Enhanced visible light photocatalysis over Pt-loaded Bi2O3: an insight into its photogenerated charge separation, transfer and capture†
Received
2nd October 2016
, Accepted 15th November 2016
First published on 15th November 2016
Abstract
In this study, we have fabricated crystalline metallic Pt nanoparticles-loaded α-Bi2O3 microrods (Pt/Bi2O3) using a precipitation method, followed by an impregnation–reduction deposition route. The Pt/Bi2O3 catalysts have much higher photocatalytic activities than that of the pure Bi2O3 for the degradation of RhB and 2,4-DCP under visible light irradiation. The photogenerated charge separation, transfer, and capture in the photocatalysis of Pt/Bi2O3 were investigated in detail according to the various characterizations and analyses of the photogenerated active species and H2O2. It was revealed that the loaded Pt plays an important mediating role in the efficient separation of photogenerated electron–hole pairs by rapidly transferring electrons from the excited Bi2O3 to the surface oxygen. As a result, the reduction of O2 to form H2O2 by the photogenerated electrons was promoted, leaving more holes on the deep valence band to drive the degradation of organic compounds and the production of ˙OH radicals, which were responsible for the enhanced photocatalysis of Pt/Bi2O3.
Introduction
For the full utilization of solar energy, great efforts have been made to develop visible light photocatalytic materials for environmental pollution remediation in the past two decades.1–5 It has been well documented that doping with anions or cations into large-band gap semiconductors, such as TiO2 and SrTiO3, can create localized levels above the valence band (VB) or below the conduction band (CB) in the forbidden band, leading to an extension of the photoactive region to visible light. A considerable number of visible light photocatalytic materials have been developed on the basis of this strategy.1,3,6–8 In addition, some small-band gap semiconductors as potential visible light photocatalytic materials have also recently received significant attention due to their intense absorption in the visible region.9–13 Among the various small-band gap semiconductors, α-Bi2O3 is an attractive material with some valuable merits including thermal stability, non-toxicity similar to TiO2, chemical inertness in neutral water, dielectric permittivity, and wide applications in gas sensors, optical coatings, and photocatalysis.14–16 Moreover, compared to TiO2, α-Bi2O3 possesses a deeper valence band (VB) maximum (∼+3.13 V vs. NHE), which infers an adequately high oxidation power of valence holes. However, α-Bi2O3 alone exhibits a poor photocatalytic performance due to the inability of its conduction band (CB) electrons (∼+0.33 V vs. NHE) to scavenge the surface oxygen molecules through the one-electron reduction reaction process (E0(O2/O2−•) = −0.33 V vs. NHE), which results in the fast recombination of photogenerated electron–hole pairs. In recent years, several approaches have been successfully developed for improving the photocatalytic performance of α-Bi2O3, which include microstructure control,16 fluorination,17 surface modification with Au or Fe(III) clusters,18,19 and combination with other semiconductors such as ZnO and NiO.20,21
Earlier investigations on the semiconductor–metal composites have revealed that semiconductor photocatalysts (TiO2 and ZnO) after Pt loading exhibit highly efficient photocatalytic redox processes since the loaded Pt can act as an electron acceptor to effectively promote the separation of electron–hole pairs with an operation mechanism different from that for loaded Au.22–24 Recently, it has also been reported that the deposition of Pt on several small bang gap semiconductors, such as Bi2WO6, g-C3N4, WO3, and Bi2O3, can enhance their visible light photocatalytic performances for the degradation of organic compounds.25–28 However, the photoinduced charge separation, transfer, and capture involved in the photocatalysis of these Pt-loaded small-band gap semiconductors have not been fully understood. Clarification of these mechanistic problems, we believe, is of great importance for the development of small-band gap semiconductors, especially α-Bi2O3 with a deeper valence band, as efficient visible light photocatalytic materials. In this work, we prepared crystalline metallic Pt nanoparticles-loaded α-Bi2O3 with different Pt loadings (Pt/Bi2O3) by the hydrolysis of bismuth nitrate under basic conditions followed by an impregnation–reduction deposition route. Pt/Bi2O3 has much higher photocatalytic activities for the degradation of Rhodamine B and 2,4-dichlorophenol in aqueous solutions than that of pure Bi2O3 upon visible light irradiation. Moreover, the role of loaded Pt in the separation of photogenerated electron–hole pairs, the transport pathway and capture of the photoinduced charge involved in the photocatalysis of Pt/Bi2O3 are well revealed according to various characterizations and analyses results.
