
Palladium-catalyzed oxidative cyclization of aniline-tethered alkylidenecyclopropanes with O2: a facile protocol to selectively synthesize 2- and 3-vinylindoles†
Bo
Cao
a,
Marwan
Simaan
 b,
Ilan
Marek
b,
Yin
Wei
*c and
Min
Shi
*ac
b,
Ilan
Marek
b,
Yin
Wei
*c and
Min
Shi
*ac
aKey Laboratory for Advanced Materials and Institute of Fine Chemicals, School of Chemistry & Molecular Engineering, East China University of Science and Technology, Meilong Road No. 130, Shanghai 200237, China
bThe Mallat Family Laboratory of Organic Chemistry, Schulich Faculty of Chemistry and Lise Meitner-Minerva Center for Computational Quantum Chemistry, Technion-Israel Institute of Technology, Technion City, Haifa 32000, Israel
cState Key Laboratory of Organometallic Chemistry, University of Chinese Academy of Sciences, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: weiyin@mail.sioc.ac.cn; mshi@mail.sioc.ac.cn
First published on 25th November 2016
Abstract
A novel palladium-catalyzed oxidative cyclization of aniline-tethered alkylidenecyclopropanes using molecular oxygen as the terminal oxidant through β-carbon elimination of aminopalladation intermediates is disclosed. The reaction opens up an effective way to obtain a series of 2- and 3-vinylindoles which are important synthetic intermediates in many natural indole derivatives.
Indole derivatives are important structural motifs in many natural products and pharmaceuticals which exhibit a wide range of promising biological activities.1 Moreover, 2- and 3-vinylindoles are key intermediates for the synthesis of biologically interesting polycyclic products.2 During the past few decades, great efforts have been devoted to the development of efficient and practical strategies for the synthesis of these synthetic intermediates.3–7 Although vinylindoles could be accessed through a variety of reactions such as Wittig,3 cross-coupling,4 SN2′-type,5 elimination6 or others,7 current synthetic methodologies lack substrate diversity as functional indoles such as formylindole or haloindole or related species, possessing a leaving group, require an advanced preparation. Therefore, the development of novel strategies leading to a broad range of 2- and 3-substituted vinyl-indoles is still an on-going challenge that one needs to address.
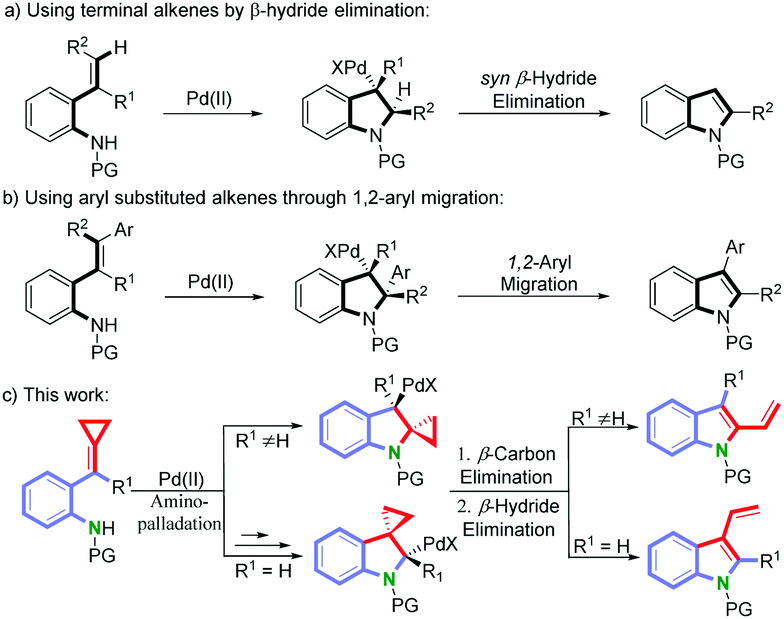
Palladium-catalyzed aza-Wacker-type cyclization has been proved to be an effective approach for the preparation of indoles.8 In this context, two distinct strategies have been explored to establish the indole structures: (a) using terminal alkenes followed by a β-hydride elimination (Scheme 1, eqn (a))8,9 and (b) using aryl substituted alkenes followed by an 1,2-aryl migratory reaction of the aminopalladated intermediate (Scheme 1, eqn (b)).10 Inspired by these methodologies, and with the hope to construct at the same time the indole framework and the vinyl group, we hypothesized that the aminopalladated intermediate may undergo a β-carbon and a β-hydride eliminations continuously to form the required vinylindole in a single-pot operation bypassing the abovementioned two-step pathway. Based on this hypothesis and our ongoing efforts to explore novel synthetic routes to indole-fused scaffolds,11 we decided to prepare ortho-aminoaryl-tethered alkylidenecyclopropanes from the important building blocks alkylidenecyclopropanes (ACPs),12 with aniline.13 Fortunately, we were delighted to observe that these substrates could undergo the above proposed cyclization to selectively give the desired 2- or 3-vinylindoles as described below (Scheme 1, eqn (c)).
