on an AXIS Ultra DLD system from Kratos with Al Kα radiation as the X-ray source.Electrochemical measurements
Each sample was mixed with carbon black (Super-P), and a polyvinylidene fluoride (PVDF) binder in 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 ratio in 1-methyl-2-pyrrolidone (NMP) solvent (Aldrich, 99.5%) then cast onto Al foil before drying in a vacuum oven at 60 °C for 12 h. All electrochemical performances were measured by using CR2016-type coin cells assembled in an argon-filled glovebox (M. BarunGlvebox) with the test electrodes by using Celgard 2400 as the separator. The electrolyte was lithium bis-(trifluoromethanesulfonyl)imide (LiTFSI, 1 M) with 0.1 M LiNO3 in a 1:1 (v/v) mixture of 1,3-dioxolane (DOL) and 1,2-dimethyl carbonate (DMC). Electrochemical charge/discharge experiments were performed using a battery cycler (LAND-CT2001A) with current rates from 0.5 to 20 A g−1 in the voltage range of 1.7–2.8 V. The cyclic voltammograms were obtained over the potential range of 1.7–2.8 V at a scanning rate of 0.1 mV s−1. Electrochemical impedance spectroscopy (EIS) measurements of the electrodes were carried out on an electrochemical workstation (PARSTAT 2273). The impedance spectra were recorded by applying a sine wave with an amplitude of 5.0 mV over the frequency range from 100 kHz to 0.01 Hz. Fitting of the impedance spectra to the proposed equivalent circuit was performed by the code Z view.
:2:1 ratio in 1-methyl-2-pyrrolidone (NMP) solvent (Aldrich, 99.5%) then cast onto Al foil before drying in a vacuum oven at 60 °C for 12 h. All electrochemical performances were measured by using CR2016-type coin cells assembled in an argon-filled glovebox (M. BarunGlvebox) with the test electrodes by using Celgard 2400 as the separator. The electrolyte was lithium bis-(trifluoromethanesulfonyl)imide (LiTFSI, 1 M) with 0.1 M LiNO3 in a 1:1 (v/v) mixture of 1,3-dioxolane (DOL) and 1,2-dimethyl carbonate (DMC). Electrochemical charge/discharge experiments were performed using a battery cycler (LAND-CT2001A) with current rates from 0.5 to 20 A g−1 in the voltage range of 1.7–2.8 V. The cyclic voltammograms were obtained over the potential range of 1.7–2.8 V at a scanning rate of 0.1 mV s−1. Electrochemical impedance spectroscopy (EIS) measurements of the electrodes were carried out on an electrochemical workstation (PARSTAT 2273). The impedance spectra were recorded by applying a sine wave with an amplitude of 5.0 mV over the frequency range from 100 kHz to 0.01 Hz. Fitting of the impedance spectra to the proposed equivalent circuit was performed by the code Z view.
Results and discussion
To ensure efficient electrolyte penetration, fast ion transfer and intimate contact between active materials and the electrolyte, a hierarchically porous carbon was constructed on the reduced graphene oxide (RGO) substrates as schematically illustrated in Fig. 1. A series of GPF-n (n = 1, 2, and 3) samples were prepared by employing the polyacrylonitrile functionalized graphene nanosheets31 (RGO-PAN) as the template and 1,4-dicyanobenzene (pDCB) as the precursor via ionothermal polymerization.32 This strategy relies on the construction of a 2D architecture induced by functionalized graphene nanosheets through the chemical bonding between templates and monomers with the introduction of nitrogen atoms throughout the porous structure. The control of porosity of the carbon matrix was achieved by adjusting the temperature and time in polymerization procedures varying from 400 °C for 40 h, 600 °C for 20 h, to 400 °C for 20 h then 600 °C for 20 h. The synthesized products were denoted as GPF-1, GPF-2, and GPF-3, respectively. For comparison, the corresponding porous carbon framework was synthesized at 400 °C for 40 h without RGO-PAN, which is denoted as PF. The sulfur impregnation of the cathode material was operated based on a facile melt-diffusion strategy39 with the resulting products denominated as GPF-S-1, GPF-S-2, GPF-S-3, and PF-S, respectively.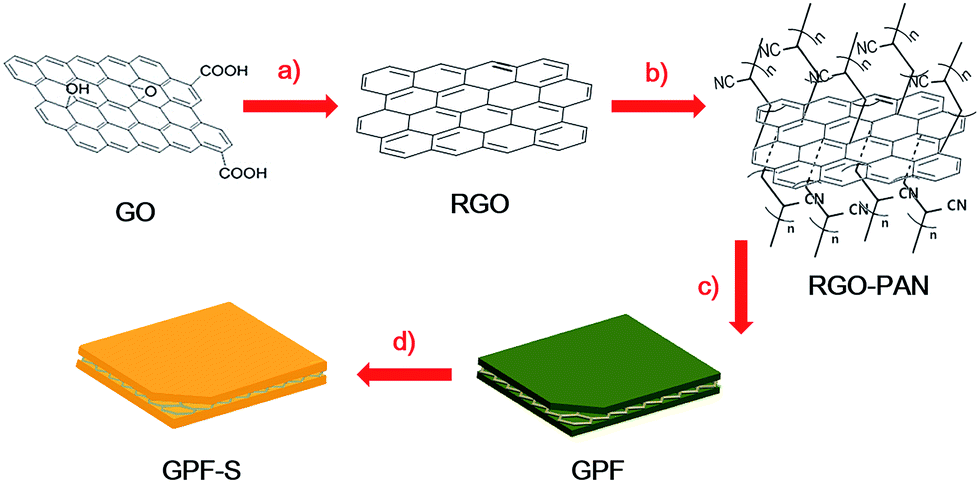 | ||
| Fig. 1 Schematic illustration of the fabrication processes of GPF-S-n (n = 1, 2 and 3) nanocomposites. (a) The preparation of RGO: SDS, N2H4·H2O, 100 °C, 10 h; (b) anionic polymerization of acrylonitrile on the surface of RGO: sodium, THF, sonication, 10 h, RT, then acrylonitrile, sonication, 10 h, RT; (c) the ionothermal polymerization of pDCB in the presence of RGO-PAN at the designed temperature and time periods in ZnCl2 (vacuum) to form GPF-n; (d) melt-diffusion of sulfur into the pores of GPF-n at 155 °C for 12 h. | ||
