
Investigation of thin/well-tunable liquid/gas diffusion layers exhibiting superior multifunctional performance in low-temperature electrolytic water splitting†
Zhenye
Kang
a,
Jingke
Mo
a,
Gaoqiang
Yang
a,
Scott T.
Retterer‡
b,
David A.
Cullen‡
b,
Todd J.
Toops‡
b,
Johney B.
Green Jr‡§
b,
Matthew M.
Mench
c and
Feng-Yuan
Zhang
 *a
*a
aDepartment of Mechanical, Aerospace & Biomedical Engineering, UT Space Institute, University of Tennessee, Knoxville, USA. E-mail: fzhang@utk.edu; Tel: +1-(931)-393-7428
bOak Ridge National Lab, USA
cDepartment of Mechanical, Aerospace & Biomedical Engineering, University of Tennessee, Knoxville, USA
First published on 11th October 2016
Abstract
Liquid/gas diffusion layers (LGDLs), which are located between the catalyst layer (CL) and bipolar plate (BP), play an important role in enhancing the performance of water splitting in proton exchange membrane electrolyzer cells (PEMECs). They are expected to transport electrons, heat, and reactants/products simultaneously with minimum voltage, current, thermal, interfacial, and fluidic losses. In this study, the thin titanium-based LGDLs with straight-through pores and well-defined pore morphologies are comprehensively investigated for the first time. The novel LGDL with a 400 μm pore size and 0.7 porosity achieved a best-ever performance of 1.66 V at 2 A cm−2 and 80 °C, as compared to the published literature. The thin/well-tunable titanium based LGDLs remarkably reduce ohmic and activation losses, and it was found that porosity has a more significant impact on performance than pore size. In addition, an appropriate equivalent electrical circuit model has been established to quantify the effects of pore morphologies. The rapid electrochemical reaction phenomena at the center of the PEMEC are observed by coupling with high-speed and micro-scale visualization systems. The observed reactions contribute reasonable and pioneering data that elucidate the effects of porosity and pore size on the PEMEC performance. This study can be a new guide for future research and development towards high-efficiency and low-cost hydrogen energy.
Broader contextHydrogen is a ‘zero’ emission energy carrier which could be an important part of environmentally friendly solutions to the global energy crisis via combustion or transformation into electricity through fuel cells without producing any greenhouse gases and pollutants. Proton exchange membrane (PEM) water splitting is one of the most practical and high-efficiency methods to produce pure hydrogen from renewable sources like wind and solar energy. The wide commercialization of PEM electrolyzer cells is still hindered by their performance and durability. Successful development of a thin titanium liquid/gas diffusion layer (LGDL) will reduce the cost, thickness, and weight of the LGDL itself and the system as a whole. This investigation demonstrates the ability to produce LGDLs with precise control of pore size, shape, distribution, and therefore overall porosity and permeability, which can aid in developing modeling routine or validating simulations. More importantly, thin titanium based LGDLs will lead to a manufacturing solution to couple the LGDL with the metallic bipolar plates, since they can be easily integrated together by top-down and bottom-up manufacturing process. Thus, one metallic part could function simultaneously as flow field, bipolar plate, current distributor/collector, and LGDL. |
1 Introduction