 | ||
| Scheme 1 Pd-catalyzed aza-Wacker-type cycloaddition for the synthesis of indole. | ||
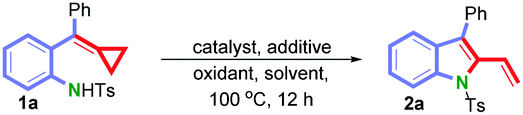
We initially investigated the reaction of 1a, as a model substrate, by employing Pd(OAc)2 as a palladium source and K2CO3 as an additive in toluene at 100 °C under an atmosphere of O2 (balloon). Although in low yield (28%), we were pleased to observe that the reaction proceeded as expected to give 2-vinylindole 2a (Table 1, entry 1). We then further screened different Pd-catalysts, such as Pd2(dba)3·CHCl3, PdCl2, Pd(dppb)Cl2, Pd(PPh3)2Cl2 (Table 1, entries 2–5), and various typical aza-Wacker oxidants including BQ, CuCl2 and AgOAc (Table 1, entries 6–8), but none of these conditions could offer a yield better than 39% (Table 1, entry 5). Additional screening of the nature of solvents and additives (for further details, see the ESI†) revealed that the presence of 1.0 equiv. of KHCO3 in toluene provided the best result (55%, Table 1, entry 9). Next, the nature of ligands on palladium was tested, such as N-heterocyclic carbene and phosphine ligands with either electron-donating or electron-withdrawing substituents, and we were pleased to observe the formation of 2a in 74% yield when the complex Pd[P(4-CF3C6H4)3]2Cl2 was used in the presence of KHCO3 (1.0 equiv.) in toluene under molecular oxygen at 100 °C within 12 h (Table 1, entry 13, for further details, see the ESI†).
|
|
|||||
|---|---|---|---|---|---|
| Entrya | Catalyst | Additive (equiv.) | Solvent | Oxidant | Yieldb (%) |
| a Reactions were performed with 1a (0.2 mmol), additive and 10 mol% of catalyst in solvent (2.0 mL) under O2 (1 atm; balloon) at 100 °C in 12 h. b Yields were determined by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard. c Yield of the isolated products. d Using 2.0 equiv. BQ under Ar. e Using 2.0 equiv. CuCl2 under Ar. f Using 2.0 equiv. AgOAc under Ar. g The reaction was performed in air. | |||||
| 1 | Pd(OAc)2 | K2CO3 (2.0) | Toluene | O2 | 28 (24)c |
| 2 | Pd2(dba)3·CHCl3 | K2CO3 (2.0) | Toluene | O2 | 8 |
| 3 | PdCl2 | K2CO3 (2.0) | Toluene | O2 | 0 |
| 4 | Pd(dppb)Cl2 | K2CO3 (2.0) | Toluene | O2 | 12 |
| 5 | Pd(PPh3)2Cl2 | K2CO3 (2.0) | Toluene | O2 | 39c |
| 6d | Pd(PPh3)2Cl2 | KaCO3 (2.0) | Toluene | BQ | 19 |
| 7e | Pd(PPh3)2Cl2 | K2CO3 (2.0) | Toluene | CuCl2 | 13 |
| 8f | Pd(PPh3)2Cl2 | K2CO3 (2.0) | Toluene | AgOAc | 13 |
| 9 | Pd(PPh3)2Cl2 | KHCO3 (1.0) | Toluene | O2 | 55 |
| 10 | PdIPrCl2 | KHCO3 (1.0) | Toluene | O2 | 38 |
| 11 | Pd[P(4-MeOC6H4)3]2Cl2 | KHCO3 (1.0) | Toluene | O2 | 52 |
| 12 | Pd[P(4-FC6H4)3]2Cl2 | KHCO3 (1.0) | Toluene | O2 | 64 |
| 13 | Pd[P(4-CF3C6H4)3]2Cl2 | KHCO3 (1.0) | Toluene | O2 | 74 (63)c |
