
A mechanistic study of AIE processes of TPE luminogens: intramolecular rotation vs. configurational isomerization†
Zhiyong
Yang
ab,
Wei
Qin
b,
Nelson L. C.
Leung
b,
Mathieu
Arseneault
b,
Jacky W. Y.
Lam
ab,
Guodong
Liang
b,
Herman H. Y.
Sung
b,
Ian D.
Williams
b and
Ben Zhong
Tang
*abc
aHKUST Shenzhen Research Institute, Hi-tech Park, Nanshan, Shenzhen 518057, China. E-mail: tangbenz@ust.hk
bDepartment of Chemistry, Division of Biomedical Engineering, Institute for Advanced Study, Division of Life Science, State Key Laboratory of Molecular Neuroscience, Institute of Molecular Functional Materials, The Hong Kong University of Science & Technology (HKUST), Clear Water Bay, Kowloon, Hong Kong, China
cGuangdong Innovative Research Team, SCUT–HKUST Joint Research Laboratory, State Key Laboratory of Luminescent Materials and Devices, South China University of Technology (SCUT), Guangzhou 510640, China
First published on 18th November 2015
Abstract
Chromophores containing olefinic double bonds are the core components of many important luminogen systems that show the novel photophysical effect of aggregation-induced emission (AIE). The role and extent of E–Z isomerization (EZI) of the double bond in affecting the solution emissions of the AIE luminogens (AIEgens), however, have not been fully understood. In this work, we verified the occurrence of EZI in the dilute solutions of TPE-cored AIEgens by NMR spectroscopy using elaborate experimental procedures. We further designed a TPE-fluorescein adduct to quantify that EZI plays a minor role whereas intramolecular rotation plays a major role in the emission quenching processes of the AIEgen solutions. This study fills the gap in the research on the restriction of the intramolecular rotation (RIR) mechanism for the AIE effect and provides a useful tool for the mechanistic investigation of photoluminescence processes.
Introduction
Organic luminophores are usually hydrophobic molecules with extended π-electron conjugations (e.g., aromatic compounds), which naturally form aggregates when they are fabricated into solid films or dispersed in aqueous media.1 Aggregation-induced emission (AIE) is a process which contrasts the notorious effect of the aggregation-caused quenching (ACQ) process,2 in which non-emissive luminogens are induced to emit light by aggregate formation.3 The AIE effect is useful in many practical situations, as many technological applications of light emitters are realized in the aggregate state, e.g., as aqueous nanosuspensions in bioimaging systems and as thin solid films in optoelectronic devices. The unique anti-ACQ attribute of the AIE luminogens (AIEgens) has attracted much attention among scientists and technologists in the past decade.3g As a result of the enthusiastic research effort, a large variety of functional AIEgens have been developed and their practical applications as smart materials, chemical sensors, biomedical probes, and solid-state emitters have been demonstrated.3–14In addition to its practical implications, the AIE effect is also of academic value. The decipherment of the AIE mechanism, for example, will help deepen our understanding of luminescence processes and guide our endeavours in designing new efficient luminophores. In an effort to understand the AIE processes, we proposed a working mechanism of restriction of intramolecular rotation (RIR), on the basis of the experimental data collected from the silole AIEgens such as hexaphenylsilole (HPS; Scheme 1a).3,14 According to the RIR mechanism, the isolated molecules of HPS in a dilute solution are non-luminescent, because their excited species are non-radiatively annihilated by the dynamic intramolecular rotation of their phenyl rotors around the single bonds (as marked by the green-coloured curved arrows in Scheme 1). The intramolecular rotation becomes difficult or restricted when the HPS molecules are aggregated due to the involved physical constraint. This RIR process effectively blocks the radiationless relaxation pathway and opens the radiative decay channel, thus rendering the aggregates of HPS emissive. The RIR mechanism has been proven valid in many AIEgen systems, especially the molecules with robust aromatic rings, such as HPS, 1-butoxy-2,4,6-triphenylbenzene (TPB-BO) and 1,2,3,4,5-pentaphenylbenzene (PPB; Scheme 1a).3,14
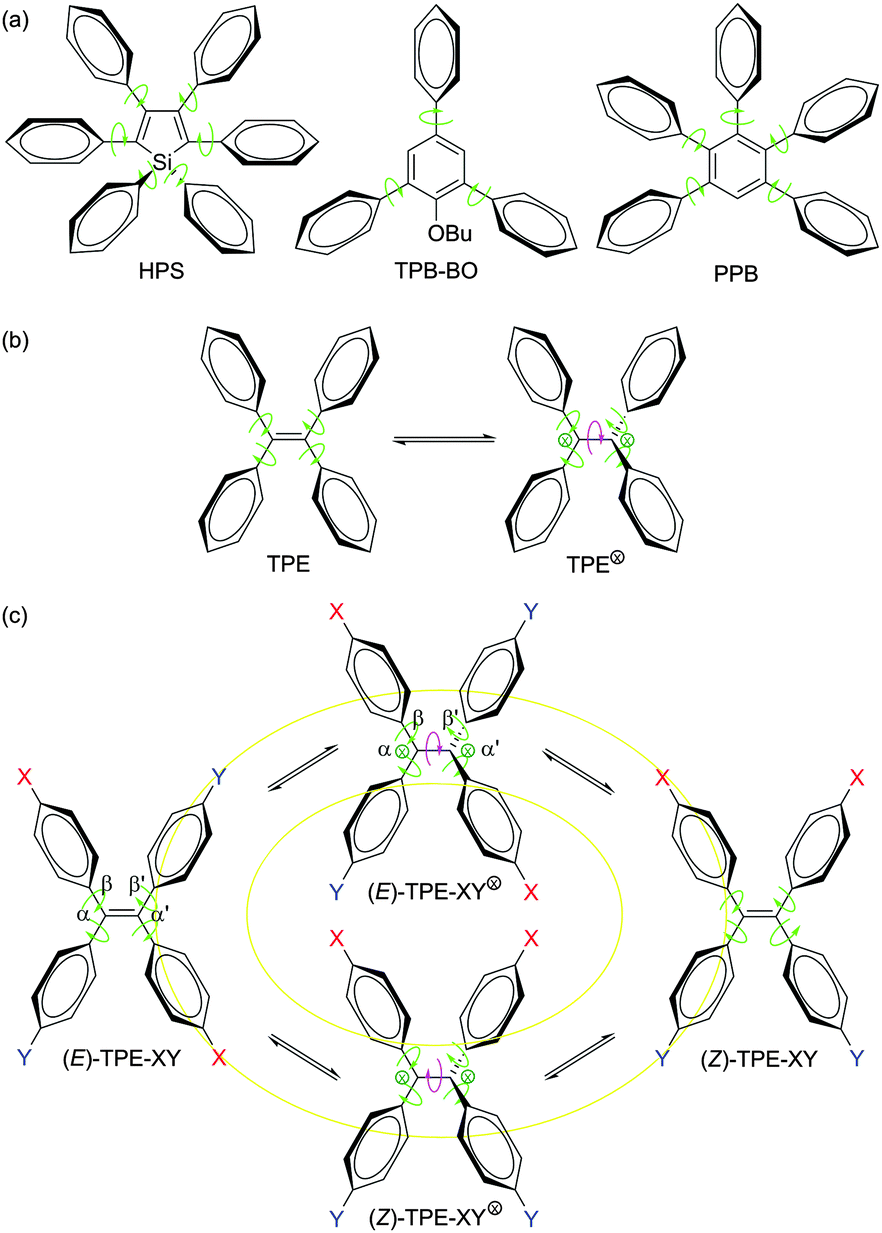 | ||
| Scheme 1 Intramolecular rotation and configurational isomerization in AIE systems. | ||
Tetraphenylethene (TPE; Scheme 1b) is another archetypical AIEgen, whose AIE process, however, is complicated by the fact that its central ethylenic double bond can be broken by UV irradiation.15,16 This bond breakage generates radical or charged species (TPE⊗, ⊗ = radical, cation and/or anion), which is often observed in the systems of stilbene and its congeners.17 The flipping over of the diphenylmethylene (DPM) units around the central single bond yielded by the breakage of the olefin double bond can cause a structural change, resulting in cis–trans or E–Z isomerization (EZI).17–19 The question is: what is the relationship between EZI and emission processes of a TPE-based AIEgen?