Experimental
Sample preparation
To prepare the pure Bi2O3 sample, 10.78 g of Bi(NO3)3·5H2O was dissolved in a 30 mL aqueous solution of 1.5 M HNO3. NaOH aqueous solution (50% w/v) was slowly added with stirring until yellow precipitate formed at about pH = 13. Subsequently, the mixture was continuously stirred at 80 °C for 2 h, and then the resulting precipitate was collected by centrifugation and washed with deionized water and ethanol several times before drying at 120 °C for 12 h. Finally, the obtained precipitate was further calcined at 450 °C for 5 h to form crystalline Bi2O3 powder.12 The deposition of Pt on Bi2O3 was carried out via an impregnation–reduction deposition route.29,30 Typically, 0.5 g of the as-prepared Bi2O3 powder was impregnated with 10 mL aqueous solution of H2PtCl6 with stirring for 0.5 h. Subsequently, the Bi2O3 powder with adsorbed H2PtCl6 was treated with 2 mL aqueous solution containing NaBH4 (0.1 M) and NaOH (0.5 M) for 0.5 h. During this process, Pt4+ in H2PtCl6 was reduced by NaBH4 to form Pt0 on the Bi2O3 surface. Then, the sample was collected by centrifugation and thoroughly washed with deionized water before drying in an oven at 80 °C for 12 h. The obtained Pt-loaded Bi2O3 samples were denoted as 0.05, 0.075, and 0.1 wt% Pt/Bi2O3, based on the amount of H2PtCl6 used in the abovementioned impregnation–reduction deposition route. For comparison, the pure Bi2O3 was also treated in the same manner in the absence of H2PtCl6.
Characterization
The polycrystalline X-ray diffraction (XRD) patterns of the as-prepared pure Bi2O3 and Pt/Bi2O3 with different Pt loadings were obtained at room temperature by an X-ray diffractometer (Shimadzu, XRD-7000) using Cu Kα X-ray radiation at 40 kV and 30 mA. The UV-vis diffuse reflectance spectra of the samples were obtained by a UV-vis spectrophotometer (Hitachi U-3900) with BaSO4 as the reference standard. The chemical states and composition of the surface elements in the samples were studied via X-ray photoelectron spectroscopy (XPS) (ESCSLAB, 250Xi) using 300 W Al Kα radiation. All binding energies were referenced to the C 1s peak (284.6 eV) of the surface adventitious carbon. Morphologies and microstructures were observed using field-emission electron microscopy (FESEM) (JEOL, JSM-7401) and transmission electron microscopy (TEM) (JEOL-2010), respectively. Photoluminescence (PL) measurements were carried out using a fluorescence spectrophotometer (Hitachi, F-4600).
Photocatalytic activity evaluation
The photocatalytic activities of the as-prepared pure Bi2O3 and Pt/Bi2O3 were evaluated with respect to the degradation of Rhodamine B (RhB) and 2,4-dichlorophenol (2,4-DCP) in aqueous solution, respectively. In a typical procedure, 100 mg of catalyst sample was suspended in 100 mL of RhB (∼10−5 M) or 2,4-DCP (∼10−4 M) aqueous solution. A 300 W Xe arc lamp (CHF-XM 150, Beijing Trusttech. Co. Ltd) with a λ > 420 nm cutoff filter was used as a visible light source. Prior to irradiation, the suspension was magnetically stirred in the dark for 1 h to ensure the establishment of an adsorption/desorption equilibrium. At the given time interval during the photoreaction, 3 mL of the reaction suspension was sampled and centrifuged to remove the catalyst particles. The RhB concentration was measured by monitoring the variation in the absorption spectrum for the absorbance at 554 nm with a spectrophotometer (Shimadzu, UV-3600). Quantitative analysis of 2,4-DCP was carried out using high-performance liquid chromatography (HPLC) (Agilent, 1100) with a C18 column. The eluent consisted of 65% acetonitrile, 35% water, and 0.1% phosphoric acid.
To study the active species involved in the photocatalysis, triethanolamine (TEOA, 10 mM) as an effective hole scavenger and tert-butyl alcohol (TBA, 10 mM) as an ˙OH radical scavenger were chosen, respectively, to participate in the photocatalytic degradation of RhB over these catalysts.
Analysis of the photogenerated H2O2
The in situ photogenerated H2O2 in the visible light irradiated (λ ≥ 420 nm) suspension of the catalysts (2.0 g L−1) was analyzed in the presence of methanol (1.0 M) as an electron donor using the colorimetric DPD-POD method.31
Results and discussion
Phase structure and morphology
XRD was used to identify the phase structure and composition of the as-prepared samples. Fig. 1 shows the XRD patterns of the pure Bi2O3 and Pt/Bi2O3 samples with different Pt loadings. All the samples exhibited the single characteristic monoclinic phase of well-crystallized α-Bi2O3 (JCPDS No. 41-1449). No apparent characteristic XRD peaks associated with Pt were observed in all the detected samples. These results suggest that loading Pt does not alter the phase structure of the substrate Bi2O3 and that the loaded Pt particles are highly dispersed on the substrate. The presence and form of the loaded Pt are further confirmed by the TEM observation and XPS analysis mentioned below.
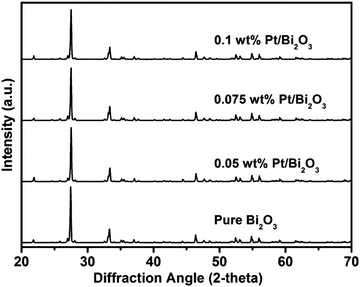 |
| | Fig. 1 Polycrystalline X-ray diffraction patterns of pure Bi2O3 and Pt/Bi2O3 with different Pt loadings. | |
The FESEM image (Fig. 2a) reveals that the pure Bi2O3 has microrod-shaped structures with diameters ranging from 500 to 800 nm and lengths of about several micrometers, which is similar to that of the previously reported α-Bi2O3.17,18 No obvious differences in both morphology and size of the substrate were observed after the Pt loading (Fig. 2b). The TEM images (Fig. 2c) of the pure Bi2O3 and 0.075 wt% Pt/Bi2O3 samples show that numerous nanoparticles with the size of several nanometers are well dispersed on the surface of the Bi2O3 microrod substrate after 0.075 wt% Pt loading. As observed in the typical high-resolution TEM (HRTEM) image (Fig. 2d) corresponding to a region of 0.075 wt% Pt/Bi2O3 in Fig. 2c, both the nanoparticles and substrate are well crystallized. The measured lattice fringes of 0.23 and 0.26 nm well match the crystallographic planes of the metallic Pt(111) and α-Bi2O3(002), respectively.32–34 These observations indicate that the crystalline metallic Pt nanoparticles were uniformly dispersed on the surface of the substrate Bi2O3.
 |
| | Fig. 2 FESEM images of pure Bi2O3 (a) and 0.075 wt% Pt/Bi2O3 (b); TEM images of pure Bi2O3 and 0.075 wt% Pt/Bi2O3 (c); and typical HRTEM image of 0.075 wt% Pt/Bi2O3 (d). | |
XPS analysis and optical property
X-ray photoelectron spectroscopy (XPS) was employed to investigate the compositions and chemical states of the surface elements in the samples. The full-scale XPS spectra of pure Bi2O3 and 0.075 wt% Pt/Bi2O3, as shown in Fig. S1 (ESI†), clearly reveal the presence of Bi, O, and C in the pure Bi2O3, and Bi, O, Pt, and C in the 0.075 wt% Pt/Bi2O3. The atomic ratios of Bi to O in both the samples were approximately 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3. These results further confirm the purity of Bi2O3 in both samples and the presence of Pt in the 0.075 wt% Pt/Bi2O3. The high-resolution XPS spectra of the Bi 4f core-level for pure Bi2O3 and 0.075 wt% Pt/Bi2O3 are displayed in Fig. 3a. It can be clearly seen that the Bi 4f spectrum of pure Bi2O3 consists of two well-defined photoelectron peaks located at 158.8 and 164.1 eV, which can be assigned to Bi 4f7/2 and Bi 4f5/2, respectively. The two peak positions and their separation (Δ = 5.3 eV) are well consistent with the characteristics of the Bi3+ species in α-Bi2O3.19 Note that the binding energies of Bi 4f7/2 and Bi 4f5/2 obviously shift to 159.0 and 164.3 eV, respectively, after Pt loading in the case of 0.075 wt% Pt/α-Bi2O3, whereas their separation value remains the same as that of the corresponding pure Bi2O3. As reported earlier,22 when a semiconductor and metal nanoparticles are in contact, electron migration between them occurs until Fermi level equilibration of the composite is established. In the present case, the substrate α-Bi2O3 is an n-type semiconductor with the Fermi level close to its conduction band (∼+0.33 V vs. NHE), whereas metal Pt has a Fermi level of about + 1.0 V vs. NHE.35,36 Therefore, electron transfer from the substrate α-Bi2O3 to the loaded Pt occurs for Fermi level equilibration, which results in the observed shift to higher binding energies. The observed shift in the binding energy also reflects a well-contacted interface between the Pt nanoparticles and the substrate in the composite. Fig. 3b presents the high-resolution XPS spectrum of the Pt 4f core-level for the 0.075 wt% Pt/Bi2O3 sample. As revealed in Fig. 3b, the Pt 4f signal can be fitted into four symmetric peaks, which suggests that the Pt species are present in two forms. The Pt 4f7/2 peak at 71.1 and Pt 4f5/2 peak at 74.5 eV originate from metallic Pt0, whereas the other pair of Pt XPS peaks with binding energies at 72.5 and 75.9 eV correspond to Pt 4f7/2 and Pt 4f5/2 of Pt2+, respectively.37 According to the relative XPS areas, the atomic percentages of Pt0 and Pt2+ in the total Pt species of the sample are estimated to be 91.2% and 8.8%, respectively, confirming that the loaded Pt is mainly present in the metallic state in Pt/Bi2O3. The presence of a small amount of Pt2+ species might be attributed to the formation of a Pt–O bond caused by the oxygen chemisorption on the surface of the Pt nanoparticles.38,39
:3. These results further confirm the purity of Bi2O3 in both samples and the presence of Pt in the 0.075 wt% Pt/Bi2O3. The high-resolution XPS spectra of the Bi 4f core-level for pure Bi2O3 and 0.075 wt% Pt/Bi2O3 are displayed in Fig. 3a. It can be clearly seen that the Bi 4f spectrum of pure Bi2O3 consists of two well-defined photoelectron peaks located at 158.8 and 164.1 eV, which can be assigned to Bi 4f7/2 and Bi 4f5/2, respectively. The two peak positions and their separation (Δ = 5.3 eV) are well consistent with the characteristics of the Bi3+ species in α-Bi2O3.19 Note that the binding energies of Bi 4f7/2 and Bi 4f5/2 obviously shift to 159.0 and 164.3 eV, respectively, after Pt loading in the case of 0.075 wt% Pt/α-Bi2O3, whereas their separation value remains the same as that of the corresponding pure Bi2O3. As reported earlier,22 when a semiconductor and metal nanoparticles are in contact, electron migration between them occurs until Fermi level equilibration of the composite is established. In the present case, the substrate α-Bi2O3 is an n-type semiconductor with the Fermi level close to its conduction band (∼+0.33 V vs. NHE), whereas metal Pt has a Fermi level of about + 1.0 V vs. NHE.35,36 Therefore, electron transfer from the substrate α-Bi2O3 to the loaded Pt occurs for Fermi level equilibration, which results in the observed shift to higher binding energies. The observed shift in the binding energy also reflects a well-contacted interface between the Pt nanoparticles and the substrate in the composite. Fig. 3b presents the high-resolution XPS spectrum of the Pt 4f core-level for the 0.075 wt% Pt/Bi2O3 sample. As revealed in Fig. 3b, the Pt 4f signal can be fitted into four symmetric peaks, which suggests that the Pt species are present in two forms. The Pt 4f7/2 peak at 71.1 and Pt 4f5/2 peak at 74.5 eV originate from metallic Pt0, whereas the other pair of Pt XPS peaks with binding energies at 72.5 and 75.9 eV correspond to Pt 4f7/2 and Pt 4f5/2 of Pt2+, respectively.37 According to the relative XPS areas, the atomic percentages of Pt0 and Pt2+ in the total Pt species of the sample are estimated to be 91.2% and 8.8%, respectively, confirming that the loaded Pt is mainly present in the metallic state in Pt/Bi2O3. The presence of a small amount of Pt2+ species might be attributed to the formation of a Pt–O bond caused by the oxygen chemisorption on the surface of the Pt nanoparticles.38,39
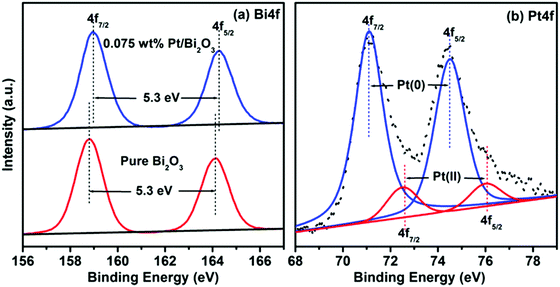 |
| | Fig. 3 (a) Bi 4f XPS spectra of pure Bi2O3 and 0.075 wt% Pt/Bi2O3. (b) Pt 4f XPS spectrum of 0.075 wt% Pt/Bi2O3. | |
The optical absorption properties of pure Bi2O3 and Pt/Bi2O3 with different Pt loadings were determined using UV-vis diffuse reflectance spectroscopy. As shown in Fig. 4, pure Bi2O3 exhibits an intense absorption with a threshold at the wavelength near 455 nm. This absorption originates from the excitation of electrons from the valence band to the conduction band in Bi2O3, which is well consistent with the intrinsic bandgap of α-Bi2O3 (Eg = 2.8 eV). Compared to the spectrum of pure Bi2O3, a broad absorption in the range of 450–800 nm is observed for all the Pt/Bi2O3 samples, which is caused by the change in the sample color from yellow to dark gray after Pt loading. Moreover, the weak surface plasmon resonance of the loaded Pt also contributes to the observed absorption based on the related report.40
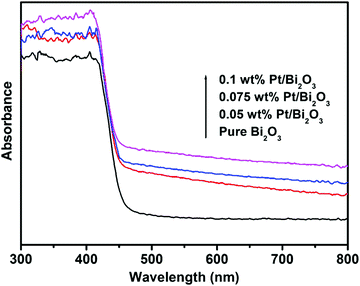 |
| | Fig. 4 UV-vis diffuse reflectance spectra of pure Bi2O3 and Pt/Bi2O3 with different Pt loadings. | |
Photocatalytic property
To explore the effects of the Pt loading on the photocatalytic properties, the photocatalytic activities of pure Bi2O3 and Pt/Bi2O3 with different Pt loadings were examined by the degradation of RhB and 2,4-DCP in aqueous solution under visible light irradiation (λ > 420 nm). Fig. 5a displays the photocatalytic degradation of RhB over these samples under visible light irradiation. The control experiment shows that RhB in an aqueous solution is stable against direct photolysis. With the excitation of Bi2O3 under visible light irradiation, all the Pt/Bi2O3 samples exhibit photocatalytic activities much higher than that of the pure Bi2O3 for the degradation of RhB, which indicates that Pt loading is an efficient method for improving the photocatalysis of Bi2O3. Moreover, it can be found that the enhanced activity is also dependent on the amount of loaded Pt. The 0.075 wt% Pt loading displays optimum activity. Approximately 85% of RhB was degraded over 0.075 wt% Pt/Bi2O3 under irradiation for 3 h. After this, the activity starts to drop when the Pt loading is 0.1 wt%. The observed decrease in the activity over 0.1 wt% Pt/Bi2O3 can be ascribed to the excess Pt nanoparticles that shield the incident light reaching Bi2O3 and diminish the active sites on Bi2O3. Similar enhancement in the photocatalytic activity after Pt loading was observed from the photocatalytic degradation of 2,4-DCP, which is an organic compound that does not absorb visible light, over 0.075 wt% Pt/Bi2O3 (Fig. 5b).
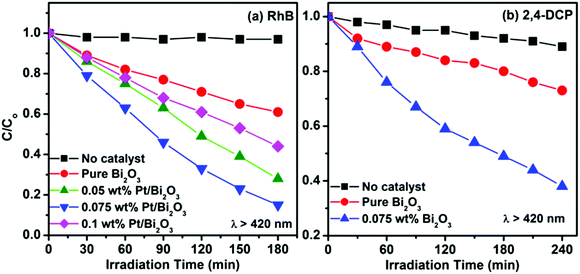 |
| | Fig. 5 (a) Photocatalytic degradation of RhB over pure Bi2O3 and Pt/Bi2O3 with different Pt loadings under visible light irradiation. (b) Photocatalytic degradation of 2,4-dichlorophenol (2,4-DCP) over pure Bi2O3 and 0.075 wt% Pt/Bi2O3 under visible light irradiation. | |
The photocatalytic stability and reusability of the Pt/Bi2O3 catalyst was determined by conducting cyclic degradation experiments with fresh RhB solution and the used catalyst from the previous runs under the same conditions. As shown in Fig. S2 (ESI†), no obvious decrease in the photocatalytic activity for RhB degradation over 0.075 wt% Pt/Bi2O3 is observed after recycle use for four times, which demonstrates the good stability of the catalyst.
Analysis of the photogenerated active species and H2O2
The active species in the photocatalysis of pure Bi2O3 and Pt/Bi2O3 was investigated by the addition of TEOA as an effective hole scavenger or TBA as an ˙OH radical scavenger (with a rate constant of k = 6 × 108) in the photocatalytic reaction of RhB degradation over both catalysts.41 As depicted in Fig. 6a and b, over both catalysts, the degradation efficiencies of RhB obviously decreased with the addition of the ˙OH radical scavenger TBA, whereas no degradation of RhB occurred in the presence of TEOA, as an effective hole scavenger. These results reveal that both ˙OH radicals and photogenerated holes are responsible for the degradation of RhB over pure Bi2O3 and Pt/Bi2O3. From the viewpoint of thermodynamics, the photogenerated electrons on the conduction band of Bi2O3 (∼+0.33 V vs. NHE) can energetically reduce the surface oxygen molecules to form H2O2 through a two-electron reduction process (reaction (1), E0(O2/H2O2) = +0.695 V vs. NHE). Thus, with the excitation of two catalysts under visible light irradiation, there are two possible production paths for ˙OH radicals over the two catalysts. One is the reductive path of the conduction band electron through reactions (1) and (2), the other is the oxidative path of the valence band hole through reaction (3).| | | 2eCB− + O2 + 2H+ → H2O2 | (1) |
| | | H2O2 + eCB−→ OH− + ˙OH | (2) |
| | | hVB+ + H2O (or –OH) → ˙OH | (3) |
No degradation of RhB over the two catalysts in the presence of the hole scavenger TEOA, as evidenced in Fig. 6, clearly reveals that reaction (3) is the sole source of ˙OH radical generation, whereas the production path through reaction (2) is negligible in both systems. This indicates that the photogenerated holes play a determining role in the photocatalytic activity by directly degrading RhB and oxidizing water or hydroxyl groups to form ˙OH radicals. The effective production yield of the photogenerated holes entirely depends on the separation extent of the photogenerated electron–hole pairs. According to the results of the photocatalytic experiments, as shown in Fig. 5a and b, it can be concluded that efficient separation over Bi2O3 is successfully achieved after Pt loading. This conclusion is also well supported by the photoluminescence (PL) spectra of pure Bi2O3 and 0.075 wt% Pt/Bi2O3 (Fig. 7). It is well known that PL emission is the result of the combination of photogenerated electrons and holes. From Fig. 7, the strong peak at around 455 nm for pure Bi2O3 is ascribed to the emission of the band gap transition. The intensity of the PL emission peak is dramatically reduced after Pt loading, indicating that Pt nanoparticles diminish the recombination of the photogenerated electron–hole pairs and that an efficient separation is achieved over Pt/Bi2O3.
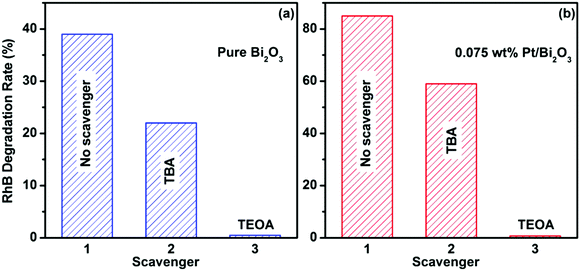 |
| | Fig. 6 Photocatalytic degradation of RhB over pure Bi2O3 (a) and 0.075 wt% Pt/Bi2O3 (b) in the presence of different scavengers under visible light irradiation for 3 h. | |
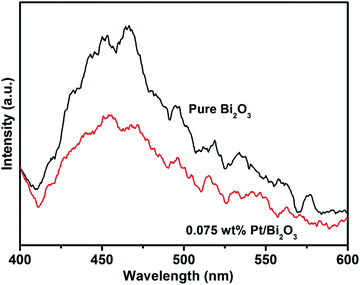 |
| | Fig. 7 PL spectra of pure Bi2O3 and 0.075 wt% Pt/Bi2O3. | |
Previous studies on the semiconductor–noble metals (Au, Ag and Pt) indicate that loading noble metals on the semiconductors can enhance the separation of electron–hole pairs photogenerated on the semiconductor.22,23 These noble metal nanoparticles play a mediating role to store and shuttle or directly transfer the electrons from the photoexcited semiconductor to an acceptor in a photocatalytic process. Therefore, in the present study, we need to understand that where the electrons go after they are transferred to Pt nanoparticles from the excited Bi2O3. The Fermi level of Pt (∼+1.0 V vs. NHE) is more positive than that of the reaction (1) (E0(O2/H2O2) = +0.695 V vs. NHE); however, it shifts to a more negative position due to the establishment of Fermi level equilibration between Bi2O3 and Pt, as evidenced by the abovementioned XPS measurements. Thus, after the photoinduced electron transfer from the excited Bi2O3 to Pt nanoparticles, it becomes possible for the Pt nanoparticles to act as a reduction site to reduce the surface oxygen molecules to form H2O2 through reaction (1). The analysis of the in situ photogenerated H2O2 in the visible light irradiated suspensions of pure Bi2O3 and 0.075 wt% Pt/Bi2O3 (Fig. 8) confirms the production of H2O2 in both systems. The production of H2O2 is much more over Pt/Bi2O3 than that over pure Bi2O3, indicating that more photogenerated electron–hole pairs are efficiently separated in Pt/Bi2O3. In contrast, in the case of pure Bi2O3, although the conduction band level (∼+0.33 V vs. NHE) is much more negative than that of reaction (1), fast recombination of the photogenerated electron–hole pairs occurs in the absence of Pt. This result is in accordance with the photocatalytic performances of pure Bi2O3 and Pt/Bi2O3, as shown in Fig. 5a and b. Note that the Pt/Bi2O3 system shows a much higher initial production rate of H2O2 than that of pure Bi2O3, which is different from that of Au/Bi2O3 in our previous report.18 This observation is consistent with the role of Pt nanoparticles in the separation of photogenerated electron–hole pairs.23 In the presence of Pt nanoparticles, rapid transfer of electrons from Pt to surface oxygen molecule occurs without electron accumulation on Pt. However, in the presence of Au nanoparticles, the electrons from the excited Bi2O3 accumulate on Au nanoparticles and do not transfer to the surface oxygen until their Fermi level merges with the conduction band of Bi2O3. The results of the analysis of photogenerated H2O2 clarify very well the transfer and capture of the electrons photogenerated over pure Bi2O3 and Pt/Bi2O3.
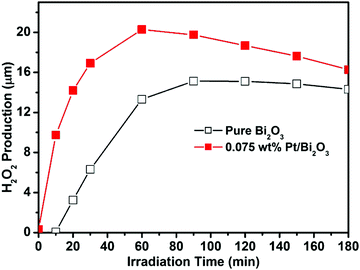 |
| | Fig. 8
In situ photogenerated H2O2 in the visible light irradiated suspensions of pure Bi2O3 and 0.075 wt% Pt/Bi2O3 in the presence of methanol as an electron donor. | |
According to the abovementioned analysis of the photogenerated active species and H2O2, the photocatalytic process over Pt/Bi2O3 is illustrated in Scheme 1. Due to the decomposition of H2O2 into H2O and O2 in the photoreaction,42 especially under the promotion of Pt catalysis,43 the concentration of H2O2 is found to gradually decrease after reaching an optimum and the decay rate is faster over Pt/Bi2O3 than that over pure Bi2O3.
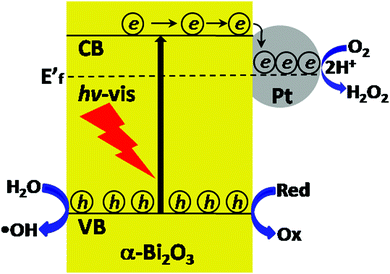 |
| | Scheme 1 Schematic for the photocatalysis over Pt/Bi2O3. | |
Conclusions
In summary, crystalline metallic Pt nanoparticles are successfully deposited on the surface of Bi2O3 microrods via an impregnation–reduction route. Upon visible light irradiation, the photocatalytic activities of Bi2O3 for the degradation of RhB and 2,4-DCP are significantly enhanced after Pt nanoparticle loading. The Pt nanoparticles loaded on Bi2O3 function as an electron mediator to effectively promote the separation of photogenerated electron–hole pairs. As a result, the reduction of O2 to H2O2 by the photogenerated electrons is greatly enhanced, leaving more holes on the conduction band of Bi2O3 for the degradation of organic compounds and production of ˙OH radicals. This work, we believe, provides some useful information for the design and development of the small-band gap semiconductor Bi2O3 with a deep valence band as an efficient visible light photocatalyst.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21273281) and National Basic Research Program of China (973 Program, No. 2013CB632405).
Notes and references
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- H. Kisch and W. Macyk, ChemPhysChem, 2002, 3, 399–400 CrossRef CAS PubMed.
- X. Qiu, M. Miyauchi, H. Yu, H. Irie and K. Hashimoto, J. Am. Chem. Soc., 2010, 132, 15259–15267 CrossRef CAS PubMed.
- P. Madhusudan, J. Ran, J. Zhang, J. Yu and J. Liu, Appl. Catal., B, 2011, 110, 286–295 CrossRef CAS.
- X. Sun and J. Lin, J. Phys. Chem. C, 2009, 113, 4970–4975 CAS.
- D. Wang, T. Kato and J. Ye, J. Am. Chem. Soc., 2008, 130, 2724–2725 CrossRef CAS PubMed.
- K. Yang, Y. Dai, B. Huang and M.-H. Whangbo, Chem. Mater., 2008, 20, 6528–6534 CrossRef CAS.
- K. Yang, Y. Dai and B. Huang, ChemPhysChem, 2009, 10, 2327–2333 CrossRef CAS PubMed.
- X. C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonieti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- K. Brezseinski, R. Ostermann, P. Hartmann, J. Perlich and T. Brezseinski, Chem. Mater., 2010, 22, 3079–3085 CrossRef.
- Z.-G. Zhao and M. Miyauchi, Angew. Chem., Int. Ed., 2008, 47, 7051–7055 CrossRef CAS PubMed.
- A. Hameed, T. Montini, V. Gombac and P. Fornasiero, J. Am. Chem. Soc., 2008, 130, 9658–9659 CrossRef CAS PubMed.
- J. Eberl and H. Kisch, Photochem. Photobiol. Sci., 2008, 7, 1400–1406 CAS.
- J. N. Clifford, E. Palomares, M. K. Nazeeruddin and M. Gratzel, J. Phys. Chem. C, 2007, 111, 6561–6567 CAS.
- L. Leontie, M. Caraman, M. Delibas and G. I. Rusu, Mater. Res. Bull., 2001, 36, 1629–1637 CrossRef CAS.
- L. Zhou, W. Wang, H. Xu, S. Sun and M. Shang, Chem. – Eur. J., 2009, 15, 1776–1782 CrossRef CAS PubMed.
- H.-Y. Jiang, J. Liu, K. Cheng, W. Sun and J. Lin, J. Phys. Chem. C, 2013, 117, 20029–20036 CAS.
- H.-Y. Jiang, K. Cheng and J. Lin, Phys. Chem. Chem. Phys., 2012, 14, 12114–12121 RSC.
- W. Sun, H. Zhang and J. Lin, J. Phys. Chem. C, 2014, 118, 17626–17632 CAS.
- A. Hameed, V. Gombac, T. Montini, L. Felisari and P. Fornasiero, Chem. Phys. Lett., 2009, 483, 254–261 CrossRef CAS.
- A. Hameed, V. Gombac, T. Montini, M. Graziani and P. Fornasiero, Chem. Phys. Lett., 2009, 472, 212–216 CrossRef CAS.
- V. Subramanian, E. E. Wolf and P. V. Kamat, J. Am. Chem. Soc., 2004, 126, 4943–4950 CrossRef CAS PubMed.
- A. Wood, M. Giersig and P. Mulvaney, J. Phys. Chem. B, 2001, 105, 8810–8815 CrossRef CAS.
- V. Subramanian, E. E. Wolf and P. V. Kamat, J. Phys. Chem. B, 2003, 107, 7479–7485 CrossRef CAS.
- R. M. Mohamed and E. S. Aazam, Mater. Res. Bull., 2013, 48, 3572–3578 CrossRef CAS.
- K. Li, Z. Zeng, L. Yan, S. Luo, X. Luo, M. Huo and Y. Guo, Appl. Catal., B, 2015, 165, 428–437 CrossRef CAS.
- J. Kim, C. W. Lee and W. Choi, Environ. Sci. Technol., 2010, 44, 6849–6854 CrossRef CAS PubMed.
- Z. Xu, I. Tabata, K. Hirogaki, K. Hisada, T. Wang, S. Wang and T. Hori, RSC Adv., 2012, 2, 103–106 RSC.
- A. V. Vorontsov, I. V. Stoyanova, D. V. Kozlov, V. I. Simagina and E. N. Savinov, J. Catal., 2000, 189, 360–369 CrossRef CAS.
- L. Qi, W. Ho, J. Wang, P. Zhang and J. Yu, Catal. Sci. Technol., 2015, 5, 2366–2377 CAS.
- H. Bader, V. Sturzenegger and J. Hoigne, Water Res., 1988, 22, 1109–1115 CrossRef CAS.
- Z. Zhang, Z. Wang, S. W. Cao and C. Xue, J. Phys. Chem. C, 2013, 117, 25939–25947 CAS.
- S. Y. Chao, Y. J. Kim, M. K. Junk, A. K. Chakraborty, D. Jung and W. Lee, J. Catal., 2009, 262, 144–149 CrossRef.
- M. Ge, Y. Li, L. Liu, Z. Zhou and W. Chen, J. Phys. Chem. C, 2011, 115, 5220–5225 CAS.
- Z. Wei, T. Hansen, M. Santella, X. Wang, C. R. Parker, X. Jiang, T. Li, M. Glyvradal, K. Jennum, E. Glibstrup, N. Bovet, X. Wang, W. Hu, G. C. Solomon, M. B. Nielsen, X. Qiu, T. Bjornholm, K. Norgaard and B. W. Laursen, Adv. Funct. Mater., 2015, 25, 1700–1708 CrossRef CAS.
- P. Pawinrat, O. Mekasuwandumrong and J. Panpranot, Catal. Commun., 2009, 10, 1380–1385 CrossRef CAS.
- Z. Jiang, Z. Wang, Y. Chu, D. Gu and G. Yin, Energy Environ. Sci., 2011, 4, 728–735 CAS.
- L. K. Ono, B. Yuan, H. Heinrich and B. R. Cuenya, J. Phys. Chem. C, 2010, 114, 22119–22133 CAS.
- P. Bera, K. R. Priolkar, A. Gayen, P. R. Sarode, M. S. Hegde, S. Emura, R. Kumashiro, V. Jayaram and G. N. Subbanna, Chem. Mater., 2003, 15, 2049–2060 CrossRef CAS.
- K. Ikeda, J. Sato, N. Fujimoto, N. Hayazawa, S. Kawata and K. Uosaki, J. Phys. Chem. C, 2009, 113, 11816–11821 CAS.
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Rose, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 CrossRef CAS.
- G. Liu, X. Li, J. Zhao, S. Horikoshi and H. Hidaka, J. Mol. Catal. A: Chem., 2000, 153, 221–229 CrossRef CAS.
- M. Kajita, K. Hikosaka, M. Iitsuka, A. Kanayama, N. Toshima and Y. Miyamoto, Free Radical Res., 2007, 41, 615–626 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full-scale XPS spectra of pure Bi2O3 and 0.075 wt% Pt/Bi2O3, cyclic use of 0.075 wt% Pt/Bi2O3 for photocatalytic degradation of RhB. See DOI: 10.1039/c6cp06755g |
|
| This journal is © the Owner Societies 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 