| 14 | Pd[P(C6F5)3]2Cl2 | KHCO3 (1.0) | Toluene | O2 | 8 |
| 15g | Pd[P(4-CF3C6H4)3]2Cl2 | KHCO3 (1.0) | Toluene | Air | 50 |
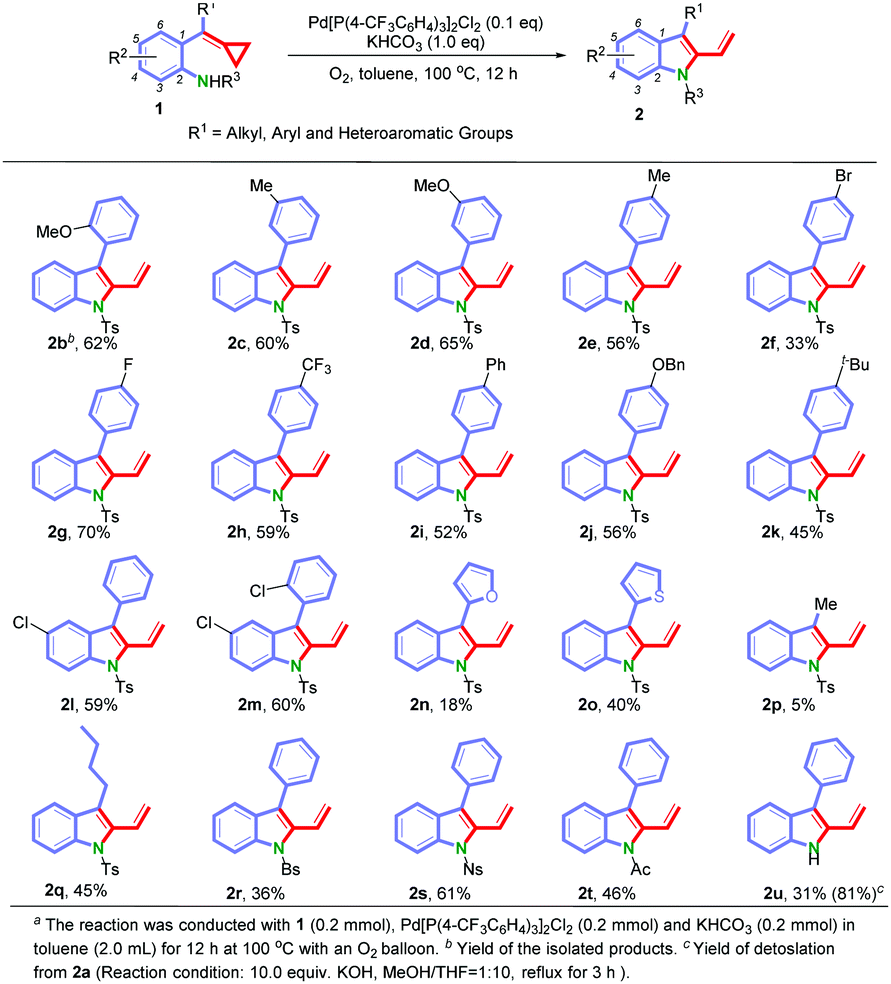
Under the optimized conditions, we next surveyed the substrate scope of this reaction and the results are shown in Scheme 2. We first examined the effect of the position of substituents on the benzene ring. As for substrates 1b–1e, the reactions proceeded smoothly to furnish the corresponding 2-vinylindoles 2b–2e in yields ranging from 56% to 65% regardless of the positions of the substituents (ortho-, meta-, or para-position on the benzene ring). Then, the electronic effect at the para-position of the benzene ring was examined. Substrates with both electron-donating and electron-withdrawing groups could afford the desired products 2f–2k in yields ranging from 33% to 70%. The structure of 2f has been confirmed by X-ray diffraction analysis.14 When substrates 1l (R1 = Ph and R2 = 5-Cl) and 1m (R1 = 2-ClC6H4 and R2 = 5-Cl) were tested, the corresponding products 2l and 2m were obtained in 59% and 60% yields, respectively. To further investigate the scope of this reaction, we prepared functionalized substrates with a heteroaromatic group (furan 1n and thiophene 1o) or with an alkyl chain (methyl group 1p and n-butyl group 1q). The experimental results showed that 1o and 1q were suitable substrates for this reaction under the current reaction conditions, giving the corresponding products 2o and 2q in 40% and 45% yields, respectively. However, the yields of 2n and 2p were low, probably due to the formation of byproducts (for detailed information, see the ESI†). Other N-sulfonyl protecting groups such as 4-bromobenzene sulfonyl (Bs) 1r, 4-nitrobenzene sulfonyl (Ns) 1s and the N-carbonyl protecting group (Ac) 1t were compatible under these reaction conditions. To our delight, when using the substrate 1u without the protecting group on the nitrogen atom, the reaction still proceeded smoothly giving 2u in 31% yield. We also got the product 2u through detosylation of 2a in 81% yield.
 | ||
| Scheme 2 Substrate scope for the synthesis of 2-vinylindoles 2.a | ||
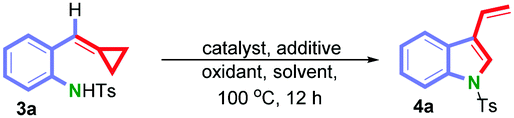
However, when using the substrate 3a, which has no substituent on the alkylidene moiety, the reaction proceeded to furnish the 3-vinylindole 4a in 48% yield (Table 2, entry 5) rather than the 2-vinylindole. Taking into account the lessons from the previous transformation (1 into 2), we reinvestigated this reaction in detail. After screening the ligands of the catalysts and the additives, we found that when 10 mol% of Pd[P(4-MeOC6H4)3]2Cl2 and 1.0 equiv. of KHCO3 were used in toluene under O2 at 100 °C, the yield of 4a could reach 60% (Table 2, entry 2).