There have been a large number of studies on photophysics, especially photoluminescence (PL), of stilbene and its analogues (noting that TPE is structurally a stilbene derivative).17–19 Many laboratories have reported that EZI quenches PL in the systems of stilbene derivatives. The photoinduced EZI transformation, for example, has been blamed to be the funnel for annihilating the PL processes of the fluorogens in biological systems, such as blue fluorescent antibodies.17h,18 Though EZI has been generally believed to be the cause of the PL quenching, some researchers have reported otherwise, revealing no causative relationship between the two processes. For example, the quantum yields of fluorescence (ΦF) and EZI (ΦEZ) for (E)-4-[N-(4-methoxy-phenyl)amino]-4′-cyanostilbene are both very low (ΦF < 0.5%, ΦEZ < 1%).17
A series of substituted TPE derivatives have been designed and developed, examples of which are given in Fig. 1. When the steric effect is increased from TPE-4mM to TPE-4oM, the ΦF of solution (ΦF,S) is increased by >640 times!17 Similarly, when the two geminal phenyl rings are replaced by two bulky anthracenyl rings, the resultant TPE-2An are emissive, showing a ΦF,S value ∼97-fold higher than that of TPE-4mM. The enhancements in ΦF,S have been rationalized to be caused by the RIR processes activated by the severe steric effects in the molecules of TPE-4oM and TPE-2An.17
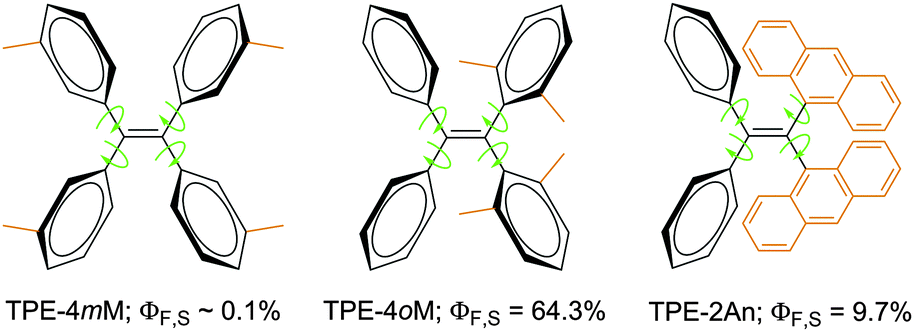 | ||
| Fig. 1 TPE derivatives carrying four methyl groups at meta (TPE-4mM) and ortho (TPE-4oM) positions as well as two geminal anthracenyl substituents (TPE-2An). | ||
Does EZI play any role in the PL process of a TPE derivative? To tackle this issue, in our previous studies, we prepared TPE derivatives with asymmetrical substituents (TPE-XY; Scheme 1c).19 Single crystals of their E and Z isomers, however, could not be grown from their solutions, preventing the identification of their absolute molecular structures. Furthermore, it is difficult to discriminate between the configurational isomerization and intramolecular rotation processes, which are extremely fast and complex.18 Because of this, the relationship between EZI and PL processes is not clear, leaving a gap in the mechanistic study of AIE processes of TPE-based AIEgens.
Incorporation of polar groups into TPE is expected to enlarge the difference in polarity between E and Z isomers and to boost their separability. A new TPE derivative with polar substituents is thus synthesized in this work (Scheme 2a). We succeeded in separating their configurational isomers and in growing their single crystals, thanks to the polarity difference between their E/Z isomers and the crystallizability endowed by their molecular structures. In our previous studies, we failed to capture the EZI signals of the TPE derivatives by NMR spectroscopy.19 In this work, through elaborate efforts, we succeeded in following the EZI process by tracing the photoirradiation-induced changes in the chemical shifts of the E and Z isomers under “normal” PL spectral measurement conditions by using the NMR technique.
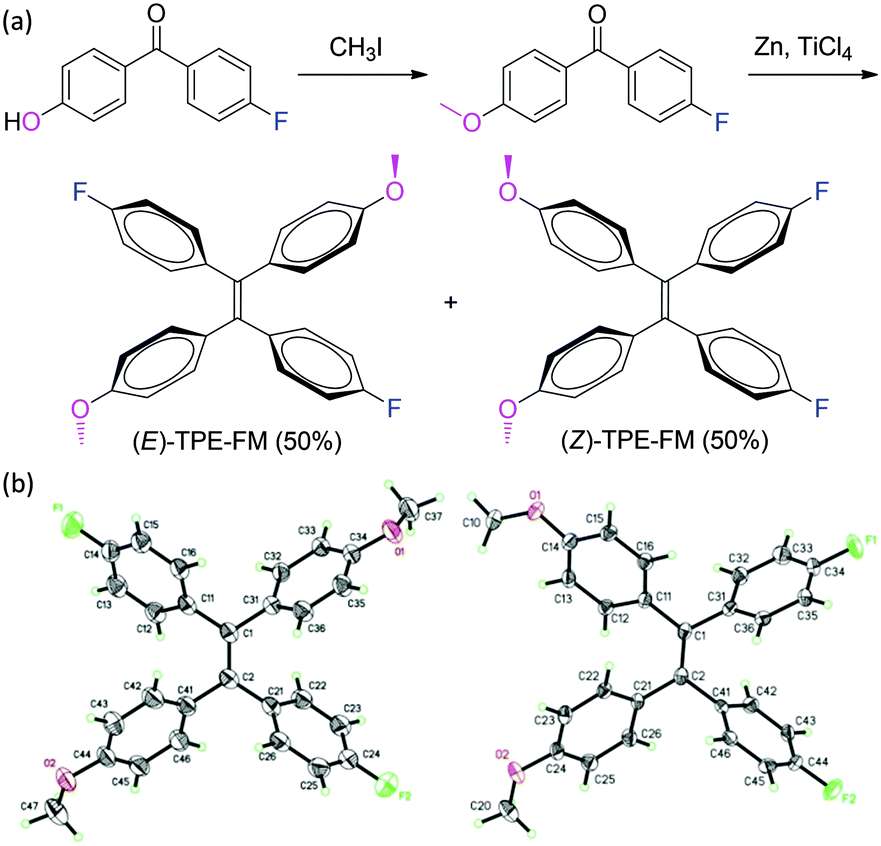 | ||
| Scheme 2 (a) Synthesis of 1,2-bis(4-fluorophenyl)-1,2-bis(4-methoxyphenyl)ethene (TPE-FM) and (b) crystal structures of its pure E and Z configurational isomers. | ||
To further discriminate between EZI and RIR processes, we designed another new luminogen by molecularly melding TPE and fluorescein (Fl) units (TPE-Fl; Fig. 2). It was anticipated that the relative contributions of EZI and RIR to the PL process of TPE-Fl could be distinguished and quantified by the excitations with two different wavelengths. The results obtained from two-colour-excitation experiments revealed that although EZI was involved, it played a minor role in the luminescence quenching process. RIR was confirmed to play a predominant role in the PL processes of the luminogens, in agreement with our proposed AIE mechanism.3 The results here offer a more comprehensive picture of emission processes in TPE-based AIEgens, filling up the gap in the mechanistic study of AIE processes.
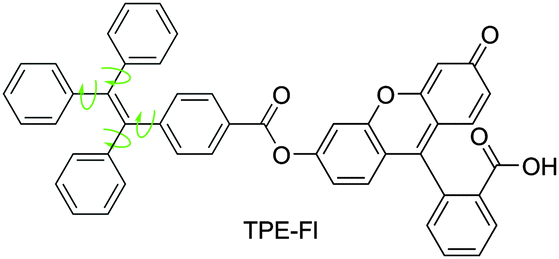 | ||
| Fig. 2 Chemical structure of the molecular adduct of TPE and fluorescein (TPE-Fl). | ||
Results and discussion
Theoretically, an asymmetric structure can enable the EZI process to be monitored and analyzed. The syntheses and isolation of such TPE derivatives, however, had been troublesome. While the Ti-catalyzed McMurry coupling is the most widely employed reaction for the syntheses of TPE derivatives, it often produces mixtures with ∼50% E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z isomers. The first asymmetric AIEgen prepared in our group was a dimethylated TPE derivative (X = CH3, Y = H in Scheme 1).19a A mixture with a Z-rich structure was obtained from a Pd-catalyzed reaction, with the E-rich mixture being out of reach. The second asymmetric structure we studied was a click product of a diethynylated TPE derivative.19b Though the mixture could be separated into E and Z isomers, single crystals could not be grown by recrystallization, preventing their absolute molecular configurations from being unambiguously determined by crystallographic analysis.
:Z isomers. The first asymmetric AIEgen prepared in our group was a dimethylated TPE derivative (X = CH3, Y = H in Scheme 1).19a A mixture with a Z-rich structure was obtained from a Pd-catalyzed reaction, with the E-rich mixture being out of reach. The second asymmetric structure we studied was a click product of a diethynylated TPE derivative.19b Though the mixture could be separated into E and Z isomers, single crystals could not be grown by recrystallization, preventing their absolute molecular configurations from being unambiguously determined by crystallographic analysis.