The 2D architecture of samples was confirmed by the FE-SEM and TEM analyses. The SEM image of GPF-S-3 displayed in Fig. 2a reveals that the 2D structure was well maintained after sulfur impregnation and the surface of the nanosheet was smooth without agglomerated sulfur (the morphology of GPF-3 before sulfur loading is shown in Fig. S1a†). All GPF-S-n nanocomposites showed similar 2D structures while the PF without the template displayed a bulk morphology (Fig. S1b†). The TEM image in Fig. 2b indicates the existence of wrinkles and folds on the GPF-S-3 nanosheet, no agglomerated sulfur can be observed either. Furthermore, the carbon, nitrogen and sulfur mapping images of GPF-S-3 suggest that nitrogen was evenly distributed throughout the carbon framework and that sulfur was homogeneously impregnated into the mirco- and meso-pores of the carbon frameworks (Fig. 2c–f).40
 | ||
| Fig. 2 (a) SEM image, (b) TEM image and (c–f) the corresponding elemental mapping images of GPF-S-3. | ||
The construction of porous carbon/sulfur nanocomposites is based on the porous nature of GPF-n (n = 1, 2, and 3) and PF, which was investigated by N2 adsorption/desorption measurements at 77 K. The N2 adsorption/desorption curve of PF in Fig. 3a shows a type-I isotherm, which declares a micropore-dominated porous structure in PF.41,42 Interestingly, the conversion from the micropore-dominated structure in PF to the mesopore-rich textures in GPF-n (n = 1, 2, and 3) occurred with the addition of the RGO-PAN template and the changing of the polymerization time and temperature. The isotherms of GPF-1, 2 and 3 displayed more evident hysteresis loops at higher relative pressure, which indicates the formation and propagation of mesopores.41 This result implies that the RGO-PAN template played a fundamental guiding role in tuning the hierarchically porous configuration in GPF-n (n = 1, 2, and 3) samples. In addition, the Brunauer–Emmett–Teller specific surface areas of PF and GPF-n rose from 1157 to 1683 m2 g−1 while adding the RGO-PAN template and changing polymerization conditions (see Table S1 in ESI†), which might be ascribed to the addition of the 2D template as well as the structural rearrangement in porous frameworks during the polymerization process with optimized temperature and reaction time.38,43–46
 | ||
| Fig. 3 (a) Nitrogen adsorption/desorption isotherms and (b) the pore size distribution of GPF-n and PF based on the density functional theory (DFT) model; (c) the pore volumes and average pore sizes of GPF-n and PF; (d) TGA curves of GPF-S-n and PF-S under nitrogen atmosphere (n = 1, 2, and 3). | ||
The evaluation of pore size distribution of GPF-n and PF samples was investigated by the density functional theory (DFT). The curves in Fig. 3b verify that a large quantity of mesopores with the size from 2 to 4 nm existed in GPF-3, while an even larger proportion of mesopores with the size from 4 to 10 nm was exhibited in GPF-2, besides micropores in both. The relatively lower specific surface areas of GPF-2 might be ascribed to the existence of more larger-sized mesopores. The fact signifies that the adjusting of polymerization conditions exerted a significant effect on the regulation of the pore size in GPF-n. After impregnation of sulfur, each nanocomposite exhibited a dramatic fall in the specific surface area and total pore volume (Table S1†). Among them, GPF-S-3 demonstrated the most remarkable reduction (specific surface area dropped from 1683 to 14 m2 g−1 and the total pore volume fell from 1.57 to 0.07 cm3 g−1). The results indicate that these carbon hosts with a hierarchically porous structure and pretty high specific surface areas possessed an efficient entrapment of elemental sulfur.47 In addition, the total pore volumes of GPF-S-n and PF-S are smaller than their theoretical values, which could be attributed to the possibility of water entrapment during the sulfur loading. Furthermore, the micro-pores in GPF-n and PF are likely to be blocked and form small enclosed spaces, which could not be reached in the N2 adsorption/desorption tests and inevitably lead to lower detected results.39 The contents of sulfur in the nanocomposites were confirmed by thermogravimetric analysis (TGA) under a nitrogen atmosphere (Fig. 3d). The obvious weight loss of PF-S and GPF-S-n (n = 1, 2, and 3) caused by sulfur evaporation happened between 250 and 350 °C, much higher than the temperature range when pure sulfur volatilizes (160–270 °C), indicating the strong adsorption of sulfur in the abundant micro-/meso-pores of carbon matrices.47–49 Furthermore, the TGA results demonstrated an increase in sulfur contents from 42 wt% to 48 wt%, 63 wt% and 72 wt% in PF-S, GPF-S-1, GPF-S-3 and GPF-S-2 with their increasing total pore volumes (see Table S1†), which were very close to their theoretical maximum values (see the Experimental section for detail information).