Renewable energy sources, including solar, wind, hydro, biomass and geothermal power, produce clean electricity in sustainable ways. However, most of these renewable sources are variable and often produce electricity intermittently (e.g., only during daylight or when windy), which present major challenges to delivering consistent power to operate today's electrical grid. In addition, the current electrical grid has very limited ability to digest the fluctuation from renewable energy sources. Thereby, a sustainable, high-efficiency, and robust electrochemical energy storage/conversion or a hybrid system to accommodate daily or even hourly changes becomes more critical.1–9 An advanced proton exchange membrane electrolyzer cell (PEMEC), which is a reverse PEM fuel cell (PEMFC), has been considered as a very attractive energy storage method for producing hydrogen/oxygen from water splitting when coupled with renewable energy sources. PEMECs have several advantages, such as distinguished efficiency, compact design, large capacity, quick startup, and low maintenance activities, and effectively connect renewable electricity supply and multiscale energy demands including stationary, transportation, and portable applications.10–16 When renewable energy resources are available, hydrogen/oxygen will be produced and stored with a PEMEC. Later, hydrogen/oxygen can be converted back to water and electricity with a PEM fuel cell (PEMFC), whether the renewable source is available or not. Additionally, surplus electricity in electric grids during off-peak periods can also be stored via the electrolyzer. This entire portfolio will make hybrid energy systems able to provide renewable and reliable energy at different scales whenever and wherever needed.17–22A PEM electrolyzer cell mainly consists of a catalyst-coated membrane sandwiched by anode and cathode electrodes, as shown in Fig. 1. Each electrode includes a catalyst layer (CL), a liquid/gas diffusion layer (LGDL), and a bipolar plate (BP), which also acts as the current distributor (CD) and the flow field. After electricity is applied, water is split into molecular oxygen, protons, and electrons at the anode side, as shown in Fig. 1. Di-oxygen, as one product on the anode CLs, is ideally transported from the CL through the LGDL back to the flow field to avoid blocking the LGDL, which can hinder the reaction. Electrons, which are also generated at anode CLs, pass through the LGDL, anode BP, and external circuit, and then back to the cathode side. Meanwhile, protons pass through the membrane to the cathode and react with electrons which come from the external circuit to form di-hydrogen. H2/O2 will be produced and stored continuously as long as water and electricity are supplied. Thus, not only should the water be supplied continuously, but also the oxygen and hydrogen should be effectively removed through the LGDLs. This is especially important at high current density, where mass transport is a dominant limiting factor of PEMEC performance.23–25
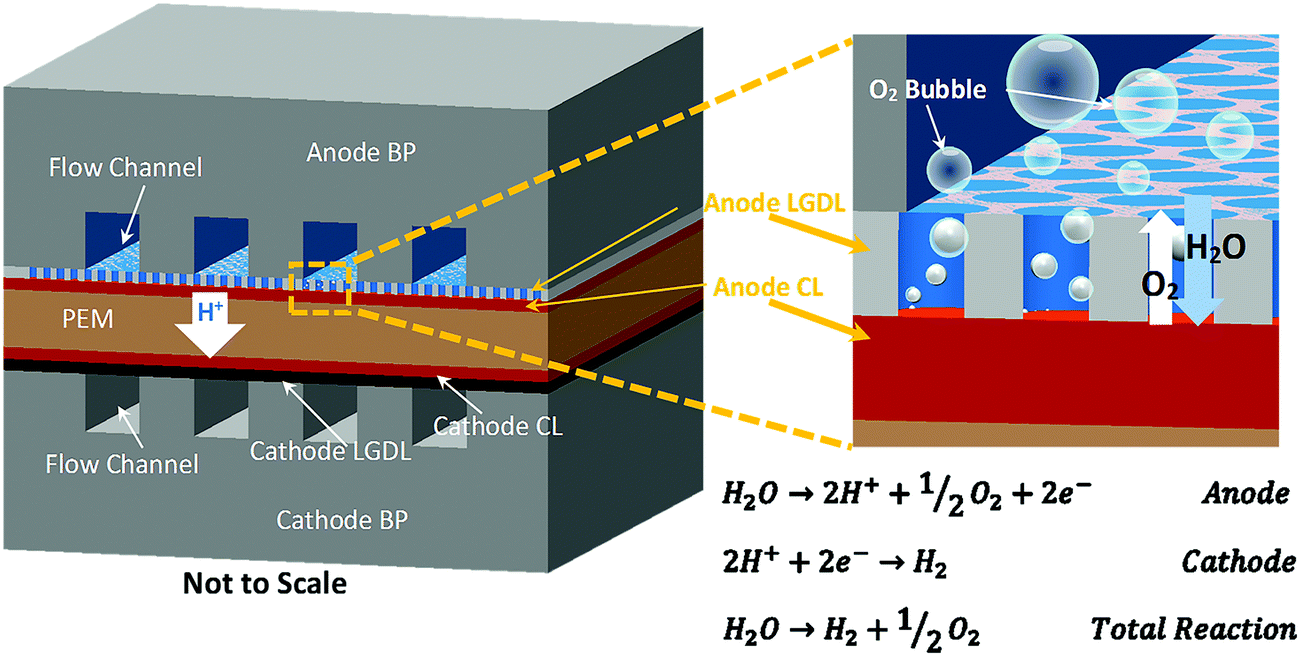 | ||
| Fig. 1 Schematic of thin titanium LGDL functions. | ||
The anode resides in a harsh environment, which is highly corrosive due to the high overpotential and humidity. Carbon materials (like carbon paper or carbon cloth), which are typically used in PEMFCs, are unsuitable on this side of the PEMECs due to the high potential of the oxygen electrode.14,26–32 Ideal anode LGDLs should have good conductivity, high corrosion resistance, good two-phase transport capability and mechanical strength. Metallic LGDLs and bipolar plates, including titanium, have attracted more interest in both PEMECs and PEMFCs due to their high conductivity, rapid production, and low cost.33–37
