
Probing the role of the backbone carbonyl interaction with the CuA center in azurin by replacing the peptide bond with an ester linkage†
Kevin M.
Clark
a,
Shiliang
Tian
b,
Wilfred A.
van der Donk
*ab and
Yi
Lu
*ab
aDepartment of Biochemistry, University of Illinois at Urbana-Champaign, 600 South Mathews Avenue Urbana, IL 61801, USA. E-mail: vddonk@illinois.edu; yi-lu@illinois.edu
bDepartment of Chemistry, University of Illinois at Urbana-Champaign, 600 South Mathews Avenue Urbana, IL 61801, USA
First published on 25th November 2016
Abstract
The role of a backbone carbonyl interaction with an engineered CuA center in azurin was investigated by developing a method of synthesis and incorporation of a depsipeptide where one of the amide bonds in azurin is replaced by an ester bond using expressed protein ligation. Studies by electronic absorption and electron paramagnetic resonance spectroscopic techniques indicate that, while the substitution does not significantly alter the geometry of the site, it weakens the axial interaction to the CuA center and strengthens the Cu–Cu bond, as evidenced by the blue shift of the near-IR absorption that has been assigned to the Cu–Cu ψ → ψ* transition. Interestingly, the changes in the electronic structure from the replacement did not result in a change in the reduction potential of the CuA center, suggesting that the diamond core structure of Cu2SCys2 is resistant to variations in axial interactions.
Exploring the structure and function of metalloproteins requires intimate knowledge about the roles of residues around the metal-binding sites. Such knowledge forms a strong basis for successful design and engineering of novel metalloproteins with tunable functional properties.1–6 The most common way to acquire such knowledge is site directed mutagenesis (SDM). Because SDM is limited to only ∼20 proteinogenic amino acids, determining the precise role of the residues is difficult, as SDM often simultaneously changes more than one factor, such as electronic and steric effects, at the same time. Therefore, it is nearly impossible to de-convolute the contributions of each individual factor in such instances. To overcome this limitation, non-proteinogenic analogs of natural amino acids have been incorporated into metalloproteins.7–11 For example, we have previously employed Expressed Protein Ligation (EPL) to replace conserved metal-binding amino acids with their iso-structural nonproteinogenic analogs.12,13 Such replacements allowed us to de-convolute steric factors from other factors such as electronic effects and hydrophobicity, thereby firmly establishing the roles of these residues in metalloprotein function.12,14–16
While most residues use side chains to interact with metal centers, increasing numbers of examples have appeared in the literature showing backbone carbonyl oxygens as metal-coordinating ligands. One such example is the CuA center found in cytochrome c oxidase (CcO) and nitrous oxide reductase (N2OR).17–20 CuA contains a dinuclear copper center bridged by two cysteine thiolates wherein each copper is also coordinated by a histidine (Fig. 1). More interestingly, perpendicular to this “diamond core” structural plane, are three carbonyl oxygens of the backbone peptide linkage that are within 2.17 to 4.07 Å of the two copper centers, suggesting that these backbone carbonyl oxygens may play a key role in the formation and fine-tuning of the CuA center.21 However, their roles in CcO, N2OR, and other metalloproteins remain a mystery, as it is not possible to replace the backbone carbonyl oxygens using SDM or most other methods that introduce non-proteinogenic amino acids into proteins. Here we describe the first investigation of the role of backbone carbonyls in the structure and function of the CuA center by developing a method of synthesis and incorporation of a depsipeptide where one of the amide (Ψ–CONHR–) bonds in azurin is replaced by an ester (Ψ–COOR–) bond by EPL, and its subsequent characterization using electronic absorption (UV-vis) and electron paramagnetic resonance (EPR) spectroscopy and cyclic voltammetry. The method developed in this work can be applied to study backbone carbonyl groups in other proteins.