|
|
|||||
|---|---|---|---|---|---|
| Entrya | Catalyst | Additive (1.0 equiv.) | Solvent | Oxidant | Yieldb (%) |
| a Reactions were performed with 1a (0.2 mmol), additive and 10 mol% of catalyst in solvent (2.0 mL) under O2 (1 atm; balloon) at 100 °C in 12 h. b Yields were determined by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard. c Yield of the isolated products. d Using 2.0 equiv. KHCO3. | |||||
| 1 | Pd(PPh3)2Cl2 | KHCO3 | Toluene | O2 | 41 |
| 2 | Pd[P(4-MeOC6H4)3]2Cl2 | KHCO3 | Toluene | O2 | 60 (58)c |
| 3 | PdIPrCl2 | KHCO3 | Toluene | O2 | 24 |
| 4 | Pd[P(4-FC6H4)3]2Cl2 | KHCO3 | Toluene | O2 | 56 |
| 5 | Pd[P(4-CF3C6H4)3]2Cl2 | KHCO3 | Toluene | O2 | 48 |
| 6 | Pd[P(C6F5)3]2Cl2 | KHCO3 | Toluene | O2 | 17 |
| 7 | Pd[P(4-MeOC6H4)3]2Cl2 | NaHCO3 | Toluene | O2 | 39 |
| 8 | Pd[P(4-MeOC6H4)3]2Cl2 | Na3PO4 | Toluene | O2 | 50 |
| 9 | Pd[P(4-MeOC6H4)3]2Cl2 | NaOAc | Toluene | O2 | 8 |
| 10d | Pd[P(4-MeOC6H4)3]2Cl2 | KHCO3 | Toluene | O2 | 45 |
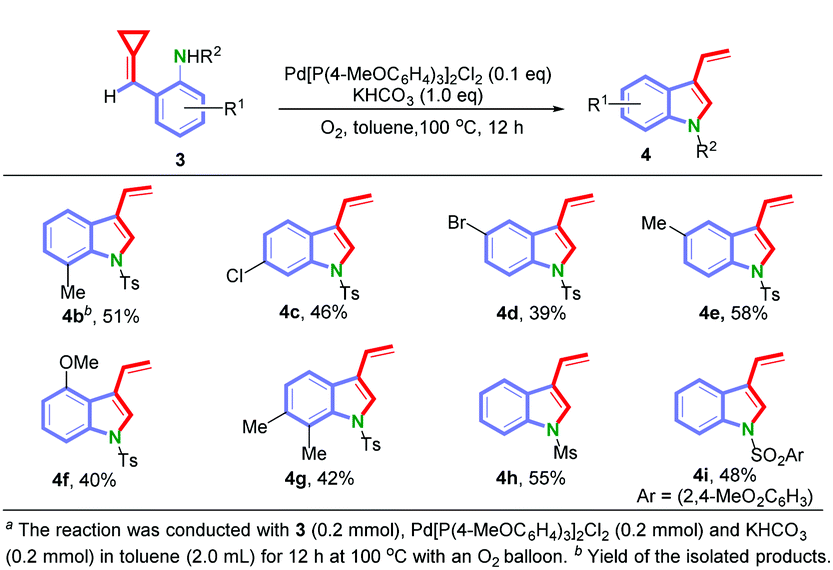
With these optimized reaction conditions in hand, the substrate scope for 3 was explored and the results are shown in Scheme 3. Substrates 3 having both electron-donating and electron-withdrawing substituents (R1) at different positions of the aromatic ring were tolerated, providing the 3-vinylindole derivatives (4b–4g) in moderate yields ranging from 39% to 58%. In addition, when methyl sulfonyl and 2,4-dimethoxybenzene sulfonyl were used as the N-protecting groups, the reactions took place efficiently to afford the corresponding 3-vinylindoles 4h and 4i in 55% and 48% yields, respectively.
 | ||
| Scheme 3 Substrate scope for the synthesis of 3-vinylindoles 4.a | ||
To further investigate the reaction mechanism, two control experiments were performed and the results are shown in Scheme 4. 2-Vinylindole 5 could not be transformed into 3-vinylindole 6 under our standard reaction conditions, suggesting that 5 was not an intermediate in this reaction. Subsequently, a deuterium-labeling experiment was conducted to obtain more information on the mechanism. The treatment of [D]-7 with Pd[P(4-MeOC6H4)3]2Cl2 produced [D]-8 in 60% yield with 66% D content. The deuterium atom at the double bond of the ACP moiety was therefore transferred to the 2-position of the 3-vinylindole, shedding some light on the reaction mechanism.