Grigoriev et al. conducted an optimization of porous current collectors. According to his research, the mean pore size of the particles and the thickness of the titanium plates have a significant effect on current–voltage performance. The study found that the optimum sphere particle sizes ranged from 50 to 75 μm and the optimum pore sizes were between 12 and 13 μm, whereas the porosity (between 0.35–0.40) and gas permeability of the porous titanium plates did not have a significant influence on electrolysis efficiency.38 Millet et al. noted that it is necessary to reduce the ohmic resistance between the separator plates and current collectors in order to improve the performance of the PEMECs.39 Ma et al. concluded that a thinner carbon paper used as an anode LGDL will improve the PEMEC performance due to its better gas diffusion properties and smaller resistance.40 Hwang et al. made microporous layers (MPL) for PEMECs by loading titanium powder over titanium felt to promote interfacial contacts and they also investigated the effect of pore properties and PTFE content for titanium felt LGDL of the anode electrode.41,42
The main efforts in the previous studies, so far, have focused on investigating conventional titanium LGDLs, including felts, woven meshes, or foams.43–45 The thickness of these LGDLs were larger than 200 μm with significantly longer electrically conductive path lengths and higher fluidic resistances. In addition, their fiber/foam-based pore morphologies result in not only nonuniform interfacial contacts, but random pore sizes and distributions. These random, nonuniform and complicated structures in conventional titanium LGDLs make it impossible to control the liquid/gas/electron/thermal distribution precisely. Therefore, novel LGDLs with tunable and controlled pore morphologies are strongly desired.
In this study, by taking advantage of advanced micro/nano-manufacturing, a new thin, planar titanium LGDL with straight-through pores and well-tunable pore morphologies is developed. The well-controllable pore size and porosity help to explicitly examine the effects of the pore morphology, and to characterize the two-phase transport through the LGDL. The effects of well-defined pore parameters on the PEMEC performance are comprehensively investigated. Both the electro-potential performance and electrochemical impedance are evaluated with the novel LGDLs, and significant improvements have been achieved. Electrochemical impedance spectroscopy (EIS) is further analyzed by equivalent electrical circuit fitting, which helps to identify the effect of each loss in the PEMEC. In addition, the LGDL thickness is reduced from greater than hundreds micrometers of conventional LGDLs to only 25 microns, which remarkably reduces the transport and ohmic resistances. More importantly, the development of thin/well-tunable LGDLs with straight pores permits direct visualizations of the electrochemical reactions, which facilitate better understanding of effects of the LGDL pore size and porosity. The visualization results reveal that the oxygen bubble only nucleates at the rim of the pores, and an appropriate assumption is proposed to analyze and explain the effects of the pore size and porosity.
2 Experimental details
2.1 Nano-manufacturing of titanium thin/well-tunable LGDLs
The thin/well-tunable titanium LGDLs are manufactured lithographically – patterned resist masks and chemical wet etching of thin foils which is shown in Fig. S1 (ESI†).46,47Fig. 2 shows a typical scanning electron microscopy (SEM) image of thin/well-tunable LGDLs (sample A1 and A2) used in this study. The thickness of all LGDLs was 25.4 μm. The pore shapes of the LGDLs were controlled to be circular and all the pores were distributed regularly and uniformly. The pore diameter and the distance between adjacent pore rims are defined as pore size and land length, which are represented by D and L, respectively. The pore morphology like pore size, pore shape, pore distribution and porosity can be well controlled through the mask design and/or etching conditions. | ||
| Fig. 2 SEM images of typical thin/well-tunable titanium LGDLs: (A) pore morphology and structure of sample A1 with approximately 100 μm pore size and 0.30 porosity. (B) Pore morphology and structure of sample A2 with approximately 200 μm pore size and 0.30 porosity. | ||
The porosity of the LGDL ε, is defined as the total pore area, AP, divided by the total area of the entity, AH, which can be given as:
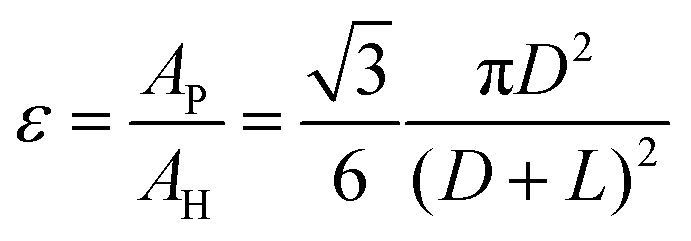 | (1) |
| Index of the LGDL | Pore size (D) [μm] | Land length (L) [μm] | Pore distance [μm] | Calculated porosity (ε) |
|---|---|---|---|---|
| A1 | 101.06 | 77.07 | 178.13 | 0.29 |
| A2 | 199.11 | 142.41 | 341.52 | 0.31 |
| A3 | 424.64 | 292.91 | 717.55 | 0.32 |
| A4 | 586.96 | 448.51 | 1035.47 | 0.29 |
| A5 | 791.61 | 589.51 | 1381.12 | 0.30 |
| B3 | 415.51 | 52.74 | 468.25 | 0.71 |
| B4 | 585.46 | 89.91 | 675.37 | 0.68 |
| B5 | 789.16 | 113.21 | 902.37 | 0.69 |
2.2 PEMEC and testing system
The thin/well-tunable LGDLs were tested in a conventional PEMEC. The two end-plates were made from commercial grade aluminum and designed to provide even compression pressure on the PEMEC. The cell was compressed by eight evenly distributed bolts which were tightened to 4.52 N m of torque. A copper plate, which was inserted between the bipolar plate and end-plate at both the anode and cathode, was used to apply current to the PEMEC. The bipolar plates were made of graphite and fabricated with a parallel flow field which was intended to introduce the reactants and products in and out of the PEMEC. Titanium thin film with 25 μm thickness and carbon paper (Toray 090 with 280 μm thickness and porosity of 0.78) were used as anode and cathode LGDLs, respectively. The catalyst-coated membrane (CCM) (Electrolyzer CCM from FuelCellsEtc, EZ-CCM) was comprised of a Nafion 115 membrane, a perfluorosulfonic polymer with a thickness of 125 μm, an anode catalyst layer with an IrRuOx catalyst loading of 3.0 mg cm−2, and a cathode layer with a platinum black (PtB) catalyst loading of 3.0 mg cm−2 with a 5 cm2 working area. Table 2 shows the detailed characteristics and experimental conditions.| Characteristics and conditions | Value |
|---|---|
| Membrane type | Nafion® 115 |
| Membrane area | 5 cm2 |
| Membrane thickness | 125 μm |
| Anode catalyst loading | 3.0 mg cm−2 (IrRuOx) |
| Cathode catalyst loading | 3.0 mg cm−2 (PtB) |
| Anode LGDL | Titanium thin film |
| Cathode LGDL | Toray 090 carbon paper |
| Operating temperature | 20, 40, 60, 80 °C |
| Operating pressure | 1 atm |
| Anode water flow rate | 20 ml min−1 |
The PEMEC was attached to an electrolyzer control system with current range up to 100 A and voltage range up to 5 V. The hardware was connected to EC-Lab, an electrochemical analysis software from Bio-Logic, which was used to evaluate performance and perform electrochemical impedance spectroscopy (EIS). For controlling the flow, a system of tubing was connected to the PEMEC. While the cathode tubing was merely intended to safely exhaust hydrogen gas that formed during electrolysis, a diaphragm liquid pump from KNF Neuberger was used to supply de-ionized (DI) water at a constant volumetric flow rate of 20 ml min−1 to the anode. The water bath (General Purpose Water Baths of Model WB10 from PolyScience) was used to pre-heat the DI water to designed temperatures. Two heaters used to heat the PEMEC were inserted into the end-plates at both anode and cathode and two thermocouples used to measure the temperature were inserted into the bipolar plates at both anode and cathode. Both of the heaters and thermocouples were connected to a temperature control system (Multi-Zone controller from OMEGA).
For performance evaluation, an increasing current density was applied to the PEMEC and the current was stepped up from a current density of 0.2 A cm−2 to 2.0 A cm−2 with a step of 10 mA s−1. Galvanostatic electrochemical impedance spectroscopy (GEIS) was used to measure the impedance of the PEMEC at different operating conditions. In this method, the current is controlled as opposed to the potential. The test station was equipped with an operating current range of −100 A to +100 A and a voltage range of 0 V to 5 V. The current precision was 100 fA. The scanning frequency was varied from 15 kHz to 10 mHz, and recorded 20 points of data per decade. For analyzing impedance data, a Nyquist plot is normally used.
3 Results and discussion
3.1 The impact of the pore size and porosity
All the titanium thin/well-tunable LGDLs were evaluated in a standard PEMEC. They were applied and tested as anode LGDLs. The performance of the PEMEC can be derived based on polarization curves of the current density and voltage. The lower voltage at a given current density indicates better PEMEC performance. Fig. 3 shows the PEMEC performance results of all the eight LGDLs with different pore sizes and porosity under the same operating conditions. As is shown in Fig. 3, increasing the pore size from 101 μm to 791 μm (A1 to A5 with the same porosity of about 0.3), the PEMEC performance decreases. The LGDL with a pore size of 791 μm (A5) results in the worst performance among these five LGDLs. For instance, at a fixed current density of 2 A cm−2, the required cell voltage increased from 1.705 V for the LGDL A1 to 1.726 V for A5, although this is still much better than conventional LGDLs.38,41,43,45,48–52 The three LGDLs with a larger porosity of about 0.7 and smaller pore distances (B3, B4, and B5), show further improved performance over LGDLs with similar pore sizes but lower porosity/larger pore distance (A3, A4, and A5), and the voltages required at a current density of 2 A cm−2 are just 1.661 V, 1.667 V, 1.675 V, respectively. With a lower porosity of about 0.3 and larger pore distances, the performance with LGDLs of A3, A4 and A5 were in the range of 1.713–1.726 V at 2 A cm−2. It was noted that porosity had a greater impact on performance than pore size.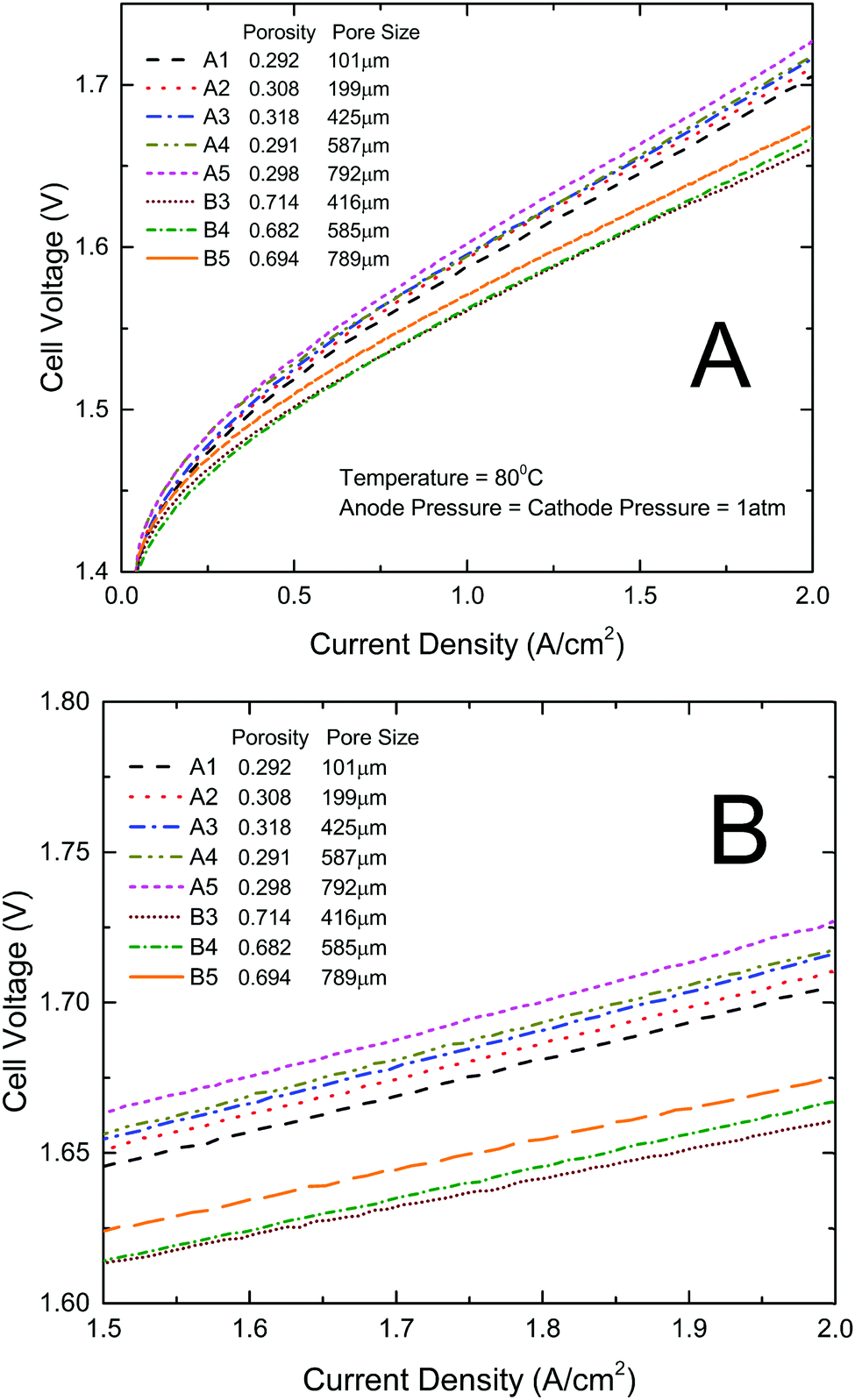 | ||
| Fig. 3 Performance comparison curves between different LGDLs. (A) Performance with a current density range from 0 to 2 A cm−2; (B) close-up of (A) with a current density range from 1.5 to 2 A cm−2. | ||
EIS is a very useful in situ method for analyzing PEMEC performance measuring the impedance of a system at different frequencies. In this study, EIS is conducted on PEMECs with different thin titanium LGDLs during performance testing at 80 °C and with a current density of 1.0 A cm−2. The scan frequency is set from high to low frequency (15 kHz to 10 mHz). As shown in Fig. 4, there are two x-intercepts: the left one (at the high frequency part) indicates the ohmic loss and the right one (at the low frequency part) is the sum of the resistance.45,53 The distance between the two intercepts indicates the sum of activation and mass transport losses.53,54 Therefore, the diameter of the first semicircle in Fig. 4 mainly indicates the activation resistance.
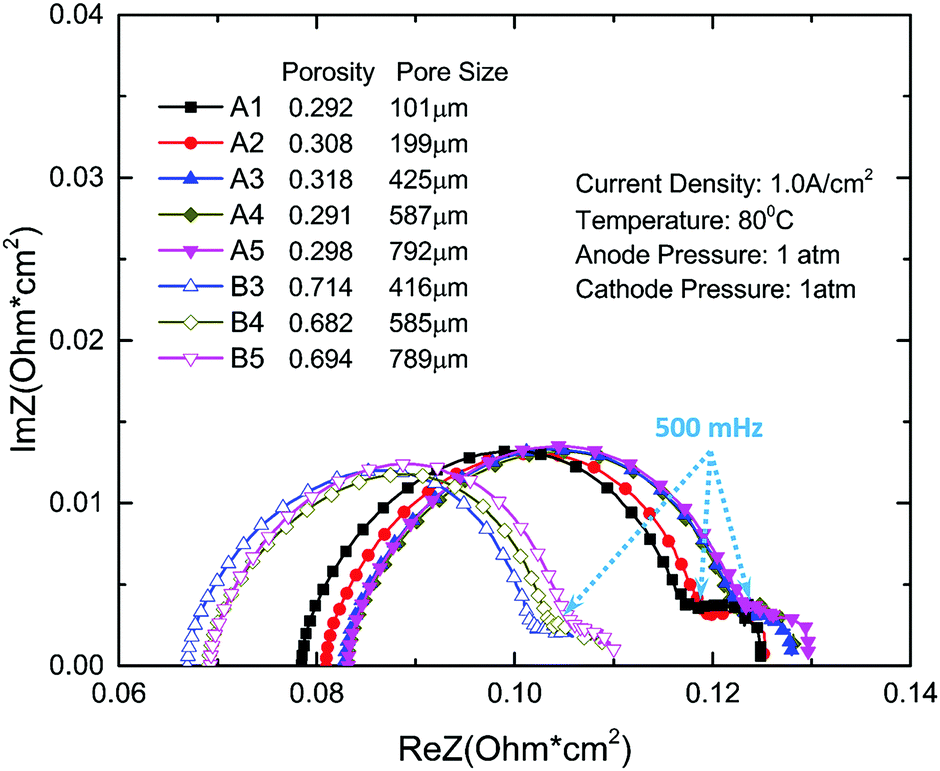 | ||
| Fig. 4 EIS comparison curves between different LGDLs. | ||
It can be found that a second arc showed up, which is very small, indicating the limited mass transport losses. The titanium well-tunable LGDLs with straight pores are thin and very hydrophilic. The water contact angle on the thin titanium foil was measured to be around 45°, while the micro pore features on LGDLs cause a wettability change. For instance, the water contact angles with air-filled pores for samples A1, A4 and B4 are about 81°, 63° and 71°, respectively. During the PEMEC operation, the anode LGDLs are immersed in liquid water, and the pores of LGDLs are water filled. Under these conditions, the LGDL contact angles were found to decrease greatly and liquid water transport through LGDLs very quickly during the measurements. These phenomena show that the titanium thin/well-tunable LGDLs exhibit very hydrophilic wettability, and significantly reduce the transport loss of liquid water from the flow field to the reaction sites in a PEMEC.
The frequency between the two arcs is about 500 mHz. The LGDL performance enhancement is closely related to the impedance changes. LGDLs with a porosity of 0.3 have larger ohmic resistance, and the value decreases with the increase of porosity. It can be seen that for LGDLs with the same porosity, the impedance spectra do not change significantly, which coincides with the result of the polarization curves. The ohmic loss decreases significantly from around 0.08 ohm cm2 for the LGDL with a porosity of 0.3 to less than 0.07 ohm cm2 for one with a porosity of 0.7. It is also obvious that the 0.7 porosity LGDLs show smaller first and second arcs, which indicates that the activation and mass transfer losses decrease with increasing porosity. The sum of activation and mass transfer losses are reduced from about 0.046 ohm cm2 for 0.3 porosity LGDLs to 0.039 ohm cm2 for 0.7 porosity LGDLs. The total losses from EIS decreased by 14% when increasing porosity from 0.3 to 0.7. The detailed quantitative analysis of the EIS will be discussed.
3.2 Temperature impact
The open circuit voltage, membrane conductive and activation of a PEMEC have a close relation with temperature.25 In order to get a full understanding of this effect on the thin titanium LGDL, the PEMEC performance and EIS were evaluated at different temperatures. It can be expected that the PEMEC operating temperature has a significant effect on the PEMEC performance. In this study, PEMEC operating temperature was varied from 20 °C to 80 °C.Fig. 5 illustrates the effect of operating temperature on the PEMEC performance and EIS. As shown in Fig. 5(A), increasing the operating temperature results in a steady improvement in PEMEC performance. At a current density of 2.0 A cm−2, the cell voltage of PEMEC assembled with LGDL A4 is reduced from 1.971 V at 20 °C to 1.715 V at 80 °C. Fig. 5(B) shows that the ohmic loss decreases significantly with the increase of PEMEC operating temperature.23–25 The ohmic loss of the PEMEC consists of the resistances of each component, including PEM, CLs, LGDLs, bipolar plates (BP), and interfacial resistances between components. With the increase of the temperature, the proton conductivity of the PEM and CLs will increase gradually, leading to decreased ohmic losses.55 The interfacial contacts between components will improve at higher temperature, which will also reduce the ohmic loss of the PEMEC. The electrical resistivities of LGDL and BP materials do not change much with the temperature range from 20 to 80 °C, and their impacts on ohmic losses would be very limited. In addition, the higher temperature will result in improved diffusion processes, catalytic activity and electrode kinetics, and promote oxygen and hydrogen evolution reactions. Both the catalytic transfer coefficient and exchange current density increase with the temperature.56 The mass transport in PEMECs will also be enhanced at higher temperature. As shown in Fig. 5(B), the second arc of the EIS becomes smaller at higher temperature. As a result, the PEMEC performance improves significantly with increasing temperature. Most importantly, it can be noted that the PEMEC with the new LGDLs has an impressive performance even at low temperature (20–40 °C) compared to the literature,57,58 which demonstrates that PEMECs with thin/well-tunable LGDLs can operate at room temperature, an exciting possibility for further applications.