In this investigation, we incorporated electron accepting (A) and donating (D) of fluoro (F) and methoxy (M) groups into TPE, in an effort to enlarge the difference in polarity between the E and Z isomers of the resultant TPE-FM (Scheme 2). It was hoped that the difference in polarity caused by the D–A interaction would enable the separation of the isomers by simple silica-gel column chromatography, enabling the acquisition of both E and Z isomers. The electronic interactions between the polarized chromophores in TPE-FM may aid the growth of single crystals for absolute structural analysis. As expected, our structural design strategy worked well. It permitted the separation of the E and Z isomers of TPE-FM as well as the acquisition of their single crystals for structural determination (CCDC ref. numbers 928304 and 928305). The crystallographic analysis data of the TPE-FM isomers are summarized in Table S1–S4 of the ESI.†
As can be clearly seen from Scheme 2b, both E and Z isomers of TPE-FM take propeller-shaped conformations and all of its phenyl rings are twisted out of the plane of the central ethylenic double bond (Cα![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Cα′) with torsion angles of 43.3° to 56.6°. This kind of twisted conformation favours intramolecular rotations of its phenyl rotors in the solution state, whereas the multiple C–H⋯π interactions in the crystalline state barricade the intramolecular motions. In this way, like other TPE derivatives, the isomers of TPE-FM both show distinct AIE activity. Examples of the PL spectra of its E isomer are shown in Fig. S1 (ESI†). Its dilute solution in THF is non-emissive but its nanoaggregates in an aqueous mixture containing a large fraction of water (fw = 95 vol%) are highly luminescent.
Cα′) with torsion angles of 43.3° to 56.6°. This kind of twisted conformation favours intramolecular rotations of its phenyl rotors in the solution state, whereas the multiple C–H⋯π interactions in the crystalline state barricade the intramolecular motions. In this way, like other TPE derivatives, the isomers of TPE-FM both show distinct AIE activity. Examples of the PL spectra of its E isomer are shown in Fig. S1 (ESI†). Its dilute solution in THF is non-emissive but its nanoaggregates in an aqueous mixture containing a large fraction of water (fw = 95 vol%) are highly luminescent.
The absorption and emission spectra of the solutions of (Z)-TPE-FM are virtually the same as those of its E counterpart. These spectra are thus of little value in terms of studying its EZI process. However, the reverse rotation of the substituted DPM units in TPE-FM and the reformation of the olefinic double bond give a structure with an opposite configuration, whose chemical shifts may be distinguished by NMR spectroscopy. This proves to be the case: the 1H NMR spectra of the TPE-FM isomers are indeed different (Fig. 3). Using single crystals of the isomers, the resonance peaks in the NMR spectra are readily identified. In the chemical shift region for aromatic protons, the E isomer gives a resonance peak at δ 6.905 (Fig. 3a), whereas the Z isomer shows two unique peaks at δ 6.974 and 6.958 (Fig. 3c). The methoxy groups in the E and Z isomers resonate at δ 3.750 and 3.753, respectively. The photoinduced EZI process of TPE-FM can thus be conveniently followed by monitoring the changes in these resonance peaks.
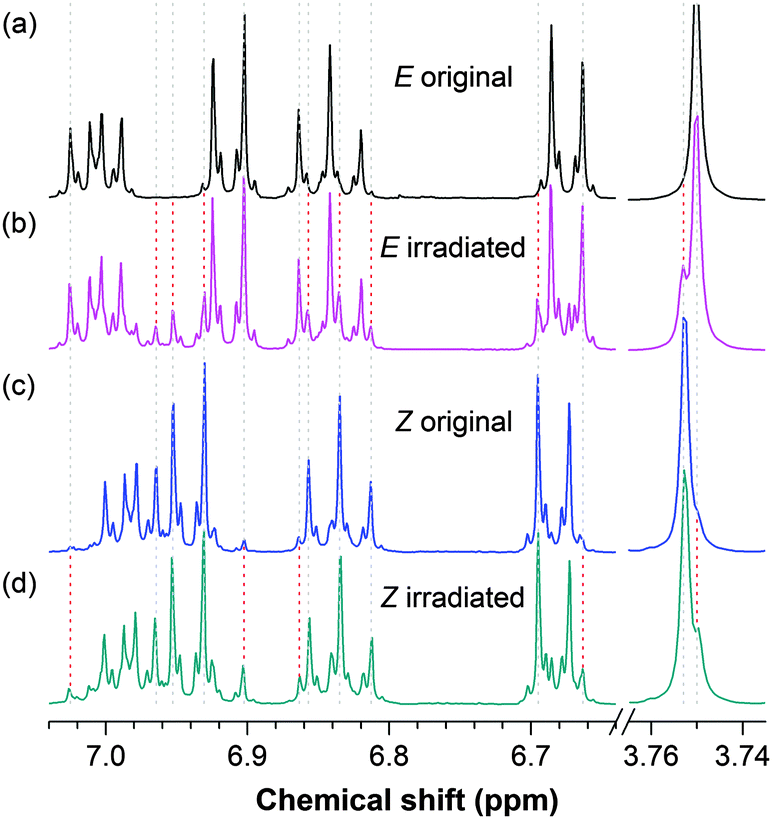 | ||
| Fig. 3 1H NMR spectra of (E)- and (Z)-isomers of TPE-FM in dichloromethane (a and c) before (original) and (b and d) after exposure to a UV lamp for 15 min (irradiated). Lamp power: ∼0.47 mW cm−2, λex = 312 nm, c ∼ 10 mM. Intensities of the resonance peaks in the chemical shift region of δ 7.1–6.6 are magnified to reveal the spectral details. | ||
TPE-FM readily undergoes EZI reaction when the solutions of its isomers (∼10 mM) are irradiated by a UV lamp (λ = 312 nm) with a high power (∼0.47 mW cm−2) for 15 min. New resonance peaks associated with the Z geometric isomer appear in the 1H NMR spectrum of the original E isomer [cf., spectra (a) and (b) in Fig. 3]. These spectral data are in good agreement with the observations in our previous studies on the photoinduced EZI processes of the TPE derivatives. Whereas the NMR technique is very useful, it is rather insensitive: it does not give discernible resonance signals if the species is low in concentration. Dye concentrations used in the routine PL measurements, however, are usually very low. In our previous investigations, we failed to capture the EZI signals of the TPE derivatives by 1H NMR spectroscopy under such conditions.19 To verify whether the EZI processes occur in such dilute solutions, great care must be taken to prepare samples with enough amounts of isomerized species that can be captured by the NMR spectrometer.
In this work, elaborate efforts were made to prepare such samples. A stock solution with a concentration common for the PL measurement (∼10 μM) was prepared by dissolving TPE-FM in a dry nitrogen-purged hexane. Cuvettes with small amounts (3 mL) of the stock solution were irradiated by the 312 nm UV light from the xenon lamp with a low power (∼0.012 mW cm−2) in the spectrofluorometer for specific periods of time. After three samples irradiated under the identical conditions were collected, the solvent was evaporated by using a dry nitrogen stream in a dark room. The sample was then re-dissolved in dichloromethane-d2 for NMR measurement. Though the sample was concentrated to ∼0.1 mM, a good spectrum still could not be obtained if it was scanned for ∼5–10 min. The sample was thus scanned for a long period of time (10 h). This allowed us to finally obtain 1H NMR spectra with high quality from the UV-irradiated dilute solutions.
Upon irradiation, the E isomer of TPE-FM starts to show new resonance peaks associated with its Z isomer at δ 6.974 and 6.958 in the 1H NMR spectra (Fig. 4). The peaks, however, are too small to be used for accurate evaluation. The change in the resonance signal of the methoxy proton at δ 3.753 is bigger and hence more useful. The NMR signal is gradually intensified by lengthening the irradiation time. The ratio of the Z isomer in the mixture after exposure to the UV irradiation by the xenon lamp inside the spectrofluorometer can be calculated by integrating the area of the resonance peak of the methoxy proton in the 1H NMR spectrum. As shown in Fig. 5, the Z ratio increased to ∼14% after the E isomer was irradiated for 5 min, revealing that the photoinduced EZI process of TPE-FM does occur even at low concentrations under low-power excitation.