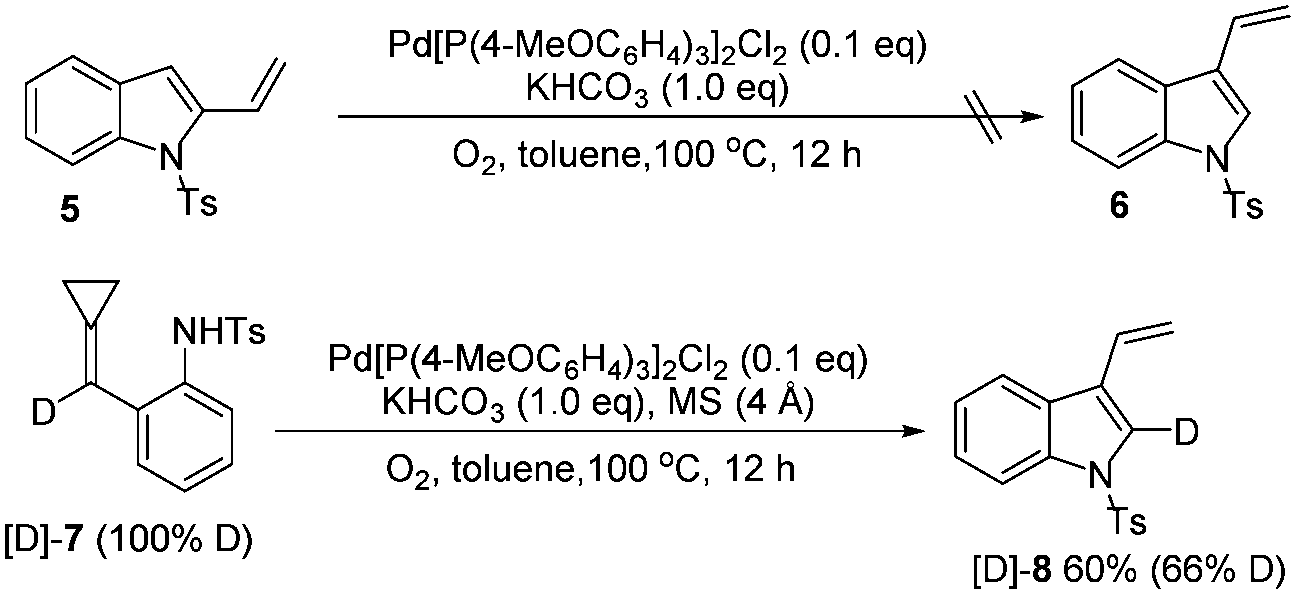 | ||
| Scheme 4 Control experiments to investigate the mechanism. | ||
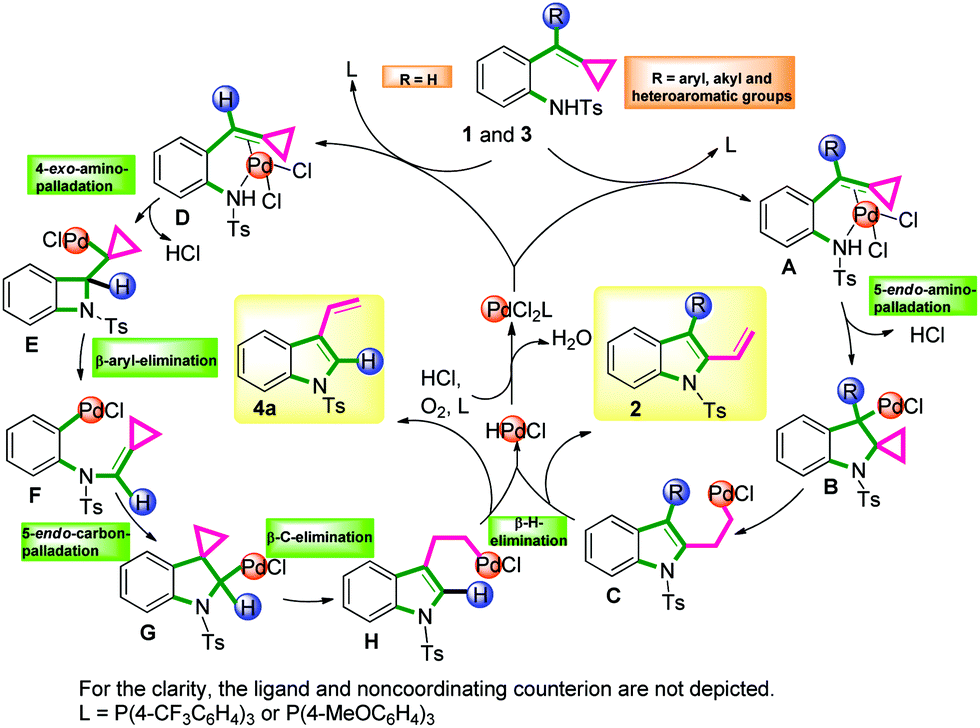
On the basis of previous reports8,9 and our control experiments, a plausible mechanism15 for this reaction is outlined in Scheme 5. First, the electrophilic Pd(II) species coordinated with the substrate 1 or 3 activates the double bond of the ACPs to form complexes A or D. If R ≠ H, A could undergo a 5-endo-aminopalladation to form B. Then following a β-carbon elimination, the Pd-alkyl intermediate C is obtained. Finally, the intermediate C undergoes a β-hydride elimination to form the product 2. The reduced Pd catalyst was then oxidized directly by molecular oxygen to regenerate the Pd(II) catalyst. However, if R = H, complex D would undergo a 4-exo-aminopalladation to yield E. Following a β-aryl elimination, the Pd-aryl intermediate F would be formed and this process transfers the alkylidene cyclopropane to the aniline nitrogen and places Pd on the ortho-position of the aniline. Next, F undergoes a 5-endo-carbonpalladation to generate G. Subsequently, G could undergo a β-carbon elimination and a β-hydride elimination to form the product 4a. Another possible mechanism for the generation of 4a is outlined in the ESI.†
 | ||
| Scheme 5 Proposed mechanism. | ||
In conclusion, a facile and versatile palladium-catalyzed oxidative cyclization of aniline-tethered alkylidenecyclopropanes with molecular oxygen could selectively provide either 2- or 3-vinylindoles in moderate to good yields. Different reaction mechanisms have also been proposed on the basis of control experiments. Further investigations on expanding the scope of this reaction toward a variety of novel and potentially useful polycyclic indole derivatives as well as the application of this protocol to natural product synthesis are in progress.