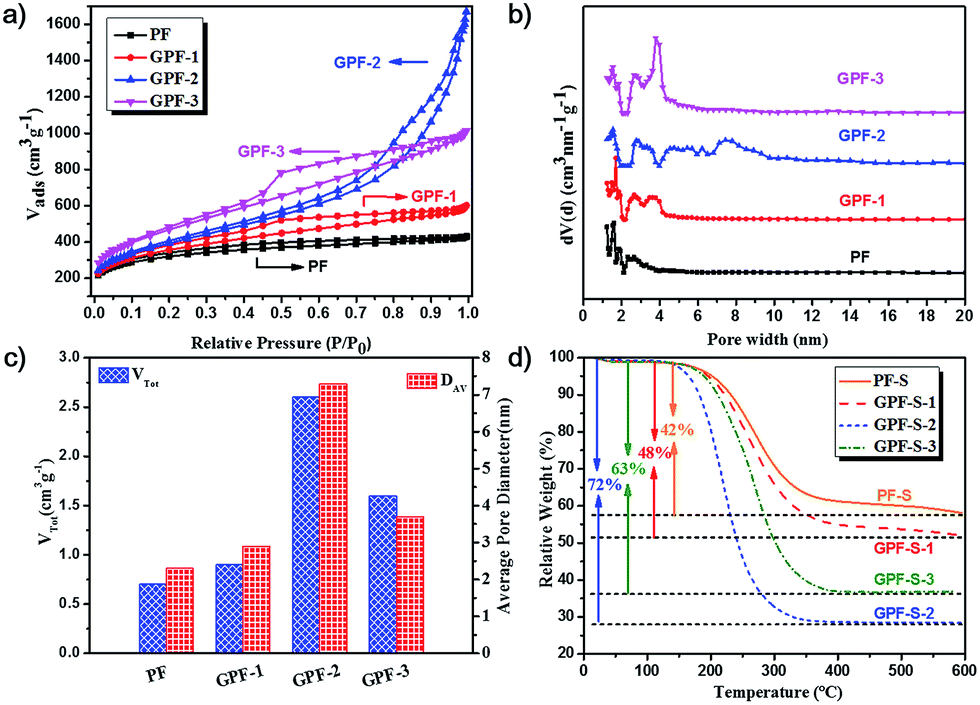
The X-ray diffraction (XRD) patterns of both GPF-n (n = 1, 2, and 3) and PF only exhibit two broad peaks, which suggest their amorphous features (Fig. S2a†). The result is consistent with the previous research studies.32,38,43,50,51 The XRD patterns of GPF-S-n (n = 1, 2, and 3) and PF-S in Fig. 4a indicate the amorphous structures by showing two broad peaks as well. Moreover, any distinct intrinsic peaks of sulfur (Fig. S2c†) is not observed from the XRD patterns (Fig. 4a), further proving that sulfur was evenly dispersed in pores of the carbon hosts,52 which agrees well with the observation in the sulfur mapping image of GPF-S-3 (Fig. 2f). Raman spectroscopy was conducted to further characterize the structural features of samples. The Raman spectra of GPF-n (n = 1, 2 and 3) and PF in Fig. S2b† displays two peaks at 1350 and 1580 cm−1, which are assigned to D (disordered, defect-activated) and G (graphitic, in-plane stretching of sp2 bonds) bands of carbon,13 respectively. After the loading of sulfur, the Raman spectra of GPF-S-n (n = 1, 2 and 3) and PF-S (Fig. 4b) exhibited the exactly similar characteristic peaks with their original samples, GPF-n (n = 1, 2 and 3) and PF. They even showed very similar integral intensity ratios, ID/IG, as labeled above each spectrum in Fig. 4b and S2b.† Hence the fact signifies that the sulfur loading did not change the structural characteristics and the graphite layer defectiveness of carbon matrices.53 Furthermore, no characteristic peaks of elemental sulfur were observed in the region between 100 and 500 cm−1 in Raman spectra of PF-S and GPF-S-n (n = 1, 2, and 3), which are related to vibration of the S–S bond in S8 species (Fig. S2d†).52 This observation suggests that sulfur particles were impregnated into pores and adsorbed on the internal surface of pores in GPF-n and PF hosts,54 which was in good accordance with the results from XRD spectra and the sulfur mapping image.