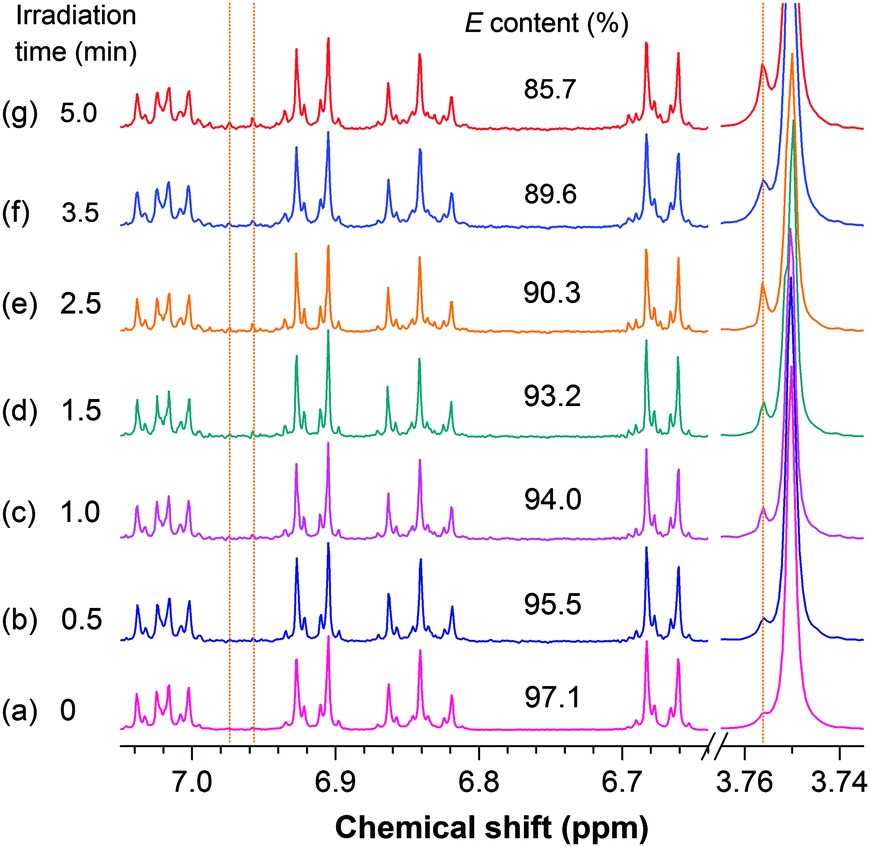 | ||
| Fig. 4 1H NMR spectra of (E)-TPE-FM solutions (∼10 μM) (a) before and (b–g) after irradiation with UV light (λex = 312 nm) from the xenon lamp (∼0.012 mW cm−2) in the PL spectrometer for defined periods of time. | ||
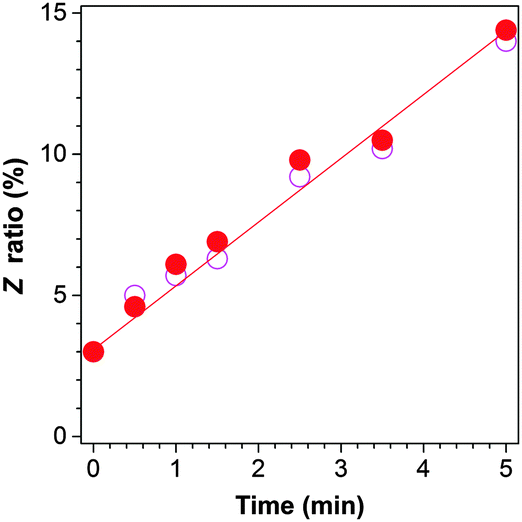 | ||
| Fig. 5 Changes in the Z ratio of TPE-FM with irradiation time; data calculated from the 1H NMR spectra shown in Fig. 4 (●) and obtained from the experiments repeated under the same conditions (○). | ||
It has become clear that EZI and intramolecular rotation both occur when a TPE-FM solution is irradiated by a UV light, but which process plays a major role in quenching the emission of the AIEgen in the solution state? Although the conformational relaxation dynamics of excited states of TPE derivatives have been extensively examined, it has been difficult to separate the EZI and rotation processes.17–19 The intramolecular rotation is a physical process that requires little energy and can occur even under thermal perturbation.18c,20 (In fact, the phenyl rings in the TPE derivatives are fast rotating in solutions even at −90 °C.21) The intramolecular rotation is active under ambient conditions, especially in dilute solutions, unless it is spatially constrained. EZI, on the other hand, is a chemical process that requires the olefinic double bond to be broken first. The chemical bond is cleaved only after the molecule is irradiated by a UV light. This difference between EZI and intramolecular rotation may be utilized to distinguish the two quenching processes.
To explore the possibility of differentiating the luminescence quenching processes, we designed and synthesized a molecular adduct of TPE and Fl, i.e., TPE-Fl (Fig. 2), by the condensation of Fl with an acid chloride of TPE, i.e., (1,2,2-triphenylvinyl)benzoyl chloride (Scheme S1, ESI†). We then carried out a two-colour excitation experiment. Due to the strong overlap between the main absorption peak of Fl and the very weak solution emission of TPE at ∼490 nm, Fl will readily absorb the emission from TPE through the fluorescence resonance energy transfer process. The TPE and Fl units absorb the photons of higher and lower energies, respectively. With the irradiation of TPE-Fl by the light with a shorter wavelength, the higher-energy light is absorbed by the TPE unit that can cleave the ethylenic double bond of TPE, causing the EZI reaction to occur. On the other hand, during the irradiation of TPE-Fl by a light with a longer wavelength, the lower-energy light is not absorbed by the TPE unit and thus is incapable of cleaving the ethylenic double bond of TPE, hence resulting in no EZI. This different response to light wavelength of TPE-Fl enables us to evaluate the relative contributions to the luminescence quenching by the EZI and intramolecular rotation processes.
As can be seen from Fig. 6a, there are two main peaks at ∼340 and ∼450 nm in the absorption spectrum of the TPE-Fl solution in an ethanol/buffer mixture.22 The former and latter peaks are assigned to the absorptions by the TPE and Fl units, respectively, through the comparison with the UV-vis spectra of the solutions of the individual TPE and Fl molecules recorded under the same conditions. The PL spectrum of the TPE-Fl solution taken under an excitation of 480 nm (Fig. 6b) is much weaker than that of Fl.22 It is further weakened when the solution was excited at 330 nm. The relative PL efficiencies of TPE-Fl obtained under different excitation wavelengths are plotted in Fig. 6c. Fl was used as the reference: its ΦF was set as 100% for comparison. The ΦF values of TPE-Fl are always lower than that of Fl under the identical measurement conditions. Evidently, the TPE moiety in TPE-Fl serves as a quenching site for the PL process of the luminogen, primarily through its intramolecular phenyl ring rotation.
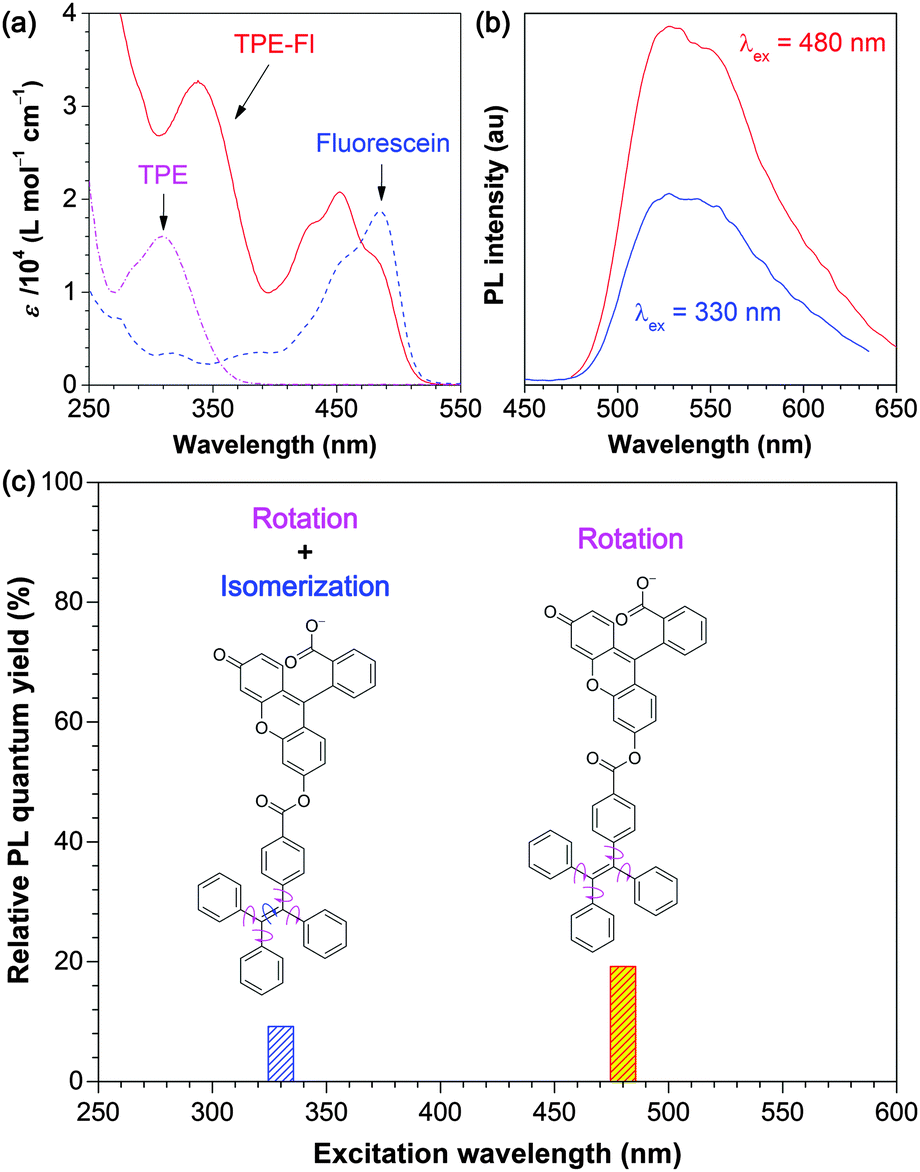 | ||
| Fig. 6 (a) UV spectra of TPE in THF solution and TPE-Fl and Fl in ethanol/buffer mixtures (1:1 v/v) with pH = 6 (keeping the Fl moiety in the monoanionic form). (b) PL spectra of a TPE-Fl solution (c ∼ 10 μM) recorded at different excitation wavelengths. (c) Relative PL quantum yields of TPE-Fl in solution measured at different excitation wavelengths using Fl as reference. | ||
The decrease in ΦF is λex dependent: the PL efficiencies of TPE-Fl are 19.2% and 9.2% (relative to that of Fl), when its solutions are excited at 480 and 330 nm, respectively (Fig. 6c). The absorption edge of TPE is located in the UV region (≤410 nm; Fig. 6a), meaning that it practically does not absorb visible light. Its ethylenic π bond can hardly be broken by the excitation of a visible light with a wavelength of 480 nm (<60 kcal mol−1). As a result, its EZI process is unlikely to occur. The loss in ΦF of TPE-Fl thus should be mainly due to the intramolecular rotation of the phenyl rotors in its TPE moiety. When excited at 330 nm, however, the higher-energy UV light (∼87 kcal mol−1) can readily break the ethylenic π bond (∼63 kcal mol−1),23 leading to the EZI reaction. The decrease in ΦF caused by EZI, however, is much smaller (ΔΦF = −10%) than that brought by the intramolecular rotation (ΔΦF = −80.8%). The molecular motion has thus played a predominant role in quenching the emission process of TPE-Fl. Through the two-colour excitation experiment, the relative contributions of the EZI and intramolecular rotation to the PL process of TPE-Fl are readily distinguished and quantified.