This work was supported by the Joint NSFC-ISF Research Program, jointly funded by the National Natural Science Foundation of China and the Israel Science Foundation. We are also grateful for the financial support from the National Basic Research Program of China ((973)-2015CB856603) and the National Natural Science Foundation of China (20472096, 21372241, 21361140350, 20672127, 21421091, 21372250, 21121062, 21302203, 20732008 and 21572052).
Notes and references
- (a) R. J. Sundberg, The Chemistry of Indoles, Academic Press, New York, 1970 Search PubMed; (b) Alkaloids Chemical and Biological Perspectives, ed. S. W. Pelletier, Wiley-Interscience, New York, 1983, vol. 4, p. 211 Search PubMed; (c) R. J. Sundberg, Indoles, Academic Press, San Diego, 1996 Search PubMed; (d) J. P. Michael, Nat. Prod. Rep., 1998, 15, 571 RSC; (e) D. J. Faulkner, Nat. Prod. Rep., 1999, 16, 155 RSC; (f) M. Lounasmaa and A. Tolvanen, Nat. Prod. Rep., 2000, 17, 175 RSC; (g) S. Cacchi and G. Fabrizi, Chem. Rev., 2005, 105, 2873 CrossRef CAS PubMed; (h) G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875 CrossRef CAS PubMed.
- (a) W. E. Noland, G.-M. Xia, K. R. Gee, M. J. Konkel, M. J. Wahlstrom, J. J. Condoluci and D. L. Rieger, Tetrahedron, 1996, 52, 4555 CrossRef CAS; (b) C.-G. Yang, J. Wang, X.-X. Tang and B. Jiang, Tetrahedron: Asymmetry, 2002, 13, 383 CrossRef CAS; (c) R. S. Kusurkar and S. K. Goswami, Tetrahedron, 2004, 60, 5315 CrossRef CAS; (d) C. Gioia, A. Hauville, L. Bernardi, F. Fini and A. Ricci, Angew. Chem., Int. Ed., 2008, 47, 9236 CrossRef CAS PubMed; (e) S. B. Jones, B. Simmons and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 13606 CrossRef CAS PubMed; (f) M. L. Bennasar, E. Zulaica, D. Sole, T. Roca, D. Garcia-Diaz and S. Alonso, J. Org. Chem., 2009, 74, 8359 CrossRef CAS PubMed; (g) M. Amat, B. Checa, N. Llor, E. Molins and J. Bosch, Chem. Commun., 2009, 2935 RSC; (h) B. Cheng, J. D. Sunderhaus and S. F. Martin, Org. Lett., 2010, 12, 3622 CrossRef CAS PubMed; (i) M. Amat, N. Llor, B. Checa, E. Molins and J. Bosch, J. Org. Chem., 2010, 75, 178 CrossRef CAS PubMed; (j) C. Gioia, L. Bernardi and A. Ricci, Synthesis, 2010, 161 CAS; (k) C.-W. Zheng, Y.-P. Lu, J.-K. Zhang, X.-K. Chen, Z. Chai, W.-Y. Ma and G. Zhao, Chem. – Eur. J., 2010, 16, 5853 CrossRef CAS PubMed; (l) B. Tan, G. Hernndez-Torres and C. F. Barbas III, J. Am. Chem. Soc., 2011, 133, 12354 CrossRef CAS PubMed; (m) M. Mizutani, F. Inagaki, T. Nakanishi, C. Yanagihara, I. Tamai and C. Mukai, Org. Lett., 2011, 13, 1796 CrossRef CAS PubMed; (n) B. D. Horning and D. W. C. MacMillan, J. Am. Chem. Soc., 2013, 135, 6442 CrossRef CAS PubMed; (o) J. Jin and F.-G. Qiu, Adv. Synth. Catal., 2014, 356, 340 CrossRef CAS; (p) M. L. Bennasar, D. Sole, T. Roca and M. Valldosera, Tetrahedron, 2015, 71, 2246 CrossRef CAS; (q) B. Cheng, J. D. Sunderhaus and S. F. Martin, Tetrahedron, 2015, 71, 7323 CrossRef CAS PubMed; (r) H. Zheng, X. Liu, C. Xu, Y. Xia, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2015, 54, 10958 CrossRef CAS PubMed.