 | ||
| Fig. 4 (a) XRD patterns of GPF-S-n and PF-S (n = 1, 2, and 3); (b) Raman spectra of GPF-S-n and PF-S (n = 1, 2, and 3). | ||
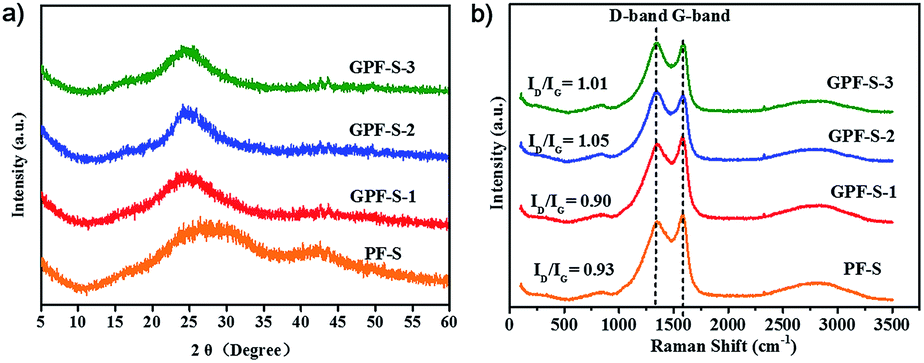
X-ray photoelectron spectroscopy (XPS) analyses (Fig. 5 and S3†) revealed that the nitrogen atom proportion in GPF-S-1, GPF-S-2, and GPF-S-3 were 6.62, 4.27 and 4.20 at%, respectively. The N 1s spectra of GPF-S-3 (Fig. 5a) and GPF-S-1, 2 (Fig. S3 in ESI†) depicted that four distinct nitrogen configurations, pyridine N (398.3 eV, N1), pyrrole N (400.1 eV, N2), quaternary N (401.1 eV, N3), and pyridine-N-oxide (403.1 eV, N4), existed in these nitrogen doped carbon matrices,32,41,55 suggesting the formation of graphitized fragments.46 Among them, the N 1s signal with binding energy (BE) at about 401.1 eV (N3) is assigned to quaternary nitrogen, namely graphite-like nitrogen, which is believed to enhance the conductivity of N-doped carbons by providing additional electrons to the delocalized π-system.56 Quantitative analysis based on XPS suggests that the ratios of N3 gradually increases while the ratio of N1 decreases in GPF-S-1, GPF-S-2 and GPF-S-3, which confirms the occurrence of rearrangement reactions and a gradual increase of graphitized fragments.46 In addition, XPS has been successfully used to identify the chemical state of sulfur molecules and distinguish the sulfur types in Li–S batteries.49,57 All three S 2p spectra in Fig. 5b and S3† contain the characteristic sulfur splitting of the S 2p signal into two components S 2p3/2 and S 2p1/2 as a result of spin–orbital coupling effect at 163.6 and 164.9 eV, which suggests the existence of S8 molecules in GPF-S-n samples.57 The two more peaks at 168.7 and 169.9 eV indicate the existence of sulfur atoms located at the chain end of the small S2–4 molecules,49,57 further proving the strengthened interaction between sulfur molecules and 2D porous carbon hosts.57,58 This finding implies that the effective adjustment of pore architecture can implement molecule level regulation of sulfur molecules in micro-/meso-pores.49
 | ||
| Fig. 5 (a) N 1s XPS spectra and (b) S 2p XPS spectra of GPF-S-3. | ||
To evaluate the potential of GPF-S-n and PF-S as Li–S cathodes, coin cells were fabricated by the method described in the experimental section. Their electrochemical performance for the Li–S cell was tested by cyclic voltammogram (CV), galvanostatic charge–discharge measurements, and electrochemical impedance spectroscopy (EIS) measurements. As shown in the first discharging scan of the GPF-S-3 cathode (Fig. 6a), two evident peaks around 2.35 and 2.0 V were presented, corresponding to the reduction of cyclo-sulfur (S8) to long chain polysulfides species (Li2Sn, 4 ≤ n < 8) and further reduction to short chain polysulfides and ultimately Li2S2 and Li2S.5,48 Another pronounced reduction peak at 1.7 V for the first cycle can be attributed to the irreversible reduction of LiNO3 in the electrolyte, this peak faded away in the subsequent cycles.59 In the charging scan of the cell, only one strong oxidation peak around 2.45 V was observed, which can be associated with the coupled conversion from lithium sulfide to lithium polysulfides, and ultimately to element sulfur.48 The shift of the oxidation peak from 2.45 V in the first cycle to 2.34 V in the following four cycles might be mainly attributed to the rearrangement of the active sulfur particles in pores from its original position to more energetically stable sites.60 Except for the initial activation in the first cycle, no obvious change of reduction and oxidation peaks was observed in the subsequent four cycles, signifying the high electrochemical stability of the GPF-S-3 cathode coupling with the electrolyte.48
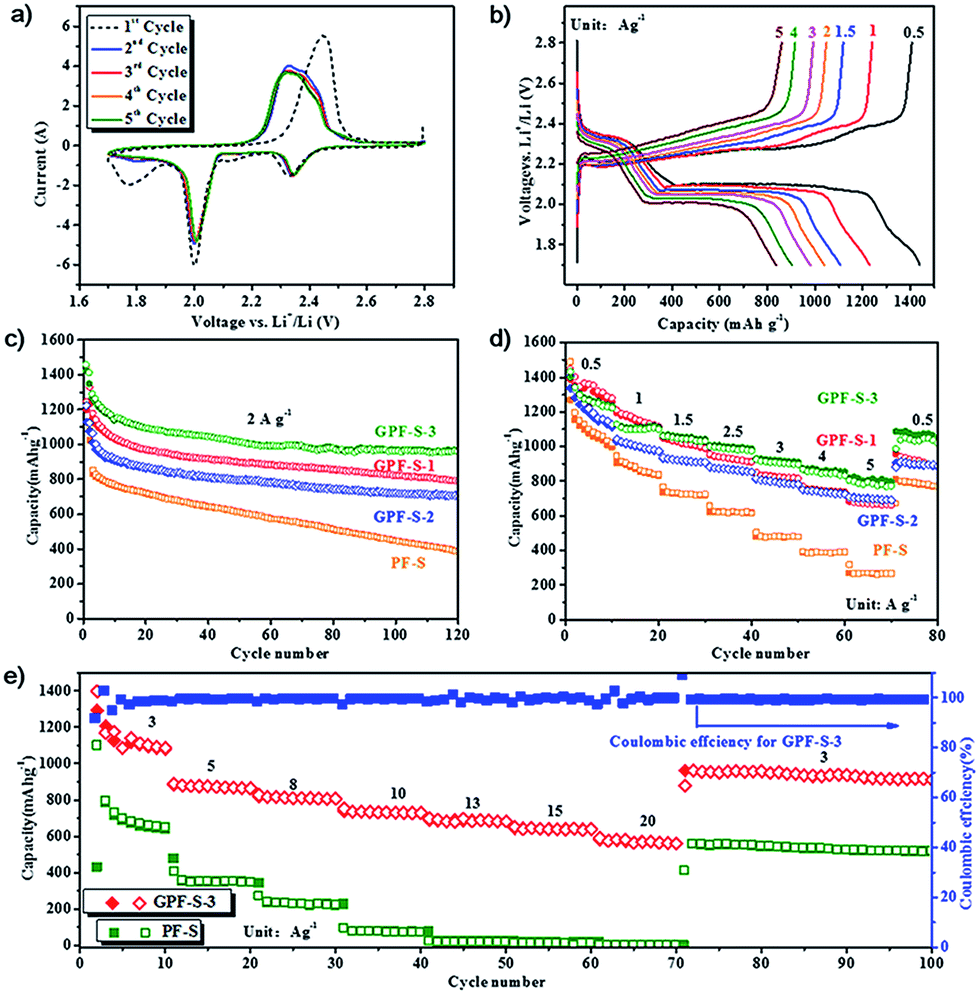 | ||