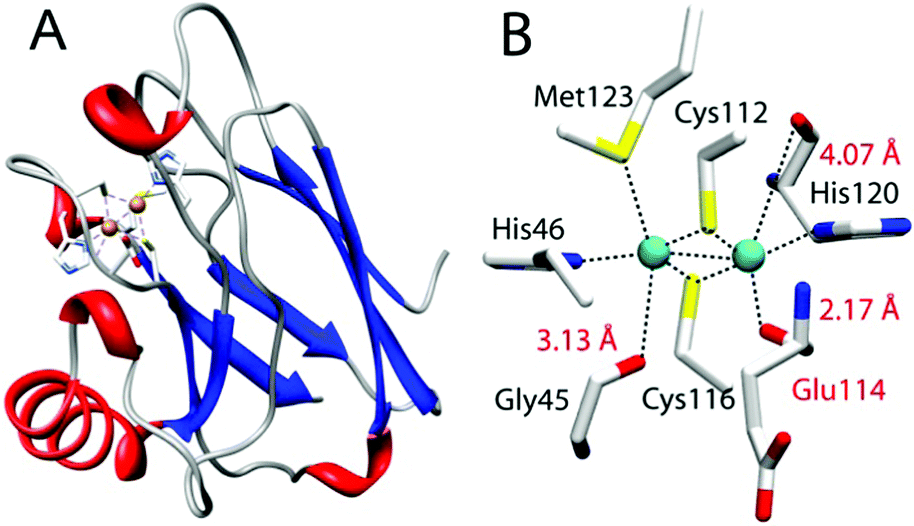 | ||
| Fig. 1 CuA azurin (PDB ID: 1CC3). (A) The full-length CuA azurin scaffold. (B) The CuA site in engineered purple CuA azurin. | ||
We chose an engineered CuA center in azurin from Pseudomonas aeruginosa (hereafter “CuA azurin”), achieved by using loop directed mutagenesis.22 Spectroscopic and X-ray crystallographic studies have confirmed that the CuA site thus engineered in azurin is nearly identical to that in CcO and N2OR, including the “diamond core” structure and the three carbonyl oxygens of the backbone peptide linkage.23–25 It also possesses the same functional properties as the native CuA, such as similar reduction potential and low reorganization energy during electron transfer. CuA azurin is an ideal model to probe the role of the backbone carbonyl oxygens involved in metal coordination. CuA azurin is much smaller, more stable and easier to express and purify with higher yield than CcO and N2OR.22,26,27 The backbone carbonyl of Glu114 is only 2.17 Å from one of the copper ions in CuA azurin (Fig. 1).22,26 This Glu residue would be accessible through EPL using Cys112 as a ligation point for transthioesterification.
Linear synthesis of depsipeptides containing ester linkages has been reported previously in the literature.28–30 However, surprisingly, application of depsipeptides in deciphering protein function is limited primarily to the study of α-helical hydrogen bonding networks or hydrogen bonds to substrates in catalysis, since esters display hydrogen bonding properties different from their amide counterparts.31–33 Depsipeptides are also known for their increased susceptibility to base hydrolysis compared to their peptide counterparts, and some studies have utilized this property within proteins.34–37 Conversely, because of their lability most reported Fmoc-based solid phase peptide synthesis (SPPS) methods for depsipeptides typically involve extensive optimization to minimize hydrolysis and reduce racemization observed when the standard protocols are used for ester synthesis. The synthesis of esters also requires longer reaction times that can further complicate the preparation of complex depsipeptides.