The UV irradiation elevates TPE-Fl to the excited state. The electron in its ethylenic unit is thus promoted from the bonding (π) orbital to the non-bonding (π*) orbital.24 According to the theoretical calculation, the length of the ethylenic CαCα′ bond in diphenyldimethylethene, a TPE derivative with two phenyl rings replaced by two methyl groups, is 1.36 Å in the ground state.24 Upon excitation, the bond length is increased to 1.46 Å, close to that of its Cα–Cβ single bond (1.50 Å). Due to the π bond cleavage by UV photoexcitation, the double bond becomes essentially a single bond (Cα–Cα′), with its bond angle enlarged from 6–7° to 49°.24 The ethylenic bond of the TPE unit in TPE-Fl should also be broken by absorbing the 330 nm UV light. The big twisting angle of the resultant Cα–Cα′ single bond enables the DPM units to flip back and forth, leading to an EZI process.3b,25,26
Clearly, two major steps are involved in the EZI reaction of a TPE derivative: (1) the breakage of the alkene double bond (CαCα′) and (2) the rotation of the aryl rings around the newly formed alkane single bond (Cα–Cα′). The double bond is broken upon absorbing the UV light, while the intramolecular rotation occurs in the excited state. The exciton energy is consumed by the 2nd step in the EZI process, or the excited species are annihilated by the molecular motion. Thus, though the decrease in ΦF of TPE-Fl by the 330 nm UV irradiation is ascribed to the EZI process, its real mechanistic cause is the Cα–Cα′ bond twisting or rotation. In the UV irradiation system, the rotation of the phenyl rotors around the Cα–Cβ single bonds should be as active as in the 480 nm excitation system. The intramolecular rotations of the aryl rings (around the original Cα–Cβ single bonds) and the DPM units (around the newly generated Cα–Cα′ single bond) collectively quench the Fl emission in TPE-Fl to a great extent (90.8%).
Concluding remarks
RIR has proven to be the main mechanistic cause for the unique AIE effects observed in many luminogen systems based on pure aromatic molecular rotors such as HPS.3 Its validity in the AIE systems containing olefinic double bonds, however, has been an issue of concern, as such AIEgens can undergo photoinduced EZI transformation. EZI has often been blamed to be the cause for quenching the PL of the stilbene-based and naturally occurring luminogens, e.g., fluorescent proteins.17h,18 Because of the easy synthetic accessibility, a significant stream of AIE research has been done on TPE derivatives.3–14 It is therefore of great importance to decipher the operating mechanism in the TPE-based AIEgen systems. To learn whether EZI is involved and what role it plays in the emission processes of TPE-based AIEgens, in this work, we designed and synthesized a new TPE derivative, i.e., TPE-FM. We efficaciously isolated its E and Z isomers by simple column chromatography and obtained their single crystals for structural analysis, particularly configurational determination.Through elaborate efforts, we verified the involvement of EZI in the emission processes of the TPE-FM isomers and monitored their EZI reactions in dilute solutions even under the normal PL measurement conditions. We also prepared a molecular adduct of TPE and Fl, i.e., TPE-Fl, and studied its emission behaviours under irradiations by UV (330 nm) and visible (480 nm) light that are capable and incapable of breaking the ethylenic π bond of the TPE unit, respectively. The study with two-colour excitation experiments has revealed that EZI plays only an insignificant role in quenching the PL of TPE-Fl. We have further looked into the major elementary steps of the EZI reaction and clarified that even in the EZI process, it is the intramolecular rotation of the DPM units around the newly formed Cα–Cα′ single bond that has caused the radiationless decay of the TPE unit.
To sum up what we have described above, through this study, we have made the following points clear: (1) EZI is involved in the PL process of TPE-FM; (2) compared with the Cα–Cβ single-bond rotation, EZI plays only a minor role in the PL quenching process of TPE-FM; and (3) in the EZI process of TPE-FM, it is not the π-bond breakage but the Cα–Cα′ single-bond rotation in the excited state that has facilitated the non-radiative deactivation of its excitons. TPE-FM is non-emissive in the solution state, due to the collective effects of the intramolecular rotations of its aryl and DPM rotors around the (existing) Cα–Cβ and (new) Cα–Cα′ single bonds. These rotations are hindered in the aggregates. The restriction of the intramolecular rotations by the physical constraint in the aggregates blocks the radiationless relaxation channels of the AIEgen molecules. This RIR mechanism activates the AIE process, thus enhancing the PL of the AIEgen molecules in the aggregate state.
The mechanistic insights gained in this work fill up a gap in the mechanistic study of AIE processes, which is instructive to the design and development of new AIEgen systems from olefinic luminogens or even chromophores carrying heteroatom double bonds. TPE has been used as a versatile building block for the construction of a wide variety of AIEgens and has served as a “magic wand” for transforming ACQ luminophores to AIEgens.3 Furthermore, many Schiff bases with aromatic rings connected by CN bridges have been found to show AIE effects.27–30 It is envisioned that more exotic fluorescent and phosphorescent AIEgens will be explored from the luminogens comprising other heteroatom double bonds, such as CO, CS, CP, BN and SO, under the guidance of the RIR working principle.31–35 The two-colour excitation approach to differentiate and quantificate PL quenching processes can be easily applied to other luminogen systems, providing a versatile tool for the mechanistic study of various luminescence processes.
Experimental
Materials and instruments
THF was distilled from sodium and benzophenone immediately prior to use. Chloroform was distilled over calcium hydride. 1-(p-Bromophenyl)-1,2,2-triphenylethene was prepared using the literature procedures.33 Iodomethane was purchased from RDH chemical Co., Ltd and fluorescein was provided by Sinopharm Chemical Reagent Co., Ltd. Other chemicals were purchased from Sigma-Aldrich. All the commercially available chemicals were used as received without further purification.
1H and 13C NMR spectra were measured on a Bruker AV400 NMR spectrometer using chloroform-d or dichloromethane-d2 as solvent and TMS as internal reference. UV-vis absorption spectra were recorded on a Milton Ray Spectronic 3000 array spectrophotometer. PL spectra were recorded on a Perkin-Elmer LS 55 spectrofluorometer. HRMS spectra were obtained from a GCT Premier CAB 048 mass spectrometer operated in LD-TOF (or Cl-TOF) mode. Elemental analysis was carried out using an ElementarVario Micro Cube. Single crystals of TPE-FM were grown from a hexane/dichloromethane (3:1, v/v) mixture at room temperature. X-ray diffraction data were collected at 100 K on a Bruker-Nonices Smart Apex CCD diffractometer with graphite monochromated Mo Kα radiation. Processing of the intensity data was conducted using the SAINT and SADABS routines, and the structure and refinement were carried out using the SHELTL suite of X-ray programs (version 6.10).
Synthesis and characterization
:1, v/v) as an eluent to separate its E and Z isomers. Global yield for the reaction was 73% (0.78 g, 1.82 mmol), while 0.31 g and 0.22 g of the E and Z isomers were obtained, respectively, after final recrystallization.
Characterization data for (E)-TPE-FM: 1H NMR (400 MHz, CDCl3) δ (TMS, ppm): 6.98 (dd, 4H), 6.89 (d, 4H), 6.80 (t, 4H), 6.65 (d, 4H), 3.75 (s, 6H). 13C NMR (400 MHz, CDCl3) δ (CDCl3, ppm): 162.5, 160.0, 158.1, 140.0, 138.7, 136.0, 135.9, 132.9, 132.8, 132.4, 114.7, 114.5, 113.2, 55.1. HRMS (CI-TOF): m/z 428.1582 [M+, calcd 428.1588]. Anal. Calcd for C28H22F2O2: C, 78.49; H, 5.18. Found: C, 78.36; H, 5.29.