| Fig. 6 The electrochemical performance of GPF-S-n and PF-S electrodes: (a) CV profiles of the GPF-S-3 electrode; (b) galvanostatic charge–discharge profiles of the GPF-S-3 electrode at various current densities in the voltage range of 1.7–2.8 V vs. Li+/Li; (c) cycle performance of PF-S and GPF-S-n electrodes at a current rate of 2 A g−1; (d) rate performance of PF-S and GPF-S-n electrodes at various current densities from 0.2 to 5 A g−1; (e) rate performance of GPF-S-3 and PF-S electrodes at a current rate from 3 to 20 A g−1. | ||
The electrochemical performances of the GPF-S-3 cathode at various current densities from 0.5 to 5 A g−1 are illustrated in its galvanostatic discharge–charge profiles (Fig. 6b). Consistent with the peak voltages in the CV curves, these discharge voltage profiles demonstrated the two-plateau characteristic of a typical Li–S cell at different current densities from 0.5 A g−1 to 5 A g−1, which further proves the excellent capacity reversibility of the GPF-S-3 cathode.48 Furthermore, the GPF-S-n cathodes exhibited favourable cycling performance at a constant current rate of 2 A g−1 (Fig. 6c). Among the samples, the GPF-S-3 cathode achieved an initial discharge and charge capacity of 1461 and 1424 mA h g−1 with the first cycle coulombic efficiency (CE) up to 97.5%. The PF-S cathode, by contrast, obtained initial discharge and charge capacities of 1435 and 1227 mA h g−1 with the first cycle CE of only 85.5%. After 120 cycles, GPF-S-1, GPF-S-2 and GPF-S-3 cathodes still sustained stabilized specific capacities of 791, 706 and 962 mA h g−1, respectively, greatly surpassing a capacity of 386 mA h g−1 for the PF-S cathode. GPF-S-1, GPF-S-2 and GPF-S-3 cathodes also exhibited better rate performance than PF-S cathode as depicted in Fig. 6d. A high reversible capacity of 810 mA h g−1 was still preserved even at a high rate of 5.0 A g−1 for the GPF-S-3 cathode, which was higher than 710 mA h g−1 for GPF-S-2, 696 mA h g−1 for GPF-S-1 and much higher than 317 mA h g−1 for PF-S. The recovery of a reversible capacity of 1040 mA h g−1 for GPF-S-3 was also achieved at 0.5 A g−1 following a series of high rate charge–discharge processes. The promising cycling and rate performance of GPF-S-n could be ascribed to their 2D architecture with RGO as the template, which guaranteed a rapid ion transfer, fast electronic conduction and intimate contact between the active materials and electrolyte. In addition, the functionalized RGO templates could provide stable frameworks to sustain the strain from the volumetric change of active sulfur and help GPF-S-n to preserve the conductive matrices well during the charge/discharge cycles. The superior electrochemical performance of GPF-S-3 among three GPF-S-n cathodes might be attributed to the abundant hierarchical micro-/mesopores with an optimal pore size in its carbon matrix and nitrogen-doped inner surface, which can increase the carbon surface affinity to active sulfur.61 This feature could contribute to the effective entrapment of active sulfur and polysulfides in carbon hosts.61 Interestingly, the GPF-S-2 cathode with the highest sulfur loading of 72 wt% presented inferior specific capacities to those of GPF-S-3 with a sulfur loading of 63 wt% under the same testing conditions, which might be attributed to the weaker entrapment of sulfur and polysulfides inside the relatively larger-sized (over 4 nm) mesopores in the GPF-2 framework (Fig. 3b) So, both 2D architecture and optimized micro-/meso-pore structure are indispensable to guarantee a fast electronic/ionic transport and enhanced reaction kinetics of sulfur in a nanocomposite skeleton.62 The improved rate charge–discharge capability and cycling performance of GPF-S-3 were further confirmed via a Nyquist plot and its fitted results of an equivalent circuit depicted in Fig. S4.† The cell with the GPF-S-3 cathode demonstrated smaller impedance of a cell component (Rf = 18.52 Ω) than that with PF-S (Rf = 56.89 Ω) as shown in Table S3,† which might be due to the stronger confinement of active materials and less dissolution of polysulfides. Moreover, the cell with the GPF-S-3 cathode exhibited a pretty low charge transfer impedance (Rct) value of 24.33 Ω compared with 146.5 Ω for the cell with the PF-S cathode, suggesting that the former one preserved highly rapid charge transportation kinetics benefited from its 2D architecture as well as the uniform distribution of sulfur in a hierarchically well-tailored porous carbon matrix.63