To investigate the interaction between the copper of the CuA center and the carbonyl oxygen of Glu114, a 19-mer peptide (H2N–CSE[ΨCOO]LCGINHALMKGTLTLK–OH) containing an ester between residues Glu114 and Leu115 (called “19-mer depsipeptide” hereafter) was required. We initially adopted a standard Fmoc-based SPPS route to access the 19-mer depsipeptide (Scheme 1). Installation of the ester moiety was completed using DIC/DMAP chemistry to minimize racemization and the reaction was monitored using a hydroxyl specific test similar to the Kaiser Test.30 Using piperidine to deprotect the Fmoc group on the residues following ester incorporation resulted exclusively in a truncated peptide spanning residues Leu114-Lys130. This truncation is likely a result of diketopiperizine formation upon deprotection of the Fmoc from Ser113 using piperidine. To reduce or eliminate diketopiperizine formation, an optimized deprotection strategy using 1% DBU (3 × 1 min) was employed. While the full length 19-mer yield was increased, truncated peptide was still prevalent necessitating an alternative route to obtain the 19-mer depsipeptide.
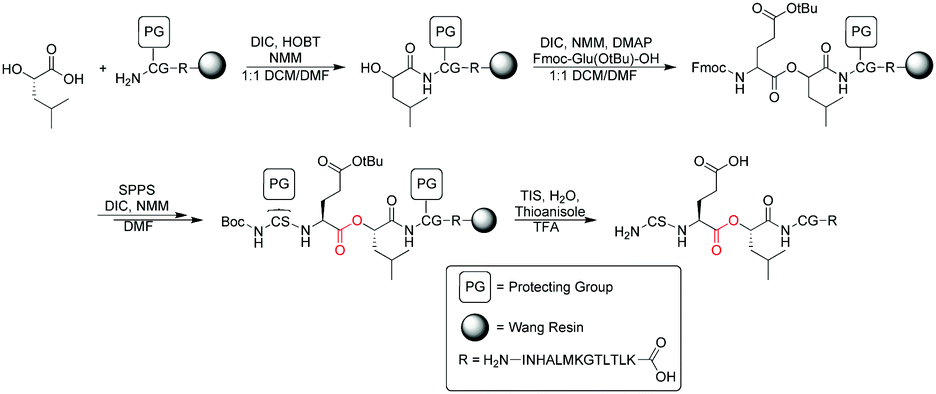 | ||
| Scheme 1 Synthesis of the 19-mer peptide for EPL. | ||
Accordingly, we synthesized the depsipeptide using Boc-based SPPS on PAM resin, thereby eliminating deprotection with a base. Removing exposure of the peptide to bases during synthesis resulted in greater peptide yields and little-to-no truncated peptide (Fig. S1, ESI†). Cys116 was protected with StBu to prevent inter- and intramolecular disulfide formation that otherwise reduced the efficiency of transthioesterification with Cys112 during EPL.38,39
After successful synthesis and purification of the 19-mer depsipeptide, it was used in EPL reactions according to previously published procedures.12,14–16 During the EPL reaction, we noticed poor protein yields with depsipeptides lacking a Cys116-StBu protection, which was attributed to disulfide bond formation with the Cys116 thiol. Furthermore, both the 19-mer depsipeptide and the ester CuA azurin protein product demonstrated high sensitivity of the ester linkage to hydrolysis at any pH above 7.5. MALDI-TOF MS analysis of EPL elutions in buffer at pH > 7.5 showed the primary product consisted of protein hydrolyzed between residues 113 and 114. Therefore, to increase the protein yield and reduce hydrolysis, buffers at pH 7.2 were used for all EPL reactions and protein storage.
Following the EPL reaction using the 19-mer depsipeptide, the product retained the Cys116-StBu protection. A refolding step was necessary to obtain the CuA azurin with the intact metal site. Full-length metal-free apo protein from EPL reactions was refolded in ammonium acetate buffer containing 4 M guanidinium chloride and 0.9 mM dithiothreitol (DTT) immediately following ligation. The protein was then exchanged into ammonium acetate buffer containing 0.9 mM DTT before a final exchange into DTT-free ammonium acetate buffer. Upon titration with Cu(II)SO4, ester CuA azurin was formed. During the titration the protein turned purple in color, which is the same as what was observed for wild type (WT) CuA azurin.