Characterization data for (Z)-TPE-FM: 1H NMR (400 MHz, CDCl3) δ (TMS, ppm): 6.96 (dd, 4H), 6.92 (d, 4H), 6.79 (t, 4H), 6.66 (d, 6H). 13C NMR (400 MHz, CDCl3) δ (CDCl3, ppm): 162.5, 160.1, 158.1, 140.0, 139.9, 138.7, 136.0, 132.9, 132.8, 132.4, 114.8, 114.6, 113.2, 55.1. HRMS (CI-TOF): m/z 428.1577 [M+, calcd 428.1588]. Anal. Calcd for C28H22F2O2: C, 78.49; H, 5.18. Found: C, 78.61; H, 5.20.
:1, v/v) as an eluent. Final yield was 85% (1.24 g, 3.31 mmol). 1H NMR (400 MHz, DMSO-d6) δ (DMSO-d6, ppm): 7.66 (d, 2H), 6.95–7.11 (m, 17H). 13C NMR (100 MHz, MeOD-d4) δ (MeOD-d4, ppm): 167.8, 148.1, 142.4, 141.8, 130.3, 128.3, 125.9. HRMS (CI-TOF): m/z 376.1458 [M+, calcd 376.1463].
:1, v/v) as an eluent to yield 0.31 g (45%) TPE-Fl as a white powder.
1H NMR (400 MHz, CDCl3) δ (TMS, ppm): 8.03 (d, 1H), 8.01 (d, 2H), 7.64 (m, 2H), 7.18 (d, 2H), 7.16–7.11 (m, 10H), 7.06–7.00 (m, 6H), 6.86 (dd, 1H), 6.81 (d, 1H), 6.70 (d, 1H), 6.63 (d, 1H), 6.53 (dd, 1H), 5.91 (s, 1H). 13C NMR (100 MHz, CDCl3) δ (CDCl3, ppm): 169.8, 165.0, 158.4, 152.2, 152.0, 150.1, 143.1, 143.0, 142.9, 139.6, 135.2, 131.6, 131.2, 129.9, 129.7, 129.2, 127.9, 127.7, 127.0, 126.4, 125.1, 124.1, 117.5, 116.7, 112.7, 110.5, 103.1. HRMS (LD-TOF): m/z 691.2040 [M+, calcd 691.2042]. Anal. Calcd for C47H30O6: C, 81.72; H, 4.38; O, 13.90. Found: C, 81.46; H, 4.29; O, 13.43.
Acknowledgements
This work was partially supported by the National Basic Research Program of China (973 Program; 2013CB834701), the University Grants Committee of Hong Kong (AoE/P-03/08), and the Research Grants Council of Hong Kong (N_HKUST620/11, 16305014 and 604913). B. Z. T. thanks the financial support from the Guangdong Innovative Research Team Program (201101C0105067115).Notes and references
- (a) J. B. Birks, Photophysics of Aromatic Molecules, Wiley, New York, 1970 Search PubMed; (b) Z. Li and A. Qin, Natl. Sci. Rev., 2014, 1, 22 CrossRef.
- (a) B. T. Nguyen, J. E. Gautrot, C. Y. Ji, P. L. Brunner, M. T. Nguyen and X. X. Zhu, Langmuir, 2006, 22, 4799 CrossRef CAS PubMed; (b) A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897 CrossRef CAS PubMed; (c) U. H. F. Bunz and V. M. Rotello, Angew. Chem., Int. Ed., 2010, 49, 3268 CrossRef CAS PubMed; (d) H. Kobayashi and P. L. Choyke, Acc. Chem. Res., 2011, 44, 83 CrossRef CAS PubMed; (e) X. H. Zhu, J. B. Peng, Y. Caoa and J. Roncali, Chem. Soc. Rev., 2011, 40, 3509 RSC; (f) M. Vendrell, D. T. Zhai, J. C. Er and Y. T. Chang, Chem. Rev., 2012, 112, 4391 CrossRef CAS PubMed.
- (a) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332 RSC; (b) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC; (c) R. Hu, N. L. C. Leung and B. Z. Tang, Chem. Soc. Rev., 2014, 43, 4494 RSC; (d) J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429 CrossRef CAS PubMed; (e) J. W. Li, Y. Li, C. Y. K. Chan, R. T. K. Kwok, H. K. Li, P. Zrazhevskiy, X. H. Gao, J. Z. Sun, A. J. Qin and B. Z. Tang, Angew. Chem., Int. Ed., 2014, 53, 13518 CrossRef CAS PubMed; (f) H. Wang, E. Zhao, J. W. Y. Lam and B. Z. Tang, Mater. Today, 2015, 18, 365 CrossRef CAS; (g) A. Qin and B. Z. Tang, Aggregation-Induced Emission: Fundamentals and Applications, ed. A. Qin and B. Z. Tang, Wiley, Singapore, 2013 Search PubMed.
- (a) W. Qin, K. Li, G. X. Feng, M. Li, Z. Y. Yang, B. Liu and B. Z. Tang, Adv. Funct. Mater., 2014, 24, 635 CrossRef CAS; (b) M. Zhang, G. X. Feng, Z. G. Song, Y. P. Zhou, H. Y. Chao, D. Q. Yuan, T. T. Y. Tan, Z. G. Guo, Z. G. Hu, B. Z. Tang, B. Liu and D. Zhao, J. Am. Chem. Soc., 2014, 136, 7241 CrossRef CAS PubMed; (c) Q. Hu, M. Gao, G. Feng and B. Liu, Angew. Chem., Int. Ed., 2014, 53, 14225 CrossRef CAS PubMed; (d) K. Li and B. Liu, Chem. Soc. Rev., 2014, 43, 6570 RSC.
- (a) Z. Li, Y. Dong, B. Mi, Y. Tang, M. Häussler, H. Tong, Y. Dong, J. W. Y. Lam, Y. Ren, H. H. Y. Sung, K. S. Wong, P. Gao, I. D. Williams, H. S. Kwok and B. Z. Tang, J. Phys. Chem. B, 2005, 109, 10061 CrossRef CAS PubMed; (b) Z. Y. Yang, Z. G. Chi, T. Yu, X. Q. Zhang, M. N. Chen, B. J. Xu, S. W. Liu, Y. Zhang and J. R. Xu, J. Mater. Chem., 2009, 19, 5541 RSC; (c) Z. A. Li, Y. Liu, G. Yu, Y. Wen, Y. Guo, L. Ji, J. Qin and Z. Li, Adv. Funct. Mater., 2009, 19, 2677 CrossRef CAS; (d) Z. G. Chi, X. Q. Zhang, B. J. Xu, X. Zhou, C. P. Ma, Y. Zhang, S. W. Liu and J. R. Xu, Chem. Soc. Rev., 2012, 41, 3878 RSC; (e) S. D. Xu, T. T. Liu, Y. X. Mu, Y.-F. Wang, Z. G. Chi, C.-C. Lo, S. W. Liu, Y. Zhang, A. Lien and J. R. Xu, Angew. Chem., Int. Ed., 2015, 54, 874 CrossRef CAS PubMed; (f) J. Huang, N. Sun, J. Yang, R. L. Tang, Q. Q. Li, D. G. Ma and Z. Li, Adv. Funct. Mater., 2014, 24, 7645 CrossRef CAS; (g) J. Zhang, B. Xu, J. Chen, S. Ma, Y. Dong, L. Wang, B. Li, L. Ye and W. Tian, Adv. Mater., 2014, 26, 739 CrossRef PubMed; (h) X. Q. Zhang, X. Y. Zhang, B. Yang, Y. L. Zhang and Y. Wei, ACS Appl. Mater. Interfaces, 2014, 6, 3600 CrossRef CAS PubMed; (i) T. Y. Han, J. W. Y. Lam, N. Zhao, M. Gao, Z. Y. Yang, E. G. Zhao, Y. P. Dong and B. Z. Tang, Chem. Commun., 2013, 49, 4848 RSC.
- (a) Q. C. Han, Q. A. Su, L. Tang, J. W. Feng, P. Lu and Y. G. Wang, J. Phys. Chem. C, 2010, 114, 18702 CrossRef CAS; (b) L. P. Heng, W. Qin, S. J. Chen, R. R. Hu, J. Li, N. Zhao, S. T. Wang, B. Z. Tang and L. Jiang, J. Mater. Chem., 2012, 22, 15869 RSC; (c) K. R. Ghosh, S. K. Saha, J. P. Gao and Z. Y. Wang, Chem. Commun., 2014, 50, 716 RSC; (d) X. R. Wang, J. M. Hu, G. Y. Zhang and S. Y. Liu, J. Am. Chem. Soc., 2014, 136, 9890 CrossRef CAS PubMed; (e) Y. Cai, K. Samedov, B. S. Dolinar, Z. Song, B. Z. Tang, C. Zhang and R. West, Organometallics, 2015, 34, 78 CrossRef CAS.