To simulate the realistic situation where a faster discharge/charge process is adopted, the high-rate performance tests for GPF-S-3 and PF-S cathodes were consequently carried out at current densities from 3 to 20 A g−1. As illustrated in Fig. 6e, GPF-S-3 displayed extremely high specific capacities of 753, 652, and 591 mA h g−1 even at very high current densities of 10, 15, and 20 A g−1. After cycling at an ultrahigh current density of 20 A g−1 and being reverted to 3 A g−1 again, GPF-S-3 still achieved both an outstanding reversible capacity of 960 mA h g−1 and excellent CE of 99.9%. The PF-S electrode, by contrast, exhibited reversible capacities of only 95, 21, and 16 mA h g−1 at 10, 15, and 20 A g−1 respectively. Such an impressive performance of the GPF-3 host for sulfur preceded most of the state-of-the-art porous carbon host materials for Li–S battery cathodes (see Table S4†), including hollow carbon spheres,64 layered graphene-based porous carbon,52 nanoarchitectured graphene/CNT@porous carbon,48 and the like.
Conclusions
In this work, 2D GPF-n (n = 1, 2 and 3) with a well-tailored porous structure and high specific surface area were synthesized via in situ ionothermal polymerization of 1,4-dicyanobenzene directed by the functionalized graphene nanosheets as sulfur hosts for the cathodes in Li–S batteries. The as-prepared GPF/sulfur cathodes exhibited a high capacity up to 962 mA h g−1 after 120 cycles at 2 A g−1. A high reversible capacity of 591 mA h g−1 was still retained even at an extremely large current density of 20 A g−1. Such an outstanding rate and cycling performance demonstrate that the construction of 2D hierarchically micro-/meso-porous carbon frameworks as sulfur hosts is an instructive and efficient strategy to fabricate Li–S batteries for the practical application of high power devices. It is expected that the synthesis strategy can provide a platform for the preparation of unprecedented hierarchical porous carbons by utilizing various monomers and diversified functionalized templates for sulfur hosts in high performance Li–S batteries.Acknowledgements
This work was financially supported by 973 Program of China (2012CB933404 and 2014CB239701), Natural Science Foundation of China (61235007, 21372155, 21320102006 and 51403126), Program for Professor of Special Appointment (Eastern Scholar), ERC project on 2DMATER and EU Graphene Flagship. We also thank the Instrumental Analysis Center of Shanghai Jiao Tong University for the characterization of materials.Notes and references
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2012, 11, 19 CrossRef CAS PubMed.
- A. Manthiram, Y. Fu and Y.-S. Su, Acc. Chem. Res., 2013, 46, 1125 CrossRef CAS PubMed.
- S. Evers and L. F. Nazar, Acc. Chem. Res., 2013, 46, 1135 CrossRef CAS PubMed.
- Y.-X. Yin, S. Xin, Y.-G. Guo and L.-J. Wan, Angew. Chem., Int. Ed., 2013, 52, 13186 CrossRef CAS PubMed.
- Y. Yang, G. Zheng and Y. Cui, Chem. Soc. Rev., 2013, 42, 3018 RSC.
- C. Liang, N. J. Dudney and J. Y. Howe, Chem. Mater., 2009, 21, 4724 CrossRef CAS.
- N. Jayaprakash, J. Shen, S. S. Moganty, A. Corona and L. A. Archer, Angew. Chem., Int. Ed., 2011, 123, 6026 CrossRef.
- Y. Zhao, W. Wu, J. Li, Z. Xu and L. Guan, Adv. Mater., 2014, 26, 5113 CrossRef CAS PubMed.
- J. Zhao, H. Lai, Z. Lyu, Y. Jiang, K. Xie, X. Wang, Q. Wu, L. Yang, Z. Jin, Y. Ma, J. Liu and Z. Hu, Adv. Mater., 2015, 27, 3541 CrossRef CAS PubMed.
- J.-Z. Wang, L. Lu, M. Choucair, J. A. Stride, X. Xu and H.-K. Liu, J. Power Sources, 2011, 196, 7030 CrossRef CAS.