- (a) U. Pindur and M. Eitel, Helv. Chim. Acta, 1988, 71, 1060 CrossRef CAS; (b) M. Eitel and U. Pindur, Synthesis, 1989, 364 CrossRef CAS; (c) U. Pindur and L. Pfeuffer, Monatsh. Chem., 1989, 120, 157 CrossRef CAS; (d) C. A. Faler and M. M. Joullie, Org. Lett., 2007, 9, 1987 CrossRef CAS PubMed; (e) P. N. Naik, A. Khan and R. S. Kusurkar, Tetrahedron, 2013, 69, 10733 CrossRef CAS; (f) S. Zhong, M. Nieger, A. Bihlmeier, M. Shi and S. Braese, Org. Biomol. Chem., 2014, 12, 3265 RSC.
- (a) H. Hagiwara, T. Choshi, H. Fujimoto, E. Sugino and S. Hibino, Chem. Pharm. Bull., 1998, 46, 1948 CrossRef CAS; (b) A. Padwa, S. M. Lynch, J. M. Mejia-Oneto and H. Zhang, J. Org. Chem., 2005, 70, 2206 CrossRef CAS PubMed; (c) Z. Cui, Y.-J. Chen, W.-Y. Gao, C.-G. Feng and G.-Q. Lin, Org. Lett., 2014, 16, 1016 CrossRef CAS PubMed.
- (a) Y. A. M. Mohamed, F. Inagaki, R. Takahashi and C. Mukai, Tetrahedron, 2011, 67, 5133 CrossRef CAS; (b) I. Ambrogio, A. Arcadi, S. Cacchi, G. Fabrizi, A. Goggiamani and F. Marinelli, Tetrahedron, 2013, 69, 9494 CrossRef CAS.
- (a) U. Pindur, M. H. Kim and M. Eitel, Tetrahedron Lett., 1990, 31, 1551 CrossRef CAS; (b) J. Sapi, Y. Grebille, J. Y. Laronze and J. Levy, Synthesis, 1992, 383 CrossRef CAS.
- (a) B. Saroja and P. C. Srinivasan, Synthesis, 1986, 748 CrossRef CAS; (b) T. Balasubramanian and K. K. Balasubramanian, J. Chem. Soc., Chem. Commun., 1994, 1237 RSC; (c) J.-Y. Merour, A.-S. Bourlot and E. Desarbre, Tetrahedron Lett., 1995, 36, 3527 CrossRef CAS; (d) H. Lebel, M. Davi, S. Diez-Gonzalez and S. P. Nolan, J. Org. Chem., 2007, 72, 144 CrossRef CAS PubMed; (e) W. E. Noland, C. L. Etienne and N. P. Lanzatella, J. Heterocycl. Chem., 2011, 48, 381 CrossRef CAS; (f) K. Suzuki, H. Iwasaki, F. Ichiyoshi, M. Tominaga, N. Kojima, M. Ozeki and M. Yamashita, Heterocycles, 2015, 91, 1244 CrossRef CAS; (g) Manisha, S. Dhiman, J. Mathew and S. S. V. Ramasastry, Org. Biomol. Chem., 2016, 14, 5563 RSC; (h) J.-b. Wang, R. Lonsdale and M. T. Reetz, Chem. Commun., 2016, 52, 8131 RSC.
- (a) L. S. Hegedus, G. F. Allen and E. L. Waterman, J. Am. Chem. Soc., 1976, 98, 2674 CrossRef CAS; (b) L. S. Hegedus, G. F. Allen, J. J. Bozell and E. L. Waterman, J. Am. Chem. Soc., 1978, 100, 5800 CrossRef CAS; (c) P. J. Harrington and L. S. Hegedus, J. Org. Chem., 1984, 49, 2657 CrossRef CAS; (d) L. S. Hegedus, Angew. Chem., Int. Ed., 1988, 27, 1113 CrossRef; (e) T. E. Müller and M. Beller, Chem. Rev., 1998, 98, 675 CrossRef; (f) P. J. Harrington, L. S. Hegedus and K. F. McDaniel, J. Am. Chem. Soc., 1987, 109, 4335 CrossRef CAS; (g) T. Kondo, T. Okada and T.-a. Mitsudo, J. Am. Chem. Soc., 2002, 124, 186 CrossRef CAS PubMed; (h) A. Minatti and K. Muñiz, Chem. Soc. Rev., 2007, 36, 1142 RSC; (i) R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111, 2981 CrossRef CAS PubMed.