- (a) W. C. Wu, C. Y. Chen, Y. Q. Tian, S. H. Jang, Y. N. Hong, Y. Liu, R. R. Hu, B. Z. Tang, Y. T. Lee, C. T. Chen, W. C. Chen and A. K. Y. Jen, Adv. Funct. Mater., 2010, 20, 1413 CrossRef CAS; (b) B. K. An, J. Gierschner and S. Y. Park, Acc. Chem. Res., 2012, 45, 544 CrossRef CAS PubMed; (c) D. D. Li, J. Z. Liu, R. T. K. Kwok, Z. Q. Liang, B. Z. Tang and J. H. Yu, Chem. Commun., 2012, 48, 7167 RSC; (d) N. B. Shustova, T.-C. Ong, A. F. Cozzolino, V. K. Michaelis, R. G. Griffin and M. Dinca, J. Am. Chem. Soc., 2012, 134, 15061 CrossRef CAS PubMed; (e) G. J. Wang, R. C. Zhang, C. Xu, R. Y. Zhou, J. Dong, H. T. Bai and X. W. Zhan, ACS Appl. Mater. Interfaces, 2014, 6, 11136 CrossRef CAS PubMed; (f) E. Preis, W. Dong, G. Brunklaus and U. Scherf, J. Mater. Chem. C, 2015, 3, 1582 RSC.
- (a) G. F. Zhang, M. P. Aldred, W. L. Gong, Q. Li and M. Q. Zhu, Chem. Commun., 2012, 48, 7711 RSC; (b) Z. Y. Yang, W. Qin, J. W. Y. Lam, S. J. Chen, H. H. Y. Sung, I. D. Williams and B. Z. Tang, Chem. Sci., 2013, 4, 3725 RSC; (c) A. Shao, Z. Guo, S. Zhu, S. Zhu, P. Shi, H. Tian and W. Zhu, Chem. Sci., 2014, 5, 1383 RSC; (d) F. Tang, C. Wang, J. S. Wang, X. Y. Wang and L. D. Li, ACS Appl. Mater. Interfaces, 2014, 6, 18337 CrossRef CAS PubMed; (e) T. Butler, W. A. Morris, J. Samonina-Kosicka and C. L. Fraser, Chem. Commun., 2015, 51, 3359 RSC.
- (a) F. Sun, G. X. Zhang, D. Q. Zhang, L. Xue and H. Jiang, Org. Lett., 2011, 13, 6378 CrossRef CAS PubMed; (b) P. Lu, R. Wei, K. Li, Z. Zhou, P. Song and A. Tong, Analyst, 2013, 138, 2068 RSC; (c) Y. Zhao, M. Cai, Y. Qian, L. Xie and W. Huang, Prog. Chem., 2013, 25, 296 CAS; (d) X. Yao, X. Ma and H. Tian, J. Mater. Chem. C, 2014, 2, 5155 RSC; (e) C. L. Tan, X. Y. Qi, X. Huang, J. Yang, B. Zheng, Z. F. An, R. F. Chen, J. Wei, B. Z. Tang, W. Huang and H. Zhang, Adv. Mater., 2014, 26, 1735 CrossRef CAS PubMed; (f) S. H. Huang, Y. W. Chiang and J. L. Hong, Polym. Chem., 2015, 6, 497 RSC.
- (a) Y. Liu, X. Tao, F. Wang, X. Dang, D. Zou, Y. Ren and M. Jiang, J. Phys. Chem. C, 2008, 112, 3975 CrossRef CAS; (b) B. R. Gao, Y. H. Wang, Z. Y. Yang, H. Wang, L. Wang, Y. Jiang, Y. W. Hao, Q. D. Chen, Y. P. Li, Y. G. Ma and H. B. Sun, J. Phys. Chem. C, 2011, 115, 16150 CrossRef CAS; (c) X. Shen, G. X. Zhang and D. Q. Zhang, Org. Lett., 2012, 14, 1744 CrossRef CAS PubMed; (d) J. Ma, T. Lin, X. Pan and W. Wang, Chem. Mater., 2014, 26, 4221 CrossRef CAS; (e) H. T. Feng, J. H. Wang and Y. S. Zheng, ACS Appl. Mater. Interfaces, 2014, 6, 20067 CrossRef CAS PubMed.
- (a) Y. Liu, Y. H. Tang, N. N. Barashkov, I. S. Irgibaeva, J. W. Y. Lam, R. R. Hu, D. Birimzhanova, Y. Yu and B. Z. Tang, J. Am. Chem. Soc., 2010, 132, 13951 CrossRef CAS PubMed; (b) M. Li, J. W. Y. Lam, M. Faisal, S. J. Chen, W. J. Zhang, Y. N. Hong, J. Xiong, Q. C. Zheng and B. Z. Tang, J. Mater. Chem. B, 2013, 1, 676 RSC; (c) S. Bhunia, N. Chatterjee, S. Das, K. Das Saha and A. Bhaumik, ACS Appl. Mater. Interfaces, 2014, 6, 22569 CrossRef CAS PubMed.
- (a) W. Z. Yuan, Y. Tan, Y. Gong, P. Lu, J. W. Y. Lam, X. Y. Shen, C. Feng, H. H. Y. Sung, Y. Lu, I. D. Williams, J. Z. Sun, Y. Zhang and B. Z. Tang, Adv. Mater., 2013, 25, 2837 CrossRef CAS PubMed; (b) W. W. Chen, Q. Z. Li, W. S. Zheng, F. Hu, G. X. Zhang, Z. Wang, D. Q. Zhang and X. Y. Jiang, Angew. Chem., Int. Ed., 2014, 53, 13734 CrossRef CAS PubMed; (c) W. Zhao, C. Y. Li, B. Liu, X. Wang, P. Li, Y. Wang, C. J. Wu, C. G. Yao, T. Tang, X. L. Liu and D. M. Cui, Macromolecules, 2014, 47, 5586 CrossRef CAS; (d) J. Chen, Y. Wang, W. Y. Li, H. P. Zhou, Y. X. Li and C. Yu, Anal. Chem., 2014, 86, 9866 CrossRef CAS PubMed.
- (a) Q. Lin, Q. P. Yang, B. Sun, Y. P. Fu, X. Zhu, T. B. Wei and Y. M. Zhang, Soft Matter, 2014, 10, 8427 RSC; (b) Y. N. Jiang, X. D. Yang, C. Ma, C. X. Wang, Y. Chen, F. X. Dong, B. Yang, K. Yu and Q. Lin, ACS Appl. Mater. Interfaces, 2014, 6, 4650 CrossRef CAS PubMed; (c) J. Zhao, D. Yang, Y. X. Zhao, X. J. Yang, Y. Y. Wang and B. Wu, Angew. Chem., Int. Ed., 2014, 53, 6632 CrossRef CAS PubMed; (d) P. Galer, R. C. Korosec, M. Vidmar and B. Sket, J. Am. Chem. Soc., 2014, 136, 7383 CrossRef CAS PubMed; (e) C. Q. Zhang, S. B. Jin, K. N. Yang, X. D. Xue, Z. P. Li, Y. G. Jiang, W. Q. Chen, L. R. Dai, G. Z. Zou and X. J. Liang, ACS Appl. Mater. Interfaces, 2014, 6, 8971 CrossRef CAS PubMed.
- (a) J. Chen, C. C. W. Law, J. W. Y. Lam, Y. P. Dong, S. M. F. Lo, I. D. Williams, D. Zhu and B. Z. Tang, Chem. Mater., 2003, 15, 1535 CrossRef CAS; (b) Q. Zeng, Z. Li, Y. Dong, C. Di, A. Qin, Y. Hong, Z. Zhu, C. K. W. Jim, G. Yu, Q. Li, Z. A. Li, Y. Liu, J. Qin and B. Z. Tang, Chem. Commun., 2007, 70 RSC; (c) H. Wang, Y. Liang, H. Xie, L. Feng, H. Lu and S. Feng, J. Mater. Chem. C, 2014, 2, 5601 RSC.
- (a) Y. Q. Dong, J. W. Y. Lam, A. J. Qin, J. Z. Liu, Z. Li and B. Z. Tang, Appl. Phys. Lett., 2007, 91, 011111 CrossRef; (b) J. Q. Shi, N. Chang, C. H. Li, J. Mei, C. M. Deng, X. L. Luo, Z. P. Liu, Z. S. Bo, Y. Q. Dong and B. Z. Tang, Chem. Commun., 2012, 48, 10675 RSC.
- (a) Z. J. Zhao, J. W. Y. Lam and B. Z. Tang, Curr. Org. Chem., 2010, 14, 2109 CrossRef CAS; (b) Z. Zhao, J. W. Y. Lam and B. Z. Tang, J. Mater. Chem., 2012, 22, 23726 RSC.