- Y. Cao, X. Li, I. A. Aksay, J. Lemmon, Z. Nie, Z. Yang and J. Liu, Phys. Chem. Chem. Phys., 2011, 13, 7660 RSC.
- L. Ji, M. Rao, H. Zheng, L. Zhang, Y. Li, W. Duan, J. Guo, E. J. Cairns and Y. Zhang, J. Am. Chem. Soc., 2011, 133, 18522 CrossRef CAS PubMed.
- A. Schneider, C. Weidmann, C. Suchomski, H. Sommer, J. Janek and T. Brezesinski, Chem. Mater., 2015, 27, 1674 CrossRef CAS.
- W. Zheng, Y. W. Liu, X. G. Hu and C. F. Zhang, Electrochim. Acta, 2006, 51, 1330 CrossRef CAS.
- W. Ahn, K.-B. Kim, K.-N. Jung, K.-H. Shin and C.-S. Jin, J. Power Sources, 2012, 202, 394 CrossRef CAS.
- L. Yuan, H. Yuan, X. Qiu, L. Chen and W. Zhu, J. Power Sources, 2009, 189, 1141 CrossRef CAS.
- L. Xiao, Y. Cao, J. Xiao, B. Schwenzer, M. H. Engelhard, L. V. Saraf, Z. Nie, G. J. Exarhos and J. Liu, Adv. Mater., 2012, 24, 1176 CrossRef CAS PubMed.
- J. Wang, J. Yang, J. Xie and N. Xu, Adv. Mater., 2002, 14, 963 CrossRef CAS.
- Z. Liang, G. Zheng, W. Li, Z. W. Seh, H. Yao, K. Yan, D. Kong and Y. Cui, ACS Nano, 2014, 8, 5249 CrossRef CAS PubMed.
- H. Yao, G. Zheng, P.-C. Hsu, D. Kong, J. J. Cha, W. Li, Z. W. Seh, M. T. McDowell, K. Yan, Z. Liang, V. K. Narasimhan and Y. Cui, Nat. Commun., 2014, 5, 3943 CAS.
- R. Demir-Cakan, M. Morcrette, F. Nouar, C. Davoisne, T. Devic, D. Gonbeau, R. Dominko, C. Serre, G. Férey and J.-M. Tarascon, J. Am. Chem. Soc., 2011, 133, 16154 CrossRef CAS PubMed.
- X. Ji, S. Evers, R. Black and L. F. Nazar, Nat. Commun., 2011, 2, 325 CrossRef PubMed.
- X. Zhuang, D. Gehrig, N. Forler, H. Liang, M. Wagner, M. R. Hansen, F. Laquai, F. Zhang and X. Feng, Adv. Mater., 2015, 27, 3789 CrossRef CAS PubMed.
- J. Schuster, G. He, B. Mandlmeier, T. Yim, K. T. Lee, T. Bein and L. F. Nazar, Angew. Chem., Int. Ed., 2012, 51, 3591 CrossRef CAS PubMed.
- J. Liu and X.-W. Liu, Adv. Mater., 2012, 24, 4097 CrossRef CAS PubMed.
- X. Huang, X. Qi, F. Boey and H. Zhang, Chem. Soc. Rev., 2012, 41, 666 RSC.
- X. Zheng, J. Luo, W. Lv, D.-W. Wang and Q.-H. Yang, Adv. Mater., 2015, 27, 5388 CrossRef CAS PubMed.
- X. Zheng, W. Lv, Y. Tao, J. Shao, C. Zhang, D. Liu, J. Luo, D.-W. Wang and Q.-H. Yang, Chem. Mater., 2014, 26, 6896 CrossRef CAS.
- H. Wang, L. Zhi, K. Liu, L. Dang, Z. Liu, Z. Lei, C. Yu and J. Qiu, Adv. Funct. Mater., 2015, 25, 5420 CrossRef CAS.
- J. Hou, C. Cao, F. Idrees and X. Ma, ACS Nano, 2015, 9, 2556 CrossRef CAS PubMed.
- C. A. Cao, X. Zhuang, Y. Su, Y. Zhang, F. Zhang, D. Wu and X. Feng, Polym. Chem., 2014, 5, 2057 RSC.
- Y. Su, Y. Liu, P. Liu, D. Wu, X. Zhuang, F. Zhang and X. Feng, Angew. Chem., Int. Ed., 2015, 54, 1812 CrossRef CAS PubMed.
- H. Wang, Y. Yang, Y. Liang, J. T. Robinson, Y. Li, A. Jackson, Y. Cui and H. Dai, Nano Lett., 2011, 11, 2644 CrossRef CAS PubMed.
- Y. Cao, X. Li, I. A. Aksay, J. Lemmon, Z. Nie, Z. Yang and J. Liu, Phys. Chem. Chem. Phys., 2011, 13, 7660 RSC.
- L. Ji, M. Rao, H. Zheng, L. Zhang, Y. Li, W. Duan, J. Guo, E. J. Cairns and Y. Zhang, J. Am. Chem. Soc., 2011, 133, 18522 CrossRef CAS PubMed.
- X. Zhuang, F. Zhang, D. Wu and X. Feng, Adv. Mater., 2014, 26, 3081 CrossRef CAS PubMed.