To verify the ester CuA azurin contained the ester moiety instead of the normal amide moiety, base hydrolysis of the protein was performed by exposing the protein to 1 M NaOH. As shown in Fig. 3, exposing the WT CuA azurin to 1 M NaOH for 20 min at room temperature resulted in no change of the protein band by SDS-PAGE (lanes 1 and 2). This result is not surprising, as native protein amide bonds are not susceptible to base hydrolysis. In contrast, exposure of the ester CuA azurin to 1 M NaOH showed partially hydrolyzed product (lanes 4 (20 min) and 5 (60 min)) that was not found in the same protein that was not exposed to 1 M NaOH (lane 3). These results strongly suggest correct incorporation of the ester moiety.
 | ||
| Fig. 2 SDS PAGE results of recombinant WT CuA azurin and EPL ester CuA azurin: (1) WT CuA azurin, (2) WT CuA azurin in the presence of 1 M NaOH for 20 min, (3) ester CuA azurin, (4) ester CuA azurin in the presence of 1 M NaOH for 20 min, (5) ester CuA azurin in the presence of 1 M NaOH for 60 min. The ester CuA azurin protein hydrolyzes at the ester bond in the presence of NaOH, whereas the WT scaffold does not show hydrolysis. | ||
 | ||
| Fig. 3 UV-vis spectra of ester CuA azurin with absorption bands at 475 nm, 530 nm, and 750 nm and WT CuA azurin with absorption bands at 480 nm, 530 nm, and 775 nm. | ||
After confirming successful preparation of CuA azurin containing an ester linkage at position 114, we collected an electronic absorption spectrum in the UV-vis range (UV-vis). As shown in Fig. 3, the ester CuA azurin had absorption bands blue shifted to 475 nm, 530 nm, and 750 nm, in comparison to WT CuA azurin (Fig. 3).26 Specifically the near-IR band, assigned to a Cu–Cu ψ → ψ* transition, shifted from 760 to 750 nm. Since the amide carbonyl group is a stronger base than the ester carbonyl, a weaker interaction would be expected between Cu and the ester carbonyl. Such a weakened Cu carbonyl interaction would result in a stronger Cu–Cu interaction in CuA azurin and a blue shift in the UV-vis spectrum. Previous mutations of the axial Met to Glu in both CuA azurin and the Thermus thermophilus CuA center resulted in a red-shift of the near-IR band from 760 to 855 nm, which was attributed to an elongated Cu–Cu distance due to a strong interaction between the Glu and the Cu.24,40 The weaker interaction as a result of the change from amide to ester carbonyl reported in this work produced an opposite effect of strengthening the Cu–Cu bond.
To investigate the effect of the ester bond on the CuA center further, we collected an X-band EPR spectrum of the ester CuA azurin. A 7-line hyperfine splitting, similar to WT CuA azurin was observed (Fig. 4). Both CuA sites exhibit the same g values and parallel hyperfine couplings, gx = 2.02, gy = 2.01, gz = 2.17 and averaged Az = 56 × 10−4 cm−1 respectively, as indicated by the simulation. The major difference between the spectra of WT CuA and ester CuA azurins is the presence of a type 2 copper species in the ester CuA azurin, which accounts for 60% of all Cu signal; a type 2 copper species is not expected to have contributed significantly to the electronic absorption spectrum in Fig. 2, as it displays very low extinction coefficient due to the forbidden d–d transitions. Interestingly, the EPR parameters (see parameters in Table S1, ESI†) are very similar to those of a C112S/C116S mutant of CuA azurin, suggesting that a portion of the ester CuA azurin contains no Cys coordination to the copper ion. This is not surprising, as the ester CuA azurin was synthesized by EPL and refolded in a high concentration of guanidinium chloride. As a result, a portion of the protein may not fold properly around the metal-binding site to allow Cys coordination to the copper ion. The interaction of the ester with the copper is similar to the native amide and therefore appears not to significantly alter either the structural or electronic integrity of the CuA site. Substitution of the amide with an ester appears to only mildly affect the CuA site and thus likely does not appear to play a significant role in maintaining the site geometry.