- (a) D. A. Shultz and M. A. Fox, J. Am. Chem. Soc., 1989, 111, 6311 CrossRef CAS; (b) W. Schuddeboom, S. A. Jonker, J. M. Warman, M. P. Dehaas, M. J. W. Vermeulen, W. F. Jager, B. Delange, B. L. Feringa and R. W. Fessenden, J. Am. Chem. Soc., 1993, 115, 3286 CrossRef CAS; (c) J. S. Ma, G. B. Dutt, D. H. Waldeck and M. B. Zimmt, J. Am. Chem. Soc., 1994, 116, 10619 CrossRef CAS; (d) R. W. J. Zijlstra, P. T. van Duijnen, B. L. Feringa, T. Steffen, K. Duppen and D. A. Wiersma, J. Phys. Chem. A, 1997, 101, 9828 CrossRef CAS; (e) C. Dugave and L. Demange, Chem. Rev., 2003, 103, 2475 CrossRef CAS PubMed; (f) J.-S. Yang, K.-L. Liau, C.-Y. Hwang and C.-M. Wang, J. Phys. Chem. A, 2006, 110, 8003 CrossRef CAS PubMed; (g) G. J. Zhao, K. L. Han, Y. B. Lei and Y. S. Dou, J. Chem. Phys., 2007, 127, 094307 CrossRef PubMed; (h) E. W. Debler, G. F. Kaufmann, M. M. Meijler, A. Heine, J. M. Mee, G. Pljevaljcic, A. J. Di Bilio, P. G. Schultz, D. P. Millar, K. D. Janda, I. A. Wilson, H. B. Gray and R. A. Lerner, Science, 2008, 319, 1232 CrossRef CAS PubMed; (i) G.-F. Zhang, Z.-Q. Chen, M. P. Aldred, Z. Hu, T. Chen, Z. Huang, X. Meng and M.-Q. Zhu, Chem. Commun., 2014, 50, 12058 RSC; (j) G.-F. Zhang, H. Wang, M. P. Aldred, T. Chen, Z.-Q. Meng, X. Chen and M.-Q. Zhu, Chem. Mater., 2014, 26, 4433 CrossRef CAS.
- (a) T. Förster and G. Hoffman, Z. Phys. Chem., 1971, 75, 63 CrossRef; (b) J. Saltiel and J. T. D'Agostino, J. Am. Chem. Soc., 1972, 94, 6445 CrossRef CAS; (c) A. Baldridge, S. R. Samanta, N. Jayaraj, V. Ramamurthy and L. M. Tolbert, J. Am. Chem. Soc., 2010, 132, 1498 CrossRef CAS PubMed; (d) C.-C. Hsieh, P.-T. Chou, C.-W. Shih, W.-T. Chuang, M.-W. Chung, J. Lee and T. Joo, J. Am. Chem. Soc., 2011, 133, 2932 CrossRef CAS PubMed.
- (a) N. W. Tseng, J. Z. Liu, J. C. Y. Ng, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams and B. Z. Tang, Chem. Sci., 2012, 3, 493 RSC; (b) J. Wang, J. Mei, R. R. Hu, J. Z. Sun, A. J. Qin and B. Z. Tang, J. Am. Chem. Soc., 2012, 134, 9956 CrossRef CAS PubMed.
- E. P. J. Parrott, N. Y. Tan, R. R. Hu, J. A. Zeitler, B. Z. Tang and E. Pickwell-MacPherson, Mater. Horiz., 2014, 1, 251 RSC.
- N. B. Shuatova, T.-C. Ong, A. F. Cozzolino, V. K. Michaelis, R. G. Griffin and H. Dincă, J. Am. Chem. Soc., 2012, 134, 15061 CrossRef PubMed.
- L. D. Lavis, T. J. Rutkoski and R. T. Raines, Anal. Chem., 2007, 79, 6775 CrossRef CAS PubMed.
- N. L. Bauld, Alkenes I: Properties of Alkenes, http://research.cm.utexas.edu/nbauld/teach/alkenes1.html, retrieved on 15 September 2015.
- G. Fischer, G. Seger, K. A. Muszkat and E. Fischer, J. Chem. Soc., Perkin Trans. 2, 1975, 1569 RSC.
- P. F. Barbara, S. D. Rand and P. M. Rentzepis, J. Am. Chem. Soc., 1981, 103, 2156 CrossRef CAS.
- T. Zhang, H. Ma, Y. Niu, W. Li, D. Wang, Q. Peng, Z. Shuai and W. Z. Liang, J. Phys. Chem. C, 2015, 119, 5040 CAS.
- (a) C. Yao, C. Niu, N. Na, D. He and J. Ouyang, Anal. Chim. Acta, 2015, 853, 375 CrossRef CAS PubMed; (b) A. Kathiravan, K. Sundaravel, M. Jaccob, G. Dhinagaran, A. Rameshkumar, D. Ananth and T. Sivasudha, J. Phys. Chem. B, 2014, 118, 13573 CrossRef CAS PubMed; (c) H. Xiao, K. Chen, D. Cui, N. Jiang, G. Yin, J. Wang and R. Wang, New J. Chem., 2014, 38, 2386 RSC; (d) L. Zang, H. Shang, D. Wei and S. Jiang, Sens. Actuators, B, 2013, 185, 389 CrossRef CAS.
- (a) Y. Cao, M. Yang, Y. Wang, H. Zhou, J. Zheng, X. Zhang, J. Wu, Y. Tian and Z. Wu, J. Mater. Chem. C, 2014, 2, 3686 RSC; (b) G. Liu, M. Yang, L. Wang, J. Zheng, H. Zhou, J. Wu and Y. Tian, J. Mater. Chem. C, 2014, 2, 2684 RSC; (c) M. Yang, D. Xu, W. Xi, L. Wang, J. Zheng, J. Huang, J. Zhang, H. Zhou, J. Wu and Y. Tian, J. Org. Chem., 2013, 78, 10344 CrossRef CAS PubMed.
- (a) H.-T. Feng and Y.-S. Zheng, Chem. – Eur. J., 2014, 20, 195 CrossRef CAS PubMed; (b) H.-T. Feng, S. Song, Y.-C. Chen, C.-H. Shen and Y.-S. Zheng, J. Mater. Chem. C, 2014, 2, 2353 RSC; (c) X. Zhang, X. Zhang, B. Yang and Y. Wei, Chin. J. Polym. Sci., 2014, 32, 1479 CrossRef CAS; (d) X. Zhang, X. Zhang, B. Yang, M. Liu, W. Liu, Y. Chen and Y. Wei, Polym. Chem., 2013, 4, 4317 RSC.
- (a) M. Gao, C. K. Sim, C. W. T. Leung, Q. Hu, G. Feng, F. Xu, B. Z. Tang and B. Liu, Chem. Commun., 2014, 50, 8312 RSC; (b) M. Gao, Q. Hu, G. Feng, B. Z. Tang and B. Liu, J. Mater. Chem. B, 2014, 2, 3438 RSC.
- (a) Z. Song, R. T. K. Kwok, E. Zhao, Z. He, Y. Hong, J. W. Y. Lam, B. Liu and B. Z. Tang, ACS Appl. Mater. Interfaces, 2014, 6, 17245 CrossRef CAS PubMed; (b) Y. Hang, L. Yang, Y. Qu and J. Hua, Tetrahedron Lett., 2014, 55, 6998 CrossRef CAS; (c) P. Mazumdar, D. Das, G. P. Sahoo, G. Salgado-Moran and A. Misra, Phys. Chem. Chem. Phys., 2015, 17, 3343 RSC.
- (a) J. Li, Y. Jiang, J. Cheng, Y. Zhang, H. Su, J. W. Y. Lam, H. H. Y. Sung, K. S. Wong, H. S. Kwok and B. Z. Tang, Phys. Chem. Chem. Phys., 2015, 17, 1134 RSC; (b) R. Yoshii, A. Hirose, K. Tanaka and Y. Chujo, J. Am. Chem. Soc., 2014, 136, 18131 CrossRef CAS PubMed; (c) Y. Gong, J. Liu, Y. Zhang, G. He, Y. Lu, W. B. Fan, W. Z. Yuan, J. Z. Sun and Y. Zhang, J. Mater. Chem. C, 2014, 2, 7552 RSC; (d) S. K. Mellerup and S. Wang, Organometallics, 2014, 33, 5483 CrossRef CAS.
- (a) W. Z. Yuan, P. Lu, S. M. Chen, J. W. Y. Lam, Z. M. Wang, Y. Liu, H. S. Kwok, Y. G. Ma and B. Z. Tang, Adv. Mater., 2010, 22, 2159 CrossRef CAS PubMed; (b) Z. Zhao, B. He and B. Z. Tang, Chem. Sci., 2015, 6, 5347 RSC; (c) Y. Q. Dong, J. W. Y. Lam and B. Z. Tang, J. Phys. Chem. Lett., 2015, 6, 3429 CrossRef CAS PubMed.
- (a) R. T. K. Kwok, C. W. T. Leung, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2015, 44, 4228 RSC; (b) R. Hu and B. Z. Tang, Adv. Polym. Sci., 2015, 269, 17 CrossRef; (c) J. Liang, B. Z. Tang and B. Liu, Chem. Soc. Rev., 2015, 44, 2798 RSC.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Text describing the experimental procedures for the synthesis of TPE-FM and TPE-Fl and their characterization data; tables summarizing the crystallographic data of TPE-FM configurational isomers. CCDC 928304 and 928305. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5tc02924d |
| This journal is © The Royal Society of Chemistry 2016 |