- (a) M. E. Krolski, A. F. Renaldo, D. E. Rudisill and J. K. Stille, J. Org. Chem., 1988, 53, 1170 CrossRef CAS; (b) A. Kasahara, T. Izumi, S. Murakami, K. Miyamoto and T. Hino, J. Heterocycl. Chem., 1989, 26, 1405 CrossRef CAS; (c) R. C. Larock, T. R. Hightower, L. A. Hasvold and K. P. Peterson, J. Org. Chem., 1996, 61, 3584 CrossRef CAS PubMed; (d) M. Yamaguchi, M. Arisawa and M. Hirama, Chem. Commun., 1998, 1399 RSC; (e) D. R. Adams, M. A. J. Duncton, J. R. A. Roffey and J. Spencer, Tetrahedron Lett., 2002, 43, 7581 CrossRef CAS; (f) D. Tsvelikhovsky and S. L. Buchwald, J. Am. Chem. Soc., 2010, 132, 14048 CrossRef CAS PubMed; (g) S. W. Youn, J. H. Bihn and B. S. Kim, Org. Lett., 2011, 13, 3738 CrossRef CAS PubMed.
- S. W. Youn and S. R. Lee, Org. Biomol. Chem., 2015, 13, 4652 CAS.
- (a) L. Huang, H.-B. Yang, D.-H. Zhang, Z. Zhang, X.-Y. Tang, Q. Xu and M. Shi, Angew. Chem., Int. Ed., 2013, 52, 6767 CrossRef CAS PubMed; (b) Z. Zhang, X.-Y. Tang, Q. Xu and M. Shi, Chem. – Eur. J., 2013, 19, 10625 CrossRef CAS PubMed; (c) L.-Y. Mei, Y. Wei, X.-Y. Tang and M. Shi, J. Am. Chem. Soc., 2015, 137, 8131 CrossRef CAS PubMed; (d) Y.-S. Zhang, X.-Y. Tang and M. Shi, Org. Chem. Front., 2015, 2, 1516 RSC.
- For selected reviews, see: (a) I. Nakamura and Y. Yamamoto, Adv. Synth. Catal., 2002, 344, 111 CrossRef CAS; (b) E. Nakamura and S. Yamago, Acc. Chem. Res., 2002, 35, 867 CrossRef CAS PubMed; (c) A. Brandi, S. Cicchi, F. M. Cordero and A. Goti, Chem. Rev., 2003, 103, 1213 CrossRef CAS PubMed; (d) M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117 CrossRef CAS PubMed; (e) L.-X. Shao and M. Shi, Curr. Org. Chem., 2007, 11, 1135 CrossRef CAS; (f) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (g) M. Shi, L.-X. Shao, J.-M. Lu, Y. Wei, K. Mizuno and H. Maeda, Chem. Rev., 2010, 110, 5883 CrossRef CAS PubMed; (h) H. Pellissier, Tetrahedron, 2010, 66, 8341 CrossRef CAS; (i) G. Audran and H. Pellissier, Adv. Synth. Catal., 2010, 352, 575 CrossRef CAS; (j) A. Masarwa and I. Marek, Chem. – Eur. J., 2010, 16, 9712 CrossRef CAS PubMed; (k) M. Shi, J.-M. Lu, Y. Wei and L.-X. Shao, Acc. Chem. Res., 2012, 45, 641 CrossRef CAS PubMed; (l) A. Brandi, S. Cicchi, F. M. Cordero and A. Goti, Chem. Rev., 2014, 114, 7317 CrossRef CAS PubMed; (m) H. Pellissier, Tetrahedron, 2014, 70, 4991 CrossRef CAS.
- (a) A. I. Siriwardana, M. Kamada, I. Nakamura and Y. Yamamoto, J. Org. Chem., 2005, 70, 5932 CrossRef CAS PubMed; (b) K. Chen, Z. Zhang, Y. Wei and M. Shi, Chem. Commun., 2012, 48, 7696 RSC; (c) K. Chen, R. Sun, Q. Xu, Y. Wei and M. Shi, Org. Biomol. Chem., 2013, 11, 3949 RSC; (d) L.-Z. Yu, Q. Xu, X.-Y. Tang and M. Shi, ACS Catal., 2016, 6, 526 CrossRef CAS; (e) L.-Z. Yu, X.-B. Hu, Q. Xu and M. Shi, Chem. Commun., 2016, 52, 2701 RSC; (f) L.-Z. Yu, Z.-Z. Zhu, X.-B. Hu, X.-Y. Tang and M. Shi, Chem. Commun., 2016, 52, 6581 RSC.
- The crystal data of 2f have been deposited in CCDC with number 1491285.
- We appreciate the referee's important suggestion for the generation of product 4 during the peer-review process.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data of new compounds. CCDC 1491285. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc08731k |
| This journal is © The Royal Society of Chemistry 2017 |