- H.-J. Peng and Q. Zhang, Angew. Chem., Int. Ed., 2015, 54, 11018 CrossRef CAS PubMed.
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450 CrossRef CAS PubMed.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500 CrossRef CAS PubMed.
- G. Zheng, Y. Yang, J. J. Cha, S. S. Hong and Y. Cui, Nano Lett., 2011, 11, 4462 CrossRef CAS PubMed.
- L. Hao, B. Luo, X. Li, M. Jin, Y. Fang, Z. Tang, Y. Jia, M. Liang, A. Thomas, J. Yang and L. Zhi, Energy Environ. Sci., 2012, 5, 9747 CAS.
- P. Katekomol, J. Roeser, M. Bojdys, J. Weber and A. Thomas, Chem. Mater., 2013, 25, 1542 CrossRef CAS.
- P. Kuhn, A. Forget, D. Su, A. Thomas and M. Antonietti, J. Am. Chem. Soc., 2008, 130, 13333 CrossRef CAS PubMed.
- P. Kuhn, A. Forget, J. Hartmann, A. Thomas and M. Antonietti, Adv. Mater., 2009, 21, 897 CrossRef CAS.
- M. J. Bojdys, J. Jeromenok, A. Thomas and M. Antonietti, Adv. Mater., 2010, 22, 2202 CrossRef CAS PubMed.
- L. Hao, J. Ning, B. Luo, B. Wang, Y. Zhang, Z. Tang, J. Yang, A. Thomas and L. Zhi, J. Am. Chem. Soc., 2015, 137, 219 CrossRef CAS PubMed.
- M. Q. Zhao, Q. Zhang, J. Q. Huang, G. L. Tian, J. Q. Nie, H. J. Peng and F. Wei, Nat. Commun., 2014, 5, 3410 Search PubMed.
- H.-J. Peng, J.-Q. Huang, M.-Q. Zhao, Q. Zhang, X.-B. Cheng, X.-Y. Liu, W.-Z. Qian and F. Wei, Adv. Funct. Mater., 2014, 24, 2772 CrossRef CAS.
- Z. Li, Y. Jiang, L. Yuan, Z. Yi, C. Wu, Y. Liu, P. Strasser and Y. Huang, ACS Nano, 2014, 8, 9295 CrossRef CAS PubMed.
- P. Kuhn, A. Thomas and M. Antonietti, Macromolecules, 2009, 42, 319 CrossRef CAS.
- X. Liu, N. Fechler and M. Antonietti, Chem. Soc. Rev., 2013, 42, 8237 RSC.
- X. Yang, L. Zhang, F. Zhang, Y. Huang and Y. Chen, ACS Nano, 2014, 8, 5208 CrossRef CAS PubMed.
- Y. Qu, Z. Zhang, X. Zhang, G. Ren, Y. Lai, Y. Liu and J. Li, Carbon, 2015, 84, 399 CrossRef CAS.
- B. Ding, Z. Chang, G. Xu, P. Nie, J. Wang, J. Pan, H. Dou and X. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 11165 CAS.
- L. Hao, J. Ning, B. Luo, B. Wang, Y. Zhang, Z. Tang, J. Yang, A. Thomas and L. Zhi, J. Am. Chem. Soc., 2015, 137, 219 CrossRef CAS PubMed.
- Z. R. Ismagilov, A. E. Shalagina, O. Y. Podyacheva, A. V. Ischenko, L. S. Kibis, A. I. Boronin, Y. A. Chesalov, D. I. Kochubey, A. I. Romanenko, O. B. Anikeeva, T. I. Buryakov and E. N. Tkachev, Carbon, 2009, 47, 1922 CrossRef CAS.
- Z. Li, L. Yuan, Z. Yi, Y. Sun, Y. Liu, Y. Jiang, Y. Shen, Y. Xin, Z. Zhang and Y. Huang, Adv. Energy Mater., 2014, 4, 1301473 Search PubMed.
- Y. Yang, G. Yu, J. J. Cha, H. Wu, M. Vosgueritchian, Y. Yao, Z. Bao and Y. Cui, ACS Nano, 2011, 5, 9187 CrossRef CAS PubMed.
- Z. Xiao, Z. Yang, L. Wang, H. Nie, M. e. Zhong, Q. Lai, X. Xu, L. Zhang and S. Huang, Adv. Mater., 2015, 27, 2891 CrossRef CAS PubMed.
- Z. Xiao, Z. Yang, H. Nie, Y. Lu, K. Yang and S. Huang, J. Mater. Chem. A, 2014, 2, 8683 CAS.
- J. Song, T. Xu, M. L. Gordin, P. Zhu, D. Lv, Y.-B. Jiang, Y. Chen, Y. Duan and D. Wang, Adv. Funct. Mater., 2014, 24, 1243 CrossRef CAS.
- S. Liu, K. Xie, Z. Chen, Y. Li, X. Hong, J. Xu, L. Zhou, J. Yuan and C. Zheng, J. Mater. Chem. A, 2015, 3, 11395 CAS.
- Y. Zhou, C. Zhou, Q. Li, C. Yan, B. Han, K. Xia, Q. Gao and J. Wu, Adv. Mater., 2015, 27, 3774 CrossRef CAS PubMed.
- N. Jayaprakash, J. Shen, S. S. Moganty, A. Corona and L. A. Archer, Angew. Chem., Int. Ed., 2011, 50, 5904 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ta08109b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |