
Photo-catalyzed surface hydrolysis of iridium(III) ions on semiconductors: a facile method for the preparation of semiconductor/IrOx composite photoanodes toward oxygen evolution reaction†
Qingyong
Wu
,
Di
Xu
,
Ning
Xue
 ,
Tengyi
Liu
,
Min
Xiang
and
Peng
Diao
*
,
Tengyi
Liu
,
Min
Xiang
and
Peng
Diao
*
Key Laboratory of Aerospace Materials and Performance (Ministry of Education), School of Materials Science and Engineering, Beihang University, Beijing 100191, P. R. China. E-mail: pdiao@buaa.edu.cn; Tel: +86-10-8233-9562
First published on 22nd November 2016
Abstract
We previously reported that the hydrolysis of Ir3+ in homogeneous solution could be triggered by irradiation with light whose energy was larger than a threshold value. In this work, we demonstrated that, by introducing Fe2O3 particles into solution, the incident light energy-restriction for the photo-catalyzed hydrolysis could be broken and the hydrolysis occurred at the Fe2O3/solution interface. The photo-generated holes on the Fe2O3 surface played a key role in oxidizing Ir(III) to Ir(IV) species and triggered the deposition of IrOx. We showed that this photo-catalyzed surface hydrolysis is a universal phenomenon that takes place on the surface of many n-type semiconductors such as Fe2O3, TiO2, and Ag3PO4. As IrOx is an efficient catalyst for oxygen evolution reaction, surface hydrolysis is a general, facile and efficient strategy to prepare semiconductor/IrOx composites, which can be used as anodic materials for photoelectrochemical water splitting.
1. Introduction
Hydrolysis of metal cations is a well-studied and documented reaction, which is not only fundamental to the chemistry of metal cations in aqueous solution, but also important for the wet chemical process for metal-based materials. Hydrolysis has long been employed as a successful strategy for the preparation of metal oxides.1–5 For example, in many wet chemical approaches, such as forced hydrolysis synthesis,1,4 hydrothermal growth,5–7 and sol–gel method,8,9 hydrolysis of metal cations is the key process for the formation of metal hydroxides and oxides. During a hydrolysis approach to prepare metal oxides, the metal inorganic salts and alkoxides usually undergo firstly a hydrolysis reaction and then a polycondensation, resulting in the formation of colloids or precipitates of metal oxides.2 Many transition metal oxides, such as manganese,10,11 copper,6,12 titanium,13,14 zinc,15,16 nickel,17 cobalt18,19 and iron20,21 oxides, have been prepared by hydrolysis.The rate of many hydrolysis reactions is very slow at room temperature under 1 atm,3,22 making these reactions impractical for the synthesis of metal oxides at ambient temperature and pressure. Raising the temperature and pressure can provide the additional energy to trigger or accelerate these reactions.22,23 Light, another form of energy that can drive chemical reactions at room temperature, should also be effective in inducing and accelerating hydrolysis reactions. However, few works have reported the photo-driven hydrolysis of metal salts even though irradiation has been successfully used in the reductive preparation of noble metal nanoparticles (NP).24–26 Recently, we demonstrated that light could drive the hydrolysis of iridium(III) chloride,27 whose rate was extremely slow at room temperature in the dark.22 We showed that UV-vis irradiation played a crucial role not only in the formation of the hydroxide complex [Ir(OH)6]3− but also in the polymerization and oxidation of Ir(III) species to form nanoparticles of iridium oxide (IrOx). More importantly, there existed a critical wavelength of ca. 500 nm for the photo-catalyzed hydrolysis reaction, and the hydrolysis occurred only when the wavelength of incident light was much shorter than 500 nm.
Iridium oxides exhibit catalytic activity for many reactions.28–30 For example, they have long been regarded as highly efficient catalysts for electrochemical oxygen evolution reaction (OER),28,31,32 which makes them a good candidate as a co-catalyst of photoanodes for OER in solar water splitting.28,31,33 This application requires iridium oxide to be deposited on the surface of semiconductors. Therefore, as a co-catalyst, it is desirable that iridium oxide is deposited on the photoactive sites of the semiconductor surface, where photoinduced holes can be easily captured by iridium oxide and then used to oxidize water. Photo-assisted deposition of co-catalysts on semiconductors is an ideal strategy to prepare semiconductor/co-catalyst systems.34–38 This is because the photo-assisted deposition was usually caused by an oxidation or reduction process, which was induced by photogenerated holes or electrons. As a result, the deposition occurred at the sites where photoinduced holes or electrons were mostly available on the semiconductor surface.34–36 Inspired by these works, we believe that the photo-catalyzed hydrolysis of Ir3+ may be an effective way to deposit IrOx on the photo-active sites of the semiconductor surface, especially if we can confine the hydrolysis at the semiconductor/solution interface.
To examine the feasibility of depositing IrOx on the semiconductor surface by using the strategy of photo-catalyzed surface hydrolysis, hematite (Fe2O3) was selected as a model semiconductor support in this work for the following two reasons. First, to generate IrOx nanoparticles from homogeneous solution via photo-catalyzed hydrolysis of Ir3+, the oxidation of Ir(III) to Ir(IV) by dissolved oxygen is crucial.27 On the other hand, for the generation of IrOx on the surface of the semiconductor, the oxidation process can be accomplished by photoinduced holes. Therefore, n-type semiconductors whose valence band edges are more positive than the potential of the Ir(IV)/Ir(III) redox couple (ca. 0.50 V vs. NHE, 1.21 V vs. RHE at pH 12, the pH value of the deposition solution)39,40 can be used as supports. Hematite (Fe2O3) meets this requirement because it is an n-type semiconductor,41,42 with the valence band edge located at 2.48 V vs. NHE (3.19 V vs. RHE, pH 12).43 This potential is more positive than that of the Ir(IV)/Ir(III) redox couple.39,40 As a result, photoinduced holes in the valence band of Fe2O3 have enough energy to oxidize Ir(III) to Ir(IV), and then trigger the hydrolysis. Second, Fe2O3 is a promising photoanode material for photoelectrochemical (PEC) OER, which suffers from a high electron–hole recombination rate.43 Deposition of OER catalysts on the surface of Fe2O3 is an effective strategy to solve this problem because OER catalysts facilitate the transfer of photo-generated holes from the semiconductor to the O2/H2O redox couple, thus retarding the recombination rate.33,44,45 As IrOx is one of the most effective OER catalysts,31,33,44 the combination of Fe2O3 with IrOx may improve the efficiency of the PEC OER.
The purposes of this work are two-fold: (1) to clarify if the photo-catalyzed hydrolysis of Ir(III) ions can be realized at the semiconductor/solution interface, and (2) to develop a method for the preparation of semiconductor/IrOx composites by using photo-catalyzed surface hydrolysis. We demonstrated that the photo-catalyzed hydrolysis of Ir3+ could occur at the Fe2O3/solution, Ag3PO4/solution, and TiO2/solution interfaces when they were illuminated with both polychromatic and monochromatic light. We also demonstrated that Fe2O3/IrOx and TiO2/IrOx composites could be prepared by using photo-catalyzed surface hydrolysis. All the obtained composite photoanodes exhibited enhanced photoactivity toward PEC OER. These results confirmed that photo-catalyzed hydrolysis of Ir3+ is a universal strategy to prepare n-type semiconductor/IrOx composites, which can be used as active photoanodes for PEC OER.
2. Experimental
2.1 Chemicals and materials
Iridium chloride trihydrate (IrCl3·3H2O) and tetraisopropyl titanate were purchased from Alfa Aesar company. Iron(III) chloride hexahydrate was purchased from Sinopharm Chemical Reagents Company. Silver nitrate (AgNO3), sodium nitrate (NaNO3), disodium hydrogen phosphate dodecahydrate (Na2HPO4·12H2O) and potassium hydroxide (KOH) were purchased from Beijing Chemical Reagents Corporation. All chemicals were used as delivered and without further purification. Fluorine-doped tin oxide substrates (F:SnO2, 8 Ω sq−1) were purchased from Asahi Glass, Japan.2.2 Preparation of Fe2O3, TiO2, and Ag3PO4 electrodes
Before use, fluorine-doped tin oxide (FTO) coated glass was firstly cleaned by ultrasonication in acetone, ethanol and deionized water, consecutively, for 15 min each and then dried under an N2 stream at room temperature.The Fe2O3 electrodes were prepared by a hydrothermal method that was a modification of a traditional approach.46 Firstly, FTO substrates were immersed in an aqueous solution of 1 M NaNO3 and 50 mM FeCl3, with the conducting side facing downward. The hydrothermal reaction was carried out at 100 °C for 18 h, during which the FeOOH films were formed on both sides of the FTO substrates. Secondly, the FeOOH film coated FTO substrates were rinsed with high purity water, placed on a stainless steel slide on the top of an Al2O3 crucible, and then annealed at 850 °C in air for 5 min. After annealing, the yellow FeOOH films changed to red Fe2O3 films. The Fe2O3 powders were obtained by a procedure similar to that used for obtaining the Fe2O3 films. After the hydrothermal reaction, the FeOOH particles in the solution were separated by centrifugation, dried in air, and annealed at 850 °C in air for 0.5 h.
The TiO2 electrodes were also fabricated by a hydrothermal method, according to a previous work.47 In detail, 15 ml of concentrated hydrochloric acid was diluted with 15 ml of high purity water, and mixed with 0.5 ml of tetraisopropyl titanate. This solution and FTO substrates were sealed in an autoclave and maintained at 150 °C for 5 h. The obtained TiO2 substrates were then annealed at 550 °C in air for 3 h to enhance the crystallinity.
The Ag3PO4 powders were fabricated following a previous work.48 The Na2HPO4 aqueous solution (20 ml, 0.2 M) was added dropwise to a AgNO3 aqueous solution (20 ml, 0.6 M) with continuous stirring in a 100 ml flask, and the mixture was kept under stirring for 0.5 h. The yellow-colored precipitate was separated by centrifugation and dried in air at 60 °C.
2.3 Photo-catalyzed hydrolysis and photo-catalyzed surface hydrolysis of Ir(III) ions
In a typical procedure, an aqueous solution of 2 mM IrCl3 was prepared in a beaker, and the pH of the solution was adjusted to 12 with concentrated KOH. Then, the beaker was placed under polychromatic or monochromatic light irradiation. The reaction system was kept at room temperature by using a circulating water bath during irradiation. UV and visible light was generated from a PS-SXE300UV xenon lamp. A UV reflection filter (Beijing Perfect Light Science and Technology Co., Ltd) was used to generate polychromatic UV light with wavelength ranging from 260 nm to 410 nm. Bandpass filters with wavelengths 365 nm and 498 nm were used to produce monochromatic light of required wavelength. The intensity of polychromatic light was 150 mW cm−2, while the intensity of monochromatic light was 4.0 mW cm−2.For photo-catalyzed surface hydrolysis, 1.5 g of Fe2O3 or Ag3PO4 powder was dispersed in 25 ml of IrCl3 precursor solution with continuous stirring, and the solution was irradiated with monochromatic light with λ = 498 nm. After irradiation, the powder was separated by centrifugation and the UV-vis absorption spectra of the solution were recorded.
2.4 Photo-catalyzed deposition of IrOx on Fe2O3 and TiO2 semiconductors
In a typical procedure, the Fe2O3 or TiO2 electrodes prepared by the hydrothermal method (1 cm × 3 cm) were immersed in a 10 ml IrCl3 aqueous solution (2 mM IrCl3, pH 12) in a 25 ml beaker. The solution was irradiated with polychromatic or monochromatic irradiation for 2 h. After irradiation, Fe2O3 and TiO2 electrodes were rinsed with high purity water and dried in air before use. Bandpass filters (365 nm and 498 nm) were used to provide monochromatic light. The intensities of polychromatic and monochromatic light were 150 mW cm−2 and 4.0 mW cm−2, respectively. The deposition of IrOx on Fe2O3 was also carried out in an O2-free system. In this experiment, the IrCl3 solution (2 mM, pH 12) was deoxygenated by bubbling high purity N2 for 0.5 h, and the reaction system was continuously purged with N2 during photo-catalyzed deposition.2.5 Characterization
X-ray diffraction (XRD) measurements were performed on a Rigaku, rint2000 advance theta-2theta powder diffractometer with Cu Kα radiation. The UV-vis absorption spectra were recorded using a UV2600 spectrophotometer (Tianmei Co., China). The morphology of Fe2O3 was characterized by using a JEOL JSM7500 field emission scanning electron microscope (SEM), operating at an accelerating voltage of 10 kV. Transmission electron microscopy (TEM) characterization was performed on a JEM-2100F TEM (JEOL, Japan, accelerating voltage: 200 kV). X-ray photoelectron spectroscopic (XPS) measurements were carried out on a PHI Quantera SXM scanning X-ray microprobe (ULVAC-PHI, Japan), with a monochromatic Al source at a power of 25 W. The XPS measurements were carried out at a pressure of 4.5 × 10−7 Pa. The binding energies were calibrated to the C1s line at 284.8 eV, and Shirley background subtraction was applied to the raw data before deconvolution.2.6 Photoelectrochemical (PEC) measurements
The PEC measurements were carried out in a traditional 3-electrode configuration on a CHI 660D work station (CH Instruments Co.). The Fe2O3 or TiO2 photoanodes (with or without IrOx) were used as working electrodes, with Pt foil and a saturated calomel electrode (SCE) as the counter and reference electrodes, respectively. All of the PEC experiments were conducted in 1 M KOH solution (pH = 13.6). The relationship between the potential with respect to the SCE and that with respect to a reversible hydrogen electrode (RHE) is described by the following equation:E(vs.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) RHE) = E(vs.SCE) + 0.0592 × pH + E0(SCE) RHE) = E(vs.SCE) + 0.0592 × pH + E0(SCE) | (1) |
2.7 Detection of oxygen evolution
The quantity of O2 evolved from the Fe2O3 and Fe2O3/IrOx composite photoanodes was measured using an O2 fluorescence detector (Ocean Optics, R-sensor, SN#J497).49,50 A three-compartment PEC cell was designed for simultaneously measuring both the photocurrent and the O2 concentration. Each electrode was placed in one compartment. The compartment containing the working electrode has a quartz window on its side for illumination. The illumination window was a circle with a diameter of 1.0 cm. The intensity of incident light on the electrode surface was 100 mW cm−2. The oxygen sensor was immersed in the solution to detect the concentration of dissolved O2. Before PEC measurements, the solution was purged with nitrogen for 40 minutes to remove O2 from both the electrolyte and the gas phase in the headspace of the PEC cell, and then the system was sealed and left undisturbed for 20 min to check for leakage. The detection of evolved O2 was conducted at a bias of 0.25 V vs. SCE (1.29 V vs. RHE, pH 13.5), and the concentration of dissolved O2 was continuously recorded at an interval of 20 s throughout the measurement.3. Results and discussion
3.1 Photo-catalyzed surface hydrolysis of Ir(III) ions
As demonstrated previously,27 the photo-catalyzed hydrolysis of Ir3+ in homogeneous solution occurred under the condition that the wavelength of incident light is much shorter than 500 nm. In other words, there exists a threshold energy (ca. 2.49 eV) of the incident light to trigger the hydrolysis of Ir3+ in aqueous solution. Only when the energy of the incident light is larger than the threshold value, can the hydrolysis take place. However, to our surprise, we found that the threshold-wavelength-restriction ceased to hold when Fe2O3 nanoparticles were introduced into the reaction system.As shown in Fig. 1a and b, after the IrCl3 solution was illuminated for 2 h with polychromatic UV light (260 nm < λ < 410 nm) or with monochromatic UV light (λ = 365 nm), the color of the solution changed from yellow to dark blue, indicating the occurrence of photo-catalyzed hydrolysis and the formation of the IrOx colloid.27 Moreover, when the IrCl3 solution was irradiated with monochromatic visible light (λ = 498 nm) that is very close to the threshold wavelength of ca. 500 nm, the color of the solution did not change even after 4 hour irradiation, suggesting that the hydrolysis did not occur (see Fig. 1c). All these observations are in good agreement with the previous results, confirming the existence of the threshold wavelength (ca. 500 nm) of light to trigger the hydrolysis of Ir3+ ions in homogeneous solution.27 However, when Fe2O3 powder was introduced into the IrCl3 solution to form an aqueous suspension under continuous stirring, the color of the mixed solution changed to blue upon illumination with monochromatic visible light (λ = 498 nm). Fig. 1d shows the photograph of the IrCl3 solution after the Fe2O3 powder was separated by centrifugation. This observation suggests that the incident light of 498 nm, which cannot drive the hydrolysis of Ir3+ in homogeneous solution, triggered the reaction with the help of Fe2O3 particles in solution. Fig. 1e shows the UV-vis absorption spectra of the IrCl3 solution after irradiation with 498 nm light for 2 h in the absence (red line) and presence of Fe2O3 powder (blue line). The shoulder peak which appeared at 320 nm after irradiation suggests the formation of [Ir(OH)6]3− in solution,51,52 while the broad absorption peak at 580 nm implies the appearance of IrOx nanoparticles (NPs).22,27 The UV-vis absorption spectra provide further evidence that, in the presence of Fe2O3 particles, the incident light of 498 nm initiated the hydrolysis reaction and resulted in the formation of IrOx NPs.
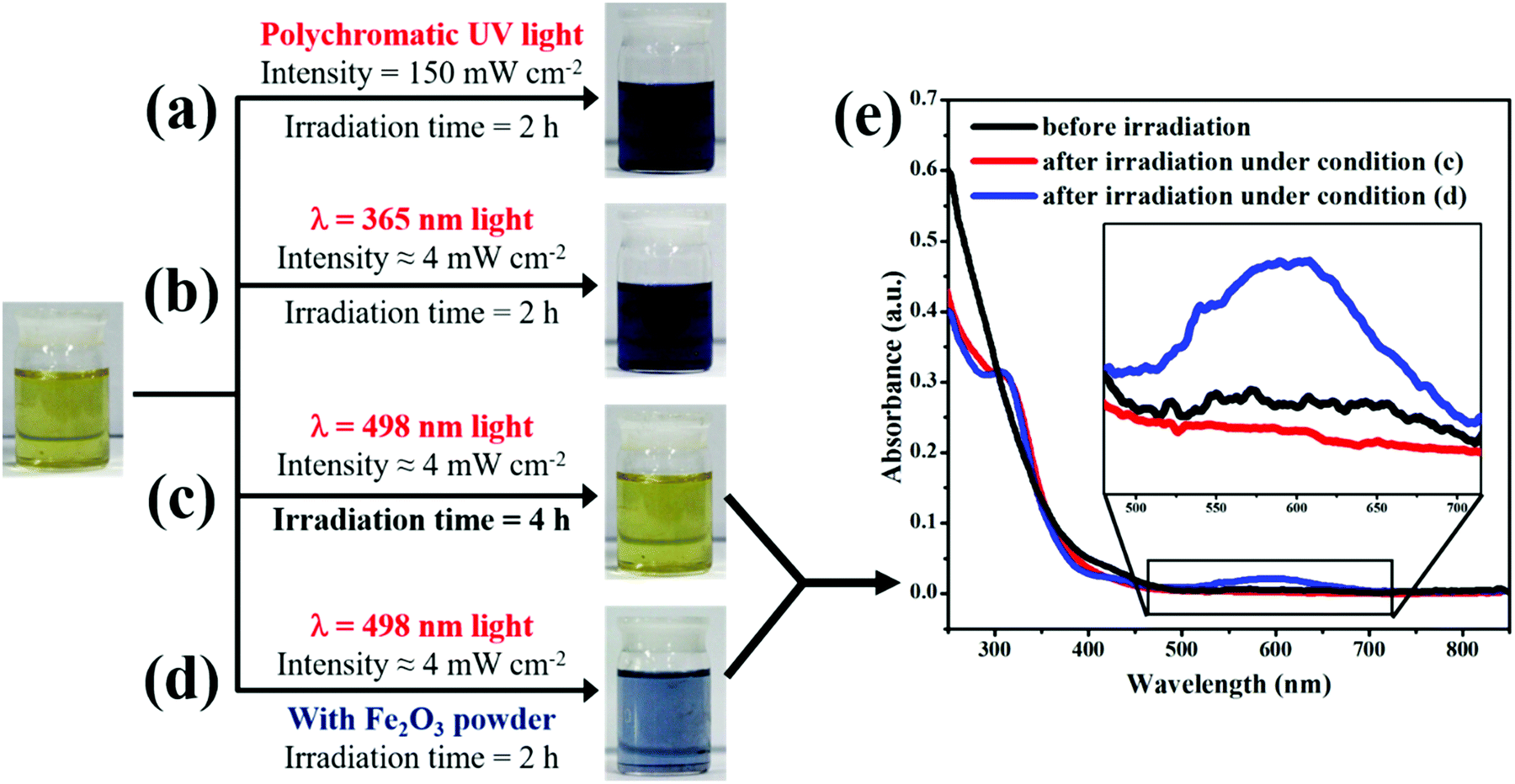 | ||
| Fig. 1 The photographs of IrCl3 solution (pH 12) before and after irradiation under polychromatic and monochromatic incident light with different wavelengths. (a) Polychromatic UV (260 nm < λ < 410 nm) irradiation for 2 h, (b) monochromatic UV irradiation (λ = 365 nm) for 2 h, (c) monochromatic visible irradiation (λ = 498 nm) for 4 h, and (d) monochromatic visible irradiation (λ = 498 nm) for 2 h, with Fe2O3 powder diffused in the solution. (e) The UV-vis absorption spectra of the solution before and after irradiation under the conditions of (c) and (d). The inset in (e) shows the local magnification of the absorption spectra. | ||
To answer the question why the presence of Fe2O3 helps to trigger the reaction, we need to review the mechanism of photo-catalyzed hydrolysis of Ir3+ in homogeneous solution. This reaction involves two consecutive steps, and the first step is the photo-driven generation of the [Ir(OH)6]3− complex, which has no restriction concerning the wavelength of incident light, as can be confirmed from the shoulder absorption peak at 320 nm in UV-vis spectra (see Fig. 3 in ref. 27 and the red line in Fig. 1e of this work).27 The second step is the polymerization of the [Ir(OH)6]3− complex, which involved the oxidation of Ir(III) species by dissolved oxygen to form IrOx nanoparticles.27 In the absence of Fe2O3 particles, only the light whose wavelength was much shorter than 500 nm could trigger the polymerization and oxidation of Ir(III). This is the reason for the existence of the threshold wavelength of incident light for the hydrolysis reaction in homogeneous solution. However, in the presence of Fe2O3 particles in solution, the incident light could be absorbed by Fe2O3 and generated electron–hole pairs as long as the light energy is larger than 2.1 eV (corresponding to 591 nm), which is the bandgap energy of Fe2O3 (see Fig. S1 in the ESI†).43,53,54 Moreover, Fe2O3 is an n-type semiconductor with its valence band edge located at ca. 2.48 V vs. NHE (3.19 V vs. RHE, pH 12),43 which is more positive than the redox potential of Ir(IV)/Ir(III) at ca. 0.50 V vs. NHE (1.21 V vs. RHE, pH 12).39,40 Therefore, the incident light with wavelength in the range of 498 ≤ λ ≤ 590 nm could generate holes in the valence band of Fe2O3. These holes were highly oxidative and played the same role as the dissolved oxygen in homogeneous solution. They could oxidize Ir(III) species to trigger the formation of IrOx NPs at the Fe2O3/solution interface. In other words, the incident light with wavelength in the range of 498 ≤ λ ≤ 590 nm can trigger the formation of IrOx NPs at the Fe2O3/solution interface though it cannot in homogeneous solution. Accordingly, the hydrolysis of Ir can be easily localized to the Fe2O3/solution interface by using an incident light with wavelength in the range of 498 ≤ λ ≤ 590 nm. The solution in Fig. 1d shows a relatively light blue color than that in Fig. 1b, though the incident light intensity was the same. The lighter color implies that the concentration of IrOx NPs of the solution in Fig. 1d is lower. This is because the photo-catalyzed surface hydrolysis of Ir3+ is a more complicated heterogeneous reaction, which involves absorption of light, generation of electron–hole pairs, transfer of holes and reaction on the surface of Fe2O3. On the other hand, the photochemical hydrolysis of Ir3+ at λ = 365 nm is a homogeneous reaction. Therefore, the reaction rate in Fig. 1d is much slower than that in Fig. 1b, resulting in a relatively light blue color.
The photo-catalyzed surface hydrolysis of Ir3+ on the surface of Fe2O3 is of great significance. (1) The presence of Fe2O3 breaks the restriction of the threshold wavelength of the incident light that can trigger the hydrolysis. (2) The photo-catalyzed hydrolysis can be limited to the Fe2O3/solution interface as long as the wavelength of incident light falls in the range 498 ≤ λ ≤ 590 nm. (3) The surface hydrolysis of Ir3+ leads to the deposition of IrOx on the surface of Fe2O3. Inspired by the above results, we developed a facile method to prepare the Fe2O3/IrOx composite OER catalyst by using the photo-catalyzed hydrolysis reaction.
3.2 The preparation of the Fe2O3/IrOx OER catalyst by photo-catalyzed hydrolysis of Ir(III)
To confirm that the photo-catalyzed surface hydrolysis of Ir3+ can be used to deposit IrOx on the surface of Fe2O3, the FTO supported Fe2O3 films prepared by a hydrothermal method were used as substrates. Fig. 2a shows the typical SEM image of a Fe2O3 film in which a porous structure composed of nanograins is clearly seen. The porous structure ensures a high specific area for Fe2O3, which is beneficial for the catalytic activity especially when Fe2O3 is used as the photoanode material for PEC OER. High resolution TEM (HR-TEM) was used to characterize the microstructure of the Fe2O3 nanograins, and the typical result is shown in Fig. 2b. The Fe2O3 film was composed of single crystalline nanograins, and the lattice spacing is 0.25 nm, which corresponds to the (110) plane of rhombohedral Fe2O3, indicating that the Fe2O3 film has a rhombohedral structure. The crystal structure of the Fe2O3 film was also characterized by XRD, and the diffraction patterns are shown in Fig. 2c. The diffraction peaks located at 35.44° and 63.70° are indexed to the (110) and (300) planes of rhombohedral Fe2O3 (JCPDS 79-7), in good agreement with the TEM results.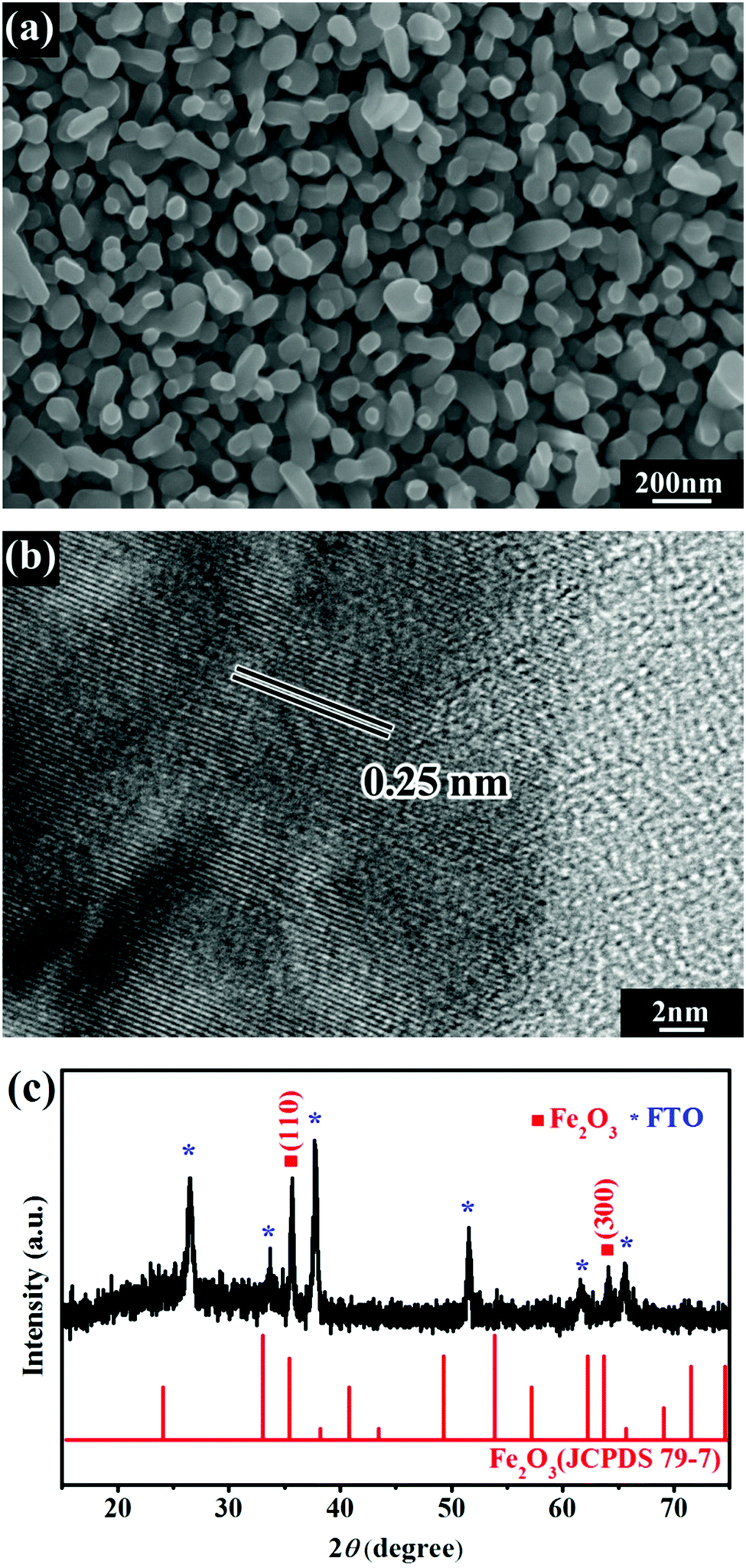 | ||
| Fig. 2 SEM image (a), high-resolution TEM image (b), and XRD pattern (c) of the FTO supported Fe2O3 film. | ||
X-ray photoelectron spectroscopy (XPS) can provide information of the elemental composition and chemical state of elements on the surface of a material. In this work, XPS was employed to examine the presence of Ir on the surface of Fe2O3 after photo-catalyzed surface hydrolysis, and the results are shown in Fig. 3a. The appearance of the Ir 4d and Ir 4f peaks after illumination provides solid evidence that Ir species were successfully deposited on the surface of Fe2O3. Fig. 3b shows the high-resolution spectrum of the Ir 4f peaks, which could be deconvoluted into two pairs of peaks. One pair of peaks was attributed to Ir(IV) (the pink line), while the other pair belonged to Ir(III). These results confirmed that the iridium oxide deposited on the surface of Fe2O3 was composed of Ir(III) and Ir(IV) oxides. Moreover, the high intensity of the Ir(IV) 4f peaks suggests that a large part of Ir(III) was oxidized to Ir(IV) during the photo-catalyzed surface hydrolysis. This result implies that photo-generated holes in Fe2O3 played a key role in the deposition of IrOx.
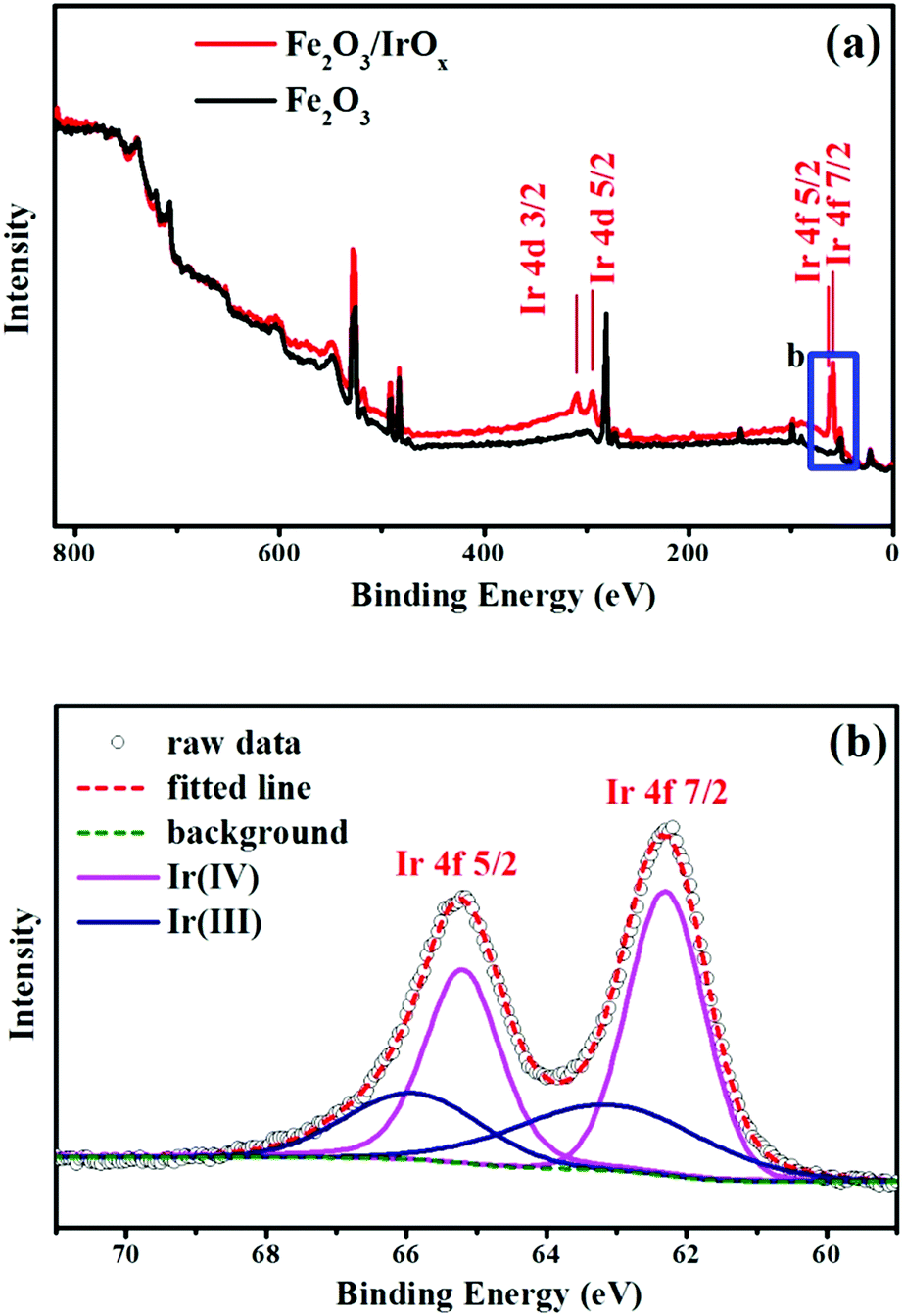 | ||
| Fig. 3 (a) XPS survey scans of Fe2O3 and Fe2O3/IrOx composite electrodes. (b) XPS spectrum detailing the Ir 4f region of the Fe2O3/IrOx composite electrode. | ||
As is well known, hematite is a promising photoanode material for solar water splitting.43,54 However, Fe2O3 suffers from a high electron–hole recombination rate43,55 due to the slow water oxidation kinetics at the Fe2O3/solution interface. The deposition of IrOx on Fe2O3 may greatly improve the photo-to-chemical conversion efficiency of the Fe2O3 photoanode because of the following two reasons. (1) IrOx is an efficient OER catalyst40,56 that may significantly improve the OER rate by accelerating the transfer of photo-generated holes from Fe2O3 to H2O. (2) Photo-catalyzed deposition ensured that IrOx was deposited on the surface sites where photo-generated holes were most available, and as a result, the photo-generated holes were easily captured by IrOx. To examine the effect of IrOx on the photoactivity of Fe2O3 toward PEC OER, the current density vs. potential (J–V) curves were recorded at the Fe2O3 photoanode under chopped illumination before and after the deposition of IrOx, and the results are shown in Fig. 4. The onset potential of photocurrent at the Fe2O3 photoanode was ca. 0.85 V vs. RHE and the photocurrent density (Jph) reached 0.59 mA cm−2 at 1.23 V vs. RHE. According to literature reports,44,45,57 this Jph is an average value among those obtained at the Fe2O3 photoanodes prepared by the hydrothermal method. We did not optimize the preparation conditions of Fe2O3 to further improve its Jph because this work focuses on the strategy of combining semiconductor materials with an OER catalyst by photo-catalyzed surface hydrolysis. Fig. 4 clearly shows that, after deposition of IrOx, the resulting Fe2O3/IrOx composite exhibited a significantly improved Jph within the entire potential sweep region. Compared to the Fe2O3 photoanode, the onset potential of the Fe2O3/IrOx composite was negatively shifted to 0.60 V vs. RHE and Jph reached 0.98 mA cm−2 at 1.23 V vs. RHE, which is an increase of ca. 66.1%. These results indicate that the deposited IrOx acted as an OER catalyst, which efficiently captured the photo-induced holes and transferred them from Fe2O3 to H2O, and thus accelerated the OER rate.
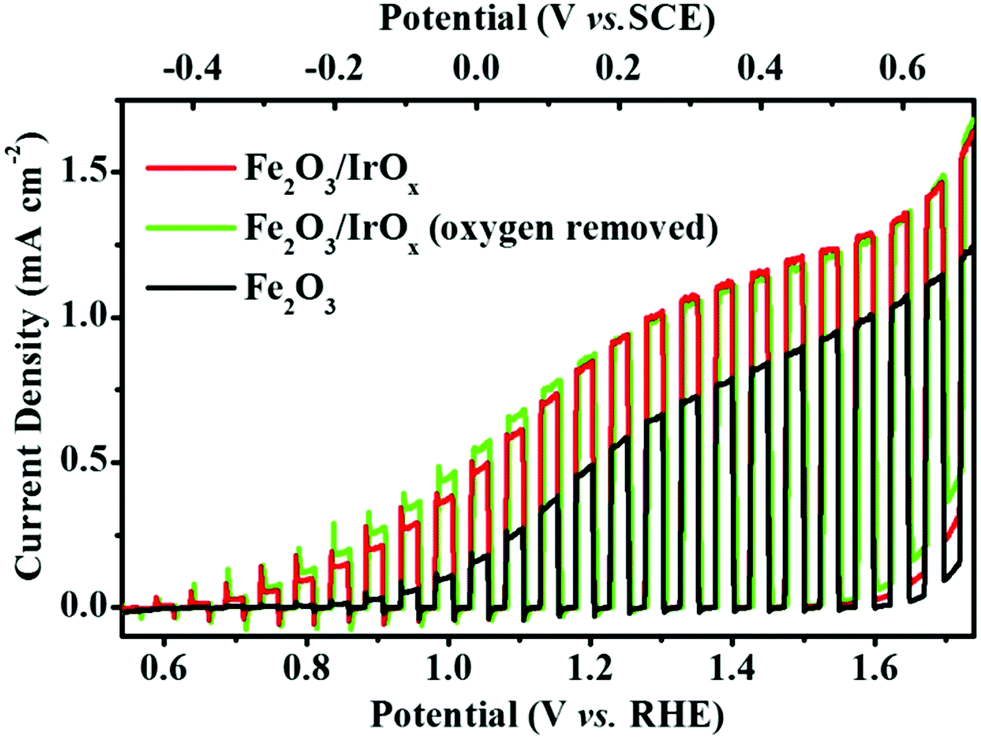 | ||
| Fig. 4 The J–V curves of the Fe2O3 (black line) and the Fe2O3/IrOx composite (red and green lines) photoanodes in 1 M KOH under chopped illumination (100 mW cm−2) at a scan rate of 5 mV s−1. Both the Fe2O3/IrOx composite photoanodes were prepared by photo-catalyzed hydrolysis of IrCl3 solution (2 mM, pH 12) under 150 mW cm−2 polychromatic UV illumination, with one of them (green line) obtained in IrCl3 solution that was deoxygenated by purging with high purity N2. | ||
Moreover, as a control experiment, Jph was also recorded at the Fe2O3 photoanode that was first immersed in 2 mM IrCl3 (pH 12) for 2 h but without UV irradiation, and then rinsed with distilled water. The J–V response of this photoanode showed no significant difference compared to that of the pure Fe2O3 electrode (Fig. S2 in the ESI†). This result confirms that illumination is the key factor for the deposition of IrOx.
As mentioned above, the oxidation of Ir(III) to Ir(IV) by dissolved O2 was crucial for the photo-catalyzed hydrolysis of Ir3+ and the formation of IrOx in homogeneous solution.27 On the other hand, for the photo-catalyzed surface hydrolysis of Ir3+, photo-generated holes were believed to act as the oxidizing agent instead of dissolved O2. To examine this assumption, a control experiment was carried out in an O2-free IrCl3 solution (2 mM, pH 12), which was deoxygenated by bubbling high purity N2 for 0.5 h before reaction and purging with N2 during the two hour reaction. The J–V response (green line) of the resulting Fe2O3/IrOx composite is also shown in Fig. 4. This Fe2O3/IrOx composite photoanode exhibited nearly the same J–V characteristics as that of the Fe2O3/IrOx composite prepared in IrCl3 solution with dissolved O2. This observation provided direct evidence that photo-generated holes participated in the photo-deposition process by oxidizing Ir(III) to Ir(IV), and then triggered the formation of IrOx.
All the above-mentioned Fe2O3/IrOx composite photoanodes were prepared by using polychromatic UV light (150 mW cm−2). In fact, the composite photoanode could also be prepared by using 498 nm monochromatic visible light even though the energy density of the incident light was only 4 mW cm−2. It should be pointed out here that, though the deposition of IrOx could be localized on the surface of Fe2O3 (with IrOx formed in solution) by using the 498 nm incident light, the deposition rate was much smaller than that achieved by using polychromatic UV light. As a result, the amount of IrOx deposited on the surface of Fe2O3 during the same deposition time was very small. We believe that the use of a monochromatic visible light (498 ≤ λ ≤ 590 nm) source with high energy density (such as a laser) can solve this problem. On the basis of the above discussion, we believe that photo-catalyzed deposition of IrOx by using polychromatic UV light was a simple and efficient way to prepare the Fe2O3/IrOx composite.
The mechanism of the photo-catalyzed deposition can be described as follows. Firstly, irradiation provided the activation energy for the Ir(H2O)3Cl3 complex, which was the product of aquation of IrCl3,27,58 to convert into the [Ir(OH)6]3− complex. Secondly, part of [Ir(OH)6]3− was oxidized to [Ir(OH)6]2− by photo-generated holes at the Fe2O3/solution interface, and the polymerization of [Ir(OH)6]2− with another [Ir(OH)6]2− or with [Ir(OH)6]3− triggered the formation of IrOx. It should be pointed out here that, when the reaction system was irradiated with polychromatic UV light, we could not exclude the possibility that the IrOx NPs generated in homogeneous solution contributed to the deposition of IrOx on the surface of Fe2O3.
3.3 Stability and Faraday efficiency of the Fe2O3/IrOx composite for PEC OER
Fig. 5a shows the current density–time curves obtained in 1 M KOH at a constant potential of 0.25 V (1.29 V vs. RHE, pH 13.5) under 100 mW cm−2 chopped illumination. Both the Fe2O3 and the Fe2O3/IrOx composite photoanodes exhibited very good stability in alkaline solution. The photocurrent density of the Fe2O3 photoanode stabilized at ca. 0.70 mA cm−2 and showed no decay throughout the 10 hour measurement. On the other hand, for the Fe2O3/IrOx composite photoanode, the photocurrent density even slowly increased in the first 8.5 h and stabilized at ca. 0.91 mA cm−2, which is 30% higher than the photocurrent density of the Fe2O3 photoanode. Although the reason for the gradual increase of photocurrent density is not quite clear, we believe that this observation was related to the activation of the IrOx co-catalyst.40The high stability of both the Fe2O3 and the Fe2O3/IrOx composite photoanodes ensured that, during PEC OER, we could measure the amount of O2 evolution, which is very important to evaluate the photoanodes and to obtain the Faraday efficiency. In this work, the amount of O2 evolution from both photoanodes was measured by using an O2 fluorescence probe immersed in solution.49,50Fig. 5b shows the variation of the detected and calculated amount of O2 evolution as a function of time at both the Fe2O3 and the Fe2O3/IrOx photoanodes under continuous illumination (100 mW cm−2). The point of zero time was taken as the time at which illumination started. The negative time values on the abscissa refer to the time duration within which a bias of 1.29 V vs. RHE was applied to the photoanodes but with no illumination. No O2 was detected in this period, confirming that (1) electrochemical OER did not occur at 1.29 V vs. RHE on both electrodes and (2) the PEC cell had good gas tightness. When illumination was turned on, O2 started to evolve from both photoanodes, as can be seen in Fig. 5b. After 40 min PEC OER, O2 production from the Fe2O3/IrOx and the Fe2O3 photoanodes was detected to be 2.70 μmol and 1.92 μmol, respectively. The high O2 evolution rate on the Fe2O3/IrOx composite provides solid evidence that the deposited IrOx acted as an OER co-catalyst and greatly accelerated the PEC OER. The amount of O2 evolved from both photoanodes could also be calculated by integrating the photocurrent and assuming that the photocurrent arose from the 4-electron OER. The calculated amount of O2 is 2.71 μmol for the Fe2O3/IrOx and 1.97 μmol for the Fe2O3 photoanodes, corresponding to a Faraday efficiency of 99.6% and 97.4%, respectively. These results indicate that nearly all of the photocurrents were engaged in OER on both the Fe2O3/IrOx and the Fe2O3 photoanodes.
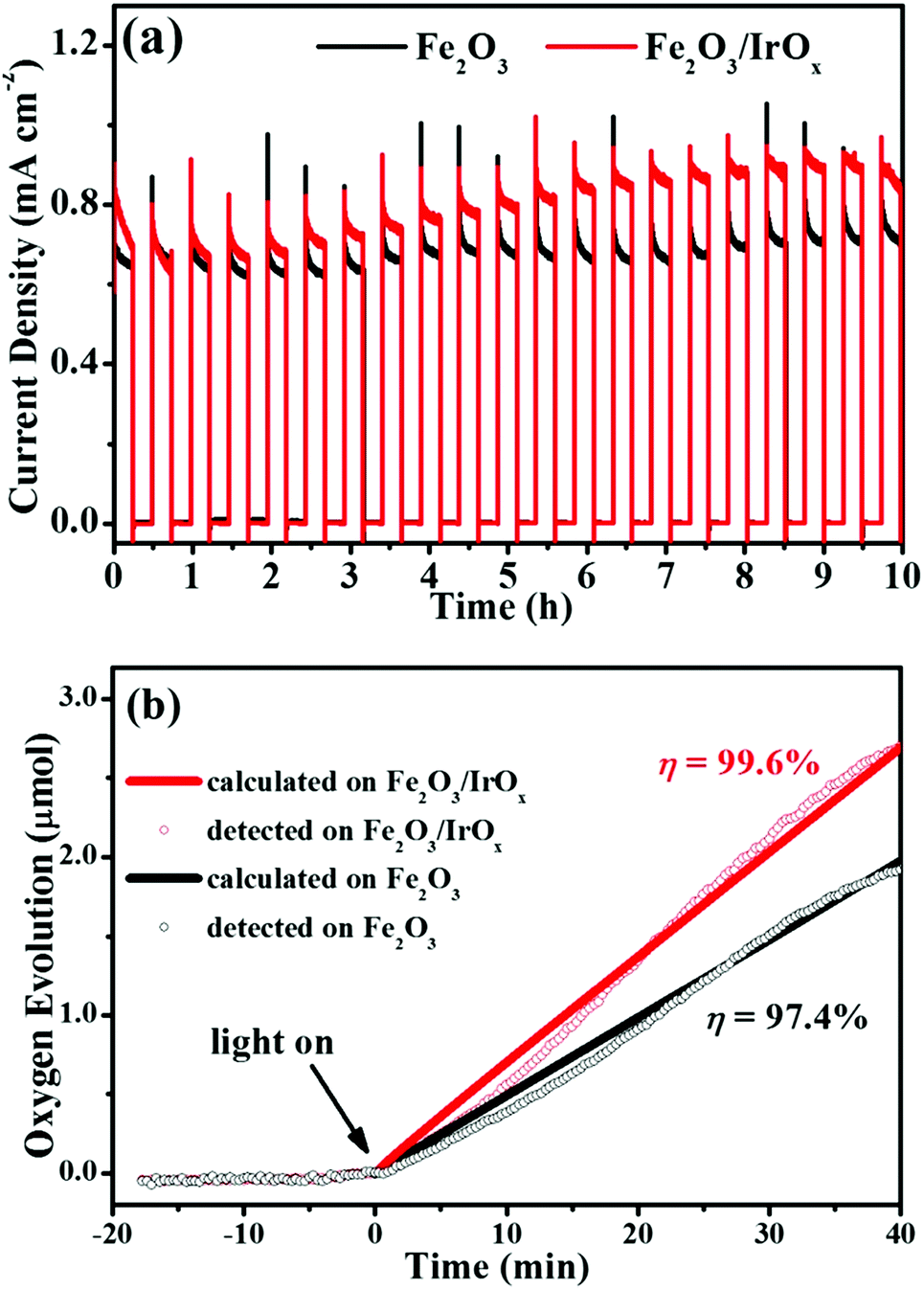 | ||
| Fig. 5 (a) The current density–time response of the Fe2O3 and Fe2O3/IrOx composite photoanodes in 1 M KOH at a constant bias of 0.25 V vs. SCE (1.29 V vs. RHE) under chopped illumination (100 mW cm−2). (b) The detected and calculated amount of oxygen evolution on the Fe2O3 and the Fe2O3/IrOx composite photoanodes in 1 M KOH at a constant bias of 0.25 V vs. SCE (1.29 V vs. RHE) under continuous illumination (100 mW cm−2). The illumination window was a circle with a diameter of 1.0 cm. | ||
3.4 Photo-catalyzed surface hydrolysis of Ir(III) as a universal strategy to prepare n-type semiconductor/IrOx composites for PEC OER
We believe that the photo-catalyzed surface hydrolysis of Ir(III) described in this work can be realized on the surface of many n-type semiconductors, as long as the valence band edge of the semiconductor is located at a more positive potential than the redox potential of Ir(IV)/Ir(III). As the redox potential of O2/H2O (1.23 V vs. RHE) is more positive than the redox potential of Ir(IV)/Ir(III), a semiconductor that is able to catalyze water oxidation is certainly suitable for the photo-catalyzed deposition of IrOx. Herein, we use two kinds of n-type semiconductors Ag3PO4 and TiO2 as examples to show that the photo-catalyzed hydrolysis is a universal strategy to prepare n-type semiconductor/IrOx composites.Ag3PO4 has been demonstrated as a possible photoanode material for PEC OER.59,60 The band-gap energy of Ag3PO4 is 2.36 eV,59 which corresponds to a band-edge absorption of λ = 530 nm. The valence band edge of Ag3PO4 is located at 2.84 V vs. NHE (3.55 V vs. RHE, pH 12),59,60 ensuring that the photo-induced holes have an energy high enough to oxidize Ir(III). In our experiments, Ag3PO4 powder was added to the IrCl3 solution (2 mM, pH 12) and the mixture was irradiated under 498 nm light (4 mW cm−2) for 4 h. The photographs and the UV-vis absorption spectra of the solution before and after irradiation are shown in Fig. 6a. The color of the solution changed from yellow to blue, implying the formation of IrOx. In addition, the peak that was characteristic of IrOx nanoparticles was observed at 580 nm in UV-vis spectra, confirming that photo-catalyzed surface hydrolysis of Ir3+ could also take place on the surface of Ag3PO4.
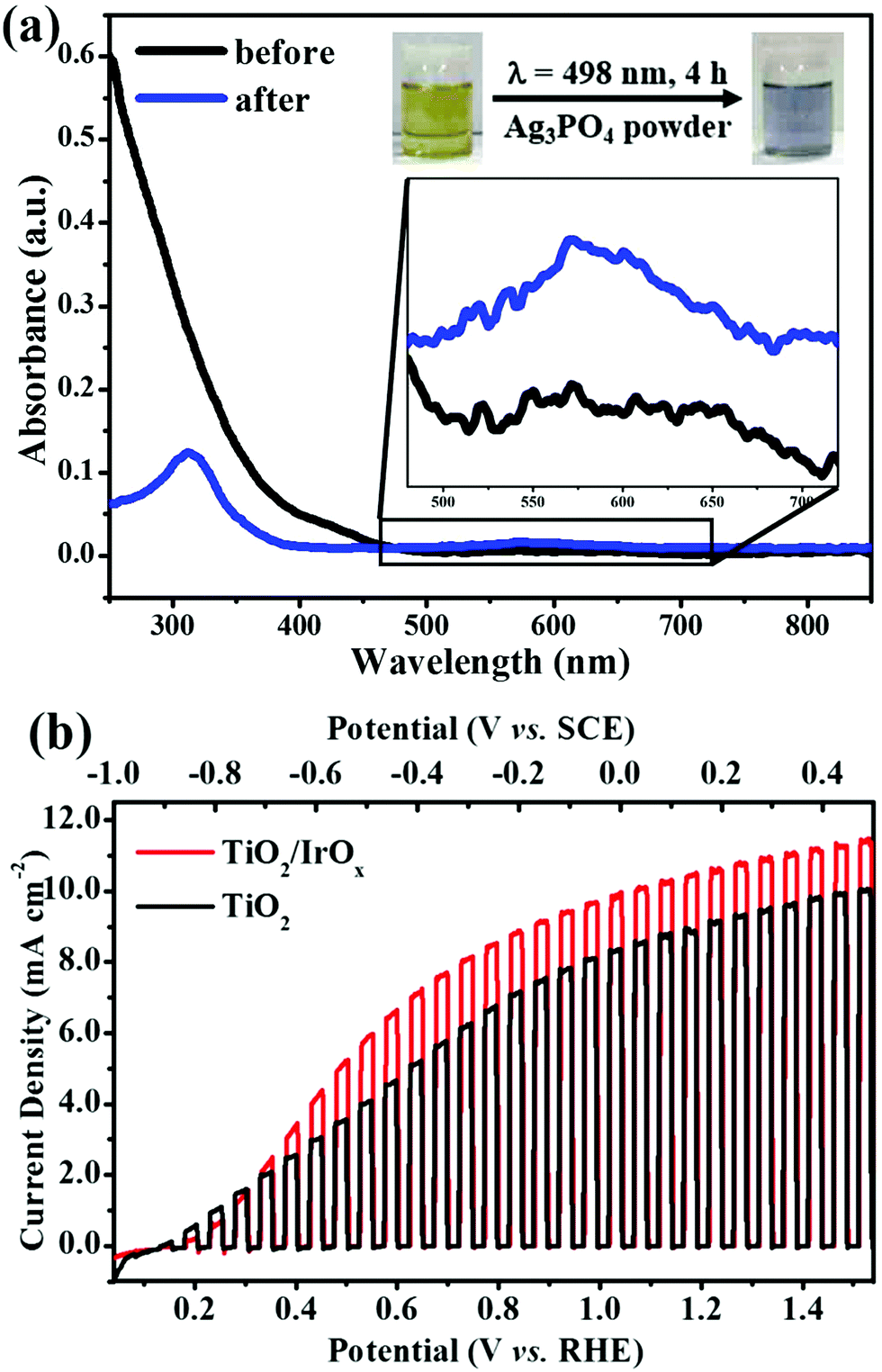 | ||
| Fig. 6 (a) Photographs and UV-vis absorption spectra of the IrCl3 solution (2 mM, pH 12) before and after irradiation under 498 nm light (4 mW cm−2) for 4 h with Ag3PO4 powder in the solution, the inset shows the local magnification of the absorption spectra. (b) The J–V curves of TiO2 and TiO2/IrOx composite photoanodes in 1 M KOH, under chopped UV light (150 mW cm−2) at a scan rate of 5 mV s−1. | ||
TiO2 has long been studied as a photo-anode material for water splitting.61,62 The valence band edge of TiO2 is located at ca. 3.0 V vs. NHE (3.7 V vs. RHE, pH 12),61 which is more positive than the Ir(IV)/Ir(III) potential. To test whether or not IrOx can be deposited on the surface of TiO2 under illumination, an FTO supported TiO2 electrode was immersed in IrCl3 solution (2 mM, pH 12) and illuminated under polychromatic UV light (150 mW cm−2) for 0.5 h. Then, the obtained electrode was used as a photoanode for water splitting. Fig. 6b shows the J–V curves of the TiO2 photoanode before and after the photo-catalyzed deposition of IrOx. It can be seen clearly in Fig. 6b that, after illumination in IrCl3 solution, the resulting photoanode exhibited a significantly improved photoactivity toward PEC OER within the entire potential sweep region. This result suggests that IrOx was successfully deposited on the surface of TiO2 and acted as an OER co-catalyst on the surface of TiO2. Similar results were obtained on the Ag3PO4/IrOx electrode. But the improvement was not obvious compared with the results obtained on Fe2O3 and TiO2. We guess this is because of the mismatch of the energy levels of Ag3PO4 and IrOx or high electrical resistance between Ag3PO4 and IrOx. Further study is needed to find out the reason and suitable conditions to deposit IrOx effectively on Ag3PO4 using the photo-catalyzed hydrolysis method.
All the above results indicate that photo-catalyzed surface hydrolysis of Ir3+ is a universal phenomenon that takes place on the surface of n-type semiconductors whose valence band edge is more positive than the redox potential of Ir(IV)/Ir(III). Moreover, the successful deposition of IrOx on the surface of Fe2O3, TiO2, and Ag3PO4 demonstrates that surface hydrolysis under illumination is a general route to couple IrOx with n-type semiconductor photoanodes.
4. Conclusions
In summary, based on our previous work on photo-driven hydrolysis of IrCl3 in homogeneous solution,27 we demonstrated in this work that the photo-catalyzed surface hydrolysis could be realized on n-type semiconductors such as Fe2O3, TiO2, and Ag3PO4. The photo-catalyzed hydrolysis follows a mechanism different from that in the homogeneous solution. For surface hydrolysis under illumination, the photo-generated holes played the key role of an oxidizing agent, which oxidized Ir(III) to Ir(IV) species and then triggered the formation of IrOx. We proved that the hydrolysis could be localized at the Fe2O3/solution interface by using monochromatic visible light of 498 nm, which could not induce hydrolysis in homogeneous solution. As IrOx is an efficient OER catalyst, the photo-catalyzed surface hydrolysis can be developed as a universal strategy to couple the OER co-catalyst IrOx with semiconductor photoanode materials, and the resulting semiconductor/IrOx composites could be employed as promising photoanodes for PEC OER. We demonstrated that, after photo-catalyzed deposition of IrOx, the Fe2O3 and the TiO2 photoanodes exhibited significantly improved photoactivity toward PEC OER. We believe that the surface hydrolysis under illumination may open a new avenue for the construction of semiconductor/co-catalyst composites for solar-powered water splitting.Acknowledgements
We gratefully acknowledge the financial support of this work by National Natural Science Foundation of China (NSFC 51672017 and 21173016) and Beijing Natural Science Foundation (2142020 and 2151001).Notes and references
- V. Fierro, G. Muñiz, G. Gonzalez-Sánchez, M. L. Ballinas and A. Celzard, J. Hazard. Mater., 2009, 168, 430–437 CrossRef CAS PubMed.
- D. W. Lee and B. R. Yoo, J. Ind. Eng. Chem., 2014, 20, 3947–3959 CrossRef CAS.
- J. Lee, S. Zhang and S. Sun, Chem. Mater., 2013, 25, 1293–1304 CrossRef CAS.
- S. Musić, S. Krehula, S. Popović and Ž. Skoko, Mater. Lett., 2003, 57, 1096–1102 CrossRef.
- Q. Yang, Z. Lu, J. Liu, X. Lei, Z. Chang, L. Luo and X. Sun, Prog. Nat. Sci., 2013, 23, 351–366 CrossRef.
- J.-Y. Li, S. Xiong, J. Pan and Y. Qian, J. Phys. Chem. C, 2010, 114, 9645–9650 CAS.
- B. Liu and H. C. Zeng, J. Am. Chem. Soc., 2003, 125, 4430–4431 CrossRef CAS PubMed.
- Z. Miao, D. Xu, J. Ouyang, G. Guo, X. Zhao and Y. Tang, Nano Lett., 2002, 2, 717–720 CrossRef CAS.
- M. Niederberger and G. Garnweitner, Chem. – Eur. J., 2006, 12, 7282–7302 CrossRef CAS PubMed.
- C. Cheng, Y. Huang, N. Wang, T. Jiang, S. Hu, B. Zheng, H. Yuan and D. Xiao, ACS Appl. Mater. Interfaces, 2015, 7, 9526–9533 CAS.
- T. D. Schladt, T. Graf, O. Köhler, H. Bauer, M. Dietzsch, J. Mertins, R. Branscheid, U. Kolb and W. Tremel, Chem. Mater., 2012, 24, 525–535 CrossRef CAS.
- G. Zhu, H. Xu, Y. Xiao, Y. Liu, A. Yuan and X. Shen, ACS Appl. Mater. Interfaces, 2012, 4, 744–751 CAS.
- X. Hong, Z. Wang, W. Cai, F. Lu, J. Zhang, Y. Yang, N. Ma and Y. Liu, Chem. Mater., 2005, 17, 1548–1552 CrossRef CAS.
- J. Zhong, F. Chen and J. Zhang, J. Phys. Chem. C, 2010, 114, 933–939 CAS.
- D. A. Schwartz, N. S. Norberg, Q. P. Nguyen, J. M. Parker and D. R. Gamelin, J. Am. Chem. Soc., 2003, 125, 13205–13218 CrossRef CAS PubMed.
- C. X. Guo, J. Xie, H. Yang and C. M. Li, Adv. Sci., 2015, 2, 1500135 CrossRef.
- C. Yuan, X. Zhang, L. Su, B. Gao and L. Shen, J. Mater. Chem., 2009, 19, 5772–5777 RSC.
- C. Justin Raj, B. C. Kim, W.-J. Cho, S. Park, H. T. Jeong, K. Yoo and K. H. Yu, J. Electroanal. Chem., 2015, 747, 130–135 CrossRef CAS.
- M. R. Buck, A. J. Biacchi and R. E. Schaak, Chem. Mater., 2014, 26, 1492–1499 CrossRef CAS.
- C. W. Wang, S. Yang, W. Q. Fang, P. Liu, H. Zhao and H. G. Yang, Nano Lett., 2016, 16, 427–433 CrossRef CAS PubMed.
- F. Ooi, J. S. DuChene, J. Qiu, J. O. Graham, M. H. Engelhard, G. Cao, Z. Gai and W. D. Wei, Small, 2015, 11, 2649–2653 CrossRef CAS PubMed.
- Y. Zhao, E. A. Hernandez-Pagan, N. M. Vargas-Barbosa, J. L. Dysart and T. E. Mallouk, J. Phys. Chem. Lett., 2011, 2, 402–406 CrossRef CAS.
- L. Chen, J. Xu, D. A. Tanner, R. Phelan, M. van der Meulen, J. D. Holmes and M. A. Morris, Chem. – Eur. J., 2009, 15, 440–448 CrossRef CAS PubMed.
- F. Kim, J. H. Song and P. Yang, J. Am. Chem. Soc., 2002, 124, 14316–14317 CrossRef CAS PubMed.
- L. Maretti, P. S. Billone, Y. Liu and J. C. Scaiano, J. Am. Chem. Soc., 2009, 131, 13972–13980 CrossRef CAS PubMed.
- M. Sakamoto and T. Majima, Bull. Chem. Soc. Jpn., 2010, 83, 1133–1154 CrossRef CAS.
- D. Xu, P. Diao, T. Jin, Q. Wu, X. Liu, X. Guo, H. Gong, F. Li, M. Xiang and Y. Ronghai, ACS Appl. Mater. Interfaces, 2015, 7, 16738–16749 CAS.
- S. Cherevko, S. Geiger, O. Kasian, N. Kulyk, J.-P. Grote, A. Savan, B. R. Shrestha, S. Merzlikin, B. Breitbach, A. Ludwig and K. J. J. Mayrhofer, Catal. Today, 2016, 262, 170–180 CrossRef CAS.
- S. W. Lee, M. Janyasupab, C.-C. Liu and R. M. Sankaran, Catal. Today, 2013, 211, 137–142 CrossRef CAS.
- J. Park, M. Kim and S. Kim, Sens. Actuators, B, 2014, 204, 197–202 CrossRef CAS.
- E. Fabbri, A. Habereder, K. Waltar, R. Kötz and T. J. Schmidt, Catal. Sci. Technol., 2014, 4, 3800–3821 CAS.
- D. Chandra, D. Takama, T. Masaki, T. Sato, N. Abe, T. Togashi, M. Kurihara, K. Saito, T. Yui and M. Yagi, ACS Catal., 2016, 6, 3946–3954 CrossRef CAS.
- S. D. Tilley, M. Cornuz, K. Sivula and M. Grätzel, Angew. Chem., Int. Ed., 2010, 122, 6549–6552 CrossRef.
- E. M. Steinmiller and K. S. Choi, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20633–20636 CrossRef CAS PubMed.
- T. Jin, P. Diao, Q. Wu, D. Xu, D. Hu, Y. Xie and M. Zhang, Appl. Catal., B, 2014, 148–149, 304–310 CrossRef CAS.
- T. Jin, P. Diao, D. Xu and Q. Wu, Electrochim. Acta, 2013, 114, 271–277 CrossRef CAS.
- A. Irshad and N. Munichandraiah, J. Electrochem. Soc., 2015, 162, H235–H243 CrossRef CAS.
- Y. Liu, Y. Zhou, G. Chen, T. Guo, L. Wang, X. Huang and W. Zeng, Mater. Lett., 2015, 148, 155–158 CrossRef CAS.
- T. Kuwabara, E. Tomita, S. Sakita, D. Hasegawa, K. Sone and M. Yagi, J. Phys. Chem. C, 2008, 112, 3774–3779 CAS.
- T. Nakagawa, C. A. Beasley and R. W. Murray, J. Phys. Chem. C, 2009, 113, 12958–12961 CAS.
- J. Sun, D. K. Zhong and D. R. Gamelin, Energy Environ. Sci., 2010, 3, 1252–1261 CAS.
- L. Xi, P. D. Tran, S. Y. Chiam, P. S. Bassi, W. F. Mak, H. K. Mulmudi, S. K. Batabyal, J. Barber, J. S. C. Loo and L. H. Wong, J. Phys. Chem. C, 2012, 116, 13884–13889 CAS.
- K. Sivula, F. Le Formal and M. Grätzel, ChemSusChem, 2011, 4, 432–449 CrossRef CAS PubMed.
- W. Li, S. W. Sheehan, D. He, Y. He, X. Yao, R. L. Grimm, G. W. Brudvig and D. Wang, Angew. Chem., Int. Ed., 2015, 54, 11428–11432 CrossRef CAS PubMed.
- Y.-S. Hu, A. Kleiman-Shwarsctein, A. J. Forman, D. Hazen, J.-N. Park and E. W. McFarland, Chem. Mater., 2008, 20, 3803–3805 CrossRef CAS.
- L. Vayssieres, N. Beermann, S.-E. Lindquist and A. Hagfeldt, Chem. Mater., 2001, 13, 233–235 CrossRef CAS.
- Y. C. Pu, G. Wang, K. D. Chang, Y. Ling, Y. K. Lin, B. C. Fitzmorris, C. M. Liu, X. Lu, Y. Tong, J. Z. Zhang, Y. J. Hsu and Y. Li, Nano Lett., 2013, 13, 3817–3823 CrossRef CAS PubMed.
- X. Yang, H. Cui, Y. Li, J. Qin, R. Zhang and H. Tang, ACS Catal., 2013, 3, 363–369 CrossRef CAS.
- D. Hu, P. Diao, D. Xu and Q. Wu, Nano Res., 2016, 9, 1735–1751 CrossRef CAS.
- W. D. Chemelewski, H. C. Lee, J. F. Lin, A. J. Bard and C. B. Mullins, J. Am. Chem. Soc., 2014, 136, 2843–2850 CrossRef CAS PubMed.
- T. Ioroi, N. Kitazawa, K. Yasuda, Y. Yamamoto and H. Takenaka, J. Electrochem. Soc., 2000, 147, 2018 CrossRef CAS.
- J.-Y. Chen, Y.-M. Chen, Y. Sun, J.-F. Lee, S.-Y. Chen, P.-C. Chen and P.-W. Wu, Ceram. Int., 2014, 40, 14983–14990 CrossRef CAS.
- N. Beermann, L. Vayssieres, S.-E. Lindquist and A. Hagfeldt, J. Electrochem. Soc., 2000, 147, 2456–2461 CrossRef CAS.
- A. Kay, I. Cesar and M. Gratzel, J. Am. Chem. Soc., 2006, 128, 15714–15721 CrossRef CAS PubMed.
- B. C. Fitzmorris, J. M. Patete, J. Smith, X. Mascorro, S. Adams, S. S. Wong and J. Z. Zhang, ChemSusChem, 2013, 6, 1907–1914 CrossRef CAS PubMed.
- S. Siracusano, N. van Dijk, E. Payne-Johnson, V. Baglio and A. S. Aricò, Appl. Catal., B, 2015, 164, 488–495 CrossRef CAS.
- E. S. Cho, M. J. Kang and Y. S. Kang, Phys. Chem. Chem. Phys., 2015, 17, 16145–16150 RSC.
- C. S. G. James and C. Chang, Inorg. Chem., 1965, 4, 209–215 CrossRef.
- Z. Yi, J. Ye, N. Kikugawa, T. Kako, S. Ouyang, H. Stuart-Williams, H. Yang, J. Cao, W. Luo, Z. Li, Y. Liu and R. L. Withers, Nat. Mater., 2010, 9, 559–564 CrossRef CAS PubMed.
- Q. Wu, P. Diao, J. Sun, D. Xu, T. Jin and M. Xiang, J. Mater. Chem. A, 2015, 3, 18991–18999 CAS.
- A. Fujishima, X. Zhang and D. Tryk, Surf. Sci. Rep., 2008, 63, 515–582 CrossRef CAS.
- M. Ni, M. K. H. Leung, D. Y. C. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401–425 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: UV-vis absorption spectrum of Fe2O3 and J–V curves of the Fe2O3 electrode before and after immersion in IrCl3 solution. See DOI: 10.1039/c6cp06821a |
| This journal is © the Owner Societies 2017 |