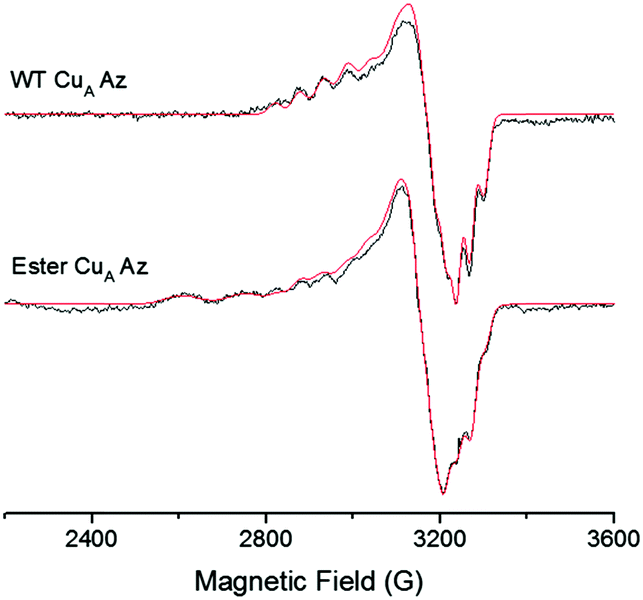 | ||
| Fig. 4 X-band EPR spectra of WT CuA and ester CuA azurin. The 7-line Cu hyperfine is present for ester CuA azurin with similar g values as those of WT CuA azurin. The simulated spectra are shown in red. Parameters for the simulations are presented in the ESI.† | ||
To investigate the effect of the amide to ester replacement on the redox properties of the CuA center, cyclic voltammetry was employed following the reported procedure for WT CuA azurin.24,41 In our hands, the reduction potential of WT CuA azurin was determined to be 277 ± 3 mV vs. NHE (Fig. S2, ESI†), matching the reported value.24,41 Interestingly, the redox potential for the ester CuA azurin under the same condition was 278 ± 5 mV (Fig. S2, ESI†). Previous studies of CuA azurin has shown that mutations of the axial Met to Glu, Gln or Leu resulted in only ∼20 mV changes of the reduction potentials of the CuA azurin.24 The lack of change in reduction potential observed for the amide to ester carbonyl change in another axial position is consistent with previous reports of mutations at the other axial Met123 position24,42 and leads us to conclude that the diamond core structure of Cu2SCys2 shown in Fig. 1B is resistant to variations of axial interactions.
In summary, a CuA azurin variant consisting of an amide-to-ester replacement in the backbone has been prepared to investigate the properties of the carbonyl interaction with the CuA center. This work marks the first such study to investigate backbone carbonyl interactions with bound metal ions in a metalloprotein. While the substitution does not significantly alter the geometry of the site, it weakens the axial interaction to the CuA center and strengthens the Cu–Cu bond, as evidenced by the blue shift of the near-IR absorption that has been assigned to Cu–Cu ψ → ψ* transition. Interestingly, the changes in the electronic structure from the replacement did not result in a change in the reduction potential of the CuA center, suggesting that the diamond core structure of Cu2SCys2 is resistant to variations of axial interactions.
We wish to thank Dr Yang Yu for helpful discussions. This work was supported by grants from CHE 14-13328 (to Y. L.) and the Howard Hughes Medical Institute (to W. A. V.). The authors would like to thank the Illinois EPR facilities including Dr Mark Nilges for help in collecting EPR spectra.
Notes and references
- William F. DeGrado, C. M. Summa, Vincenzo Pavone, F. Nastri and A. Lombardi, Annu. Rev. Biochem., 1999, 68, 779–819 CrossRef CAS PubMed.
- Y. Lu, S. M. Berry and T. D. Pfister, Chem. Rev., 2001, 101, 3047–3080 CrossRef CAS PubMed.
- C. J. Reedy and B. R. Gibney, Chem. Rev., 2004, 104, 617–650 CrossRef CAS PubMed.
- Y. Lu, N. Yeung, N. Sieracki and N. M. Marshall, Nature, 2009, 460, 855–862 CrossRef CAS PubMed.
- M. L. Zastrow and V. L. Pecoraro, Coord. Chem. Rev., 2013, 257, 2565–2588 CrossRef CAS PubMed.
- F. Yu, V. M. Cangelosi, M. L. Zastrow, M. Tegoni, J. S. Plegaria, A. G. Tebo, C. S. Mocny, L. Ruckthong, H. Qayyum and V. L. Pecoraro, Chem. Rev., 2014, 114, 3495–3578 CrossRef CAS PubMed.
- Y. Lu, Curr. Opin. Chem. Biol., 2005, 9, 118–126 CrossRef CAS PubMed.
- K. M. Clark, W. A. van der Donk and Y. Lu, in Methods Enzymol., ed. W. M. Tom and N. A. John, Academic Press, 2009, vol. 462, pp. 97–115 Search PubMed.
- E. C. Minnihan, D. G. Nocera and J. Stubbe, Acc. Chem. Res., 2013, 46, 2524–2535 CrossRef CAS PubMed.
- C. Hu, S. I. Chan, E. B. Sawyer, Y. Yu and J. Wang, Chem. Soc. Rev., 2014, 43, 6498–6510 RSC.
- J. C. Lewis, Curr. Opin. Chem. Biol., 2015, 25, 27–35 CrossRef CAS PubMed.
- D. K. Garner, M. D. Vaughan, H. J. Hwang, M. G. Savelieff, S. M. Berry, J. F. Honek and Y. Lu, J. Am. Chem. Soc., 2006, 128, 15608–15617 CrossRef CAS PubMed.
- K. M. Clark, Y. Yu, N. M. Marshall, N. A. Sieracki, M. J. Nilges, N. J. Blackburn, W. A. van der Donk and Y. Lu, J. Am. Chem. Soc., 2010, 132, 10093–10101 CrossRef CAS PubMed.
- S. M. Berry, M. D. Gieselman, M. J. Nilges, W. A. van der Donk and Y. Lu, J. Am. Chem. Soc., 2002, 124, 2084–2085 CrossRef CAS PubMed.
- S. M. Berry, M. Ralle, D. W. Low, N. J. Blackburn and Y. Lu, J. Am. Chem. Soc., 2003, 125, 8760–8768 CrossRef CAS PubMed.
- M. Ralle, S. M. Berry, M. J. Nilges, M. D. Gieselman, W. A. van der Donk, Y. Lu and N. J. Blackburn, J. Am. Chem. Soc., 2004, 126, 7244–7256 CrossRef CAS PubMed.
- S. Iwata, C. Ostermeier, B. Ludwig and H. Michel, Nature, 1995, 376, 660–669 CrossRef CAS PubMed.
- T. Tsukihara, H. Aoyama, E. Yamashita, T. Tomizaki, H. Yamaguchi, K. Shinzawa-Itoh, R. Nakashima, R. Yaono and S. Yoshikawa, Science, 1995, 669, 1069–1074 CrossRef.
- A. J. Vila and C. O. Fernández, in Handbook on metalloproteins, ed. I. Bertini, A. Sigel and H. Sigel, Dekker, New York, 2001, pp. 813–856 Search PubMed.
- M. N. Morgada, L. A. Abriata, U. Zitare, D. Álvarez-Paggi, D. H. Murgida and A. J. Vila, Angew. Chem., Int. Ed., 2014, 53, 6188–6192 CrossRef CAS PubMed.
- H. Robinson, M. C. Ang, Y.-G. Gao, M. T. Hay, Y. Lu and A. H.-J. Wang, Biochemistry, 1999, 38, 5677–5683 CrossRef CAS PubMed.
- M. Hay, J. H. Richards and Y. Lu, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 461–464 CrossRef CAS.
- S. M. Berry, X. Wang and Y. Lu, J. Inorg. Biochem., 2000, 78, 89–95 CrossRef CAS PubMed.
- H. J. Hwang, S. M. Berry, M. J. Nilges and Y. Lu, J. Am. Chem. Soc., 2005, 127, 7274–7275 CrossRef CAS PubMed.
- H. J. Hwang, N. Nagraj and Y. Lu, Inorg. Chem., 2006, 45, 102–107 CrossRef CAS PubMed.
- M. T. Hay, M. C. Ang, D. R. Gamelin, E. I. Solomon, W. E. Antholine, M. Ralle, N. J. Blackburn, P. D. Massey, X. Wang, A. H. Kwon and Y. Lu, Inorg. Chem., 1998, 37, 191–198 CrossRef CAS.
- M. T. Hay and Y. Lu, J. Biol. Inorg. Chem., 2000, 5, 699–712 CrossRef CAS PubMed.
- I. Coin, M. Beyermann and M. Bienert, Nat. Protoc., 2007, 2, 3247–3256 CrossRef CAS PubMed.
- I. Coin, R. Dölling, E. Krause, M. Bienert, M. Beyermann, C. D. Sferdean and L. A. Carpino, J. Org. Chem., 2006, 71, 6171–6177 CrossRef CAS PubMed.
- O. Kuisle, E. Quiñoá and R. Riguera, J. Org. Chem., 1999, 64, 8063–8075 CrossRef CAS PubMed.
- V. Zickermann, A. Wittershagen, B. O. Kolbesen and B. Ludwig, Biochemistry, 1997, 36, 3232–3236 CrossRef CAS PubMed.
- W. Lu, M. Randal, A. Kossiakoff and S. B. H. Kent, Chem. Biol., 1999, 6, 419–427 CrossRef CAS PubMed.
- B. J. Desai, Y. Goto, A. Cembran, A. A. Fedorov, S. C. Almo, J. Gao, H. Suga and J. A. Gerlt, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15066–15071 CrossRef CAS PubMed.
- P. M. England, H. A. Lester and D. A. Dougherty, Biochemistry, 1999, 38, 14409–14415 CrossRef CAS PubMed.
- J. Guo, J. Wang, J. C. Anderson and P. G. Schultz, Angew. Chem., Int. Ed., 2008, 47, 722–725 CrossRef CAS PubMed.
- T. Watanabe, Y. Miyata, R. Abe, N. Muranaka and T. Hohsaka, ChemBioChem, 2008, 9, 1235–1242 CrossRef CAS PubMed.
- N. A. Bindman, S. C. Bobeica, W. R. Liu and W. A. van der Donk, J. Am. Chem. Soc., 2015, 137, 6975–6978 CrossRef CAS PubMed.
- D. P. Galonić, W. A. van der Donk and D. Y. Gin, J. Am. Chem. Soc., 2004, 126, 12712–12713 CrossRef PubMed.
- J. D. Wade, J. Bedford, R. C. Sheppard and G. W. Tregear, Pept. Res., 1991, 4, 194–199 CAS.
- C. E. Slutter, I. Gromov, J. H. Richards, I. Pecht and D. Goldfarb, J. Am. Chem. Soc., 1999, 121, 5077–5078 CrossRef CAS.
- H. J. Hwang and Y. Lu, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12842–12847 CrossRef CAS PubMed.
- M.-L. Tsai, R. G. Hadt, N. M. Marshall, T. D. Wilson, Y. Lu and E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 14658–14663 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc07274g |
| This journal is © The Royal Society of Chemistry 2017 |