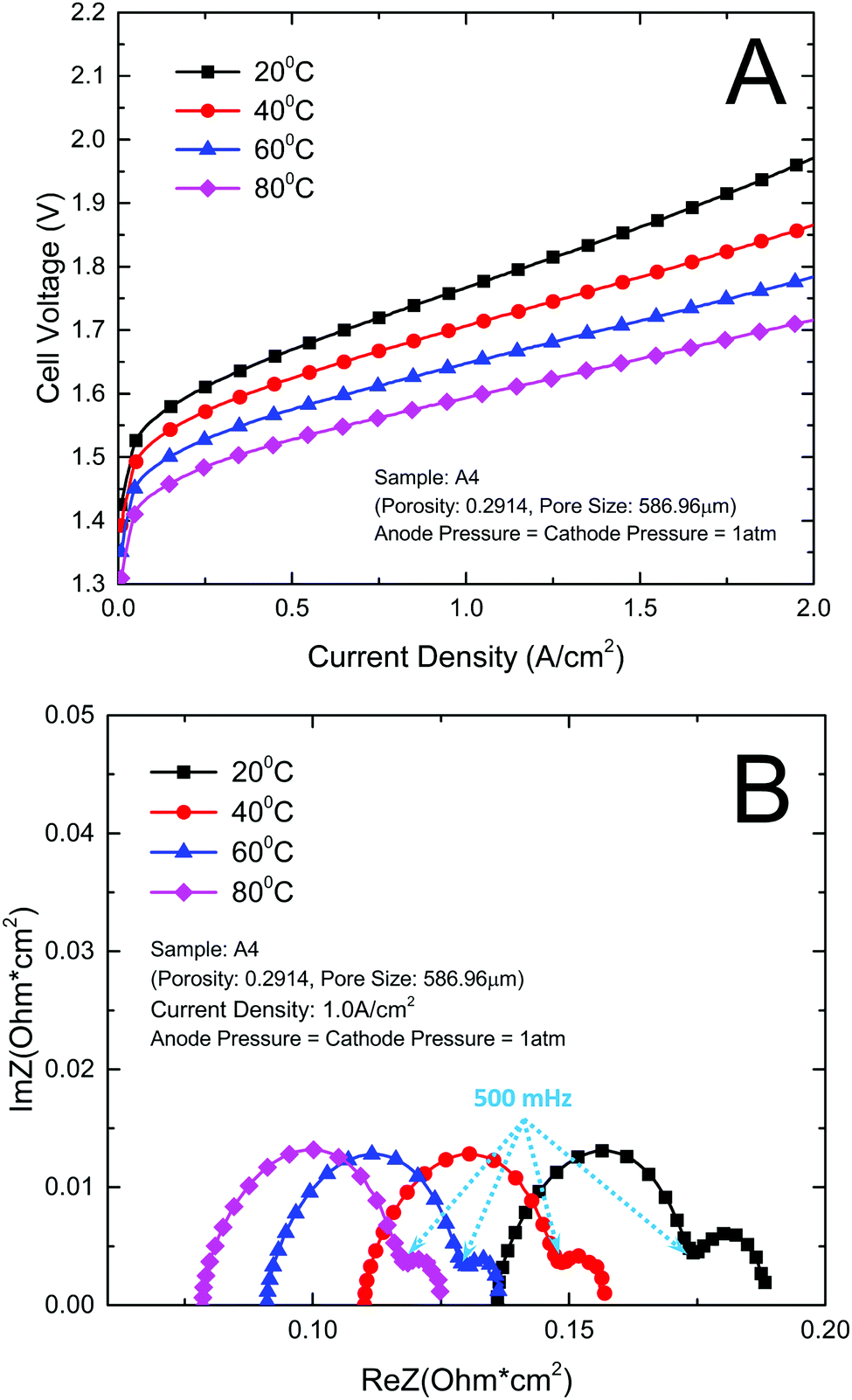 | ||
| Fig. 5 Impact of temperature change on PEMEC. (A) Performance curves comparison at different temperature; (B) EIS curves comparison at different temperature. | ||
3.3 Correlation of pore size and porosity with electrochemical reaction mechanisms
The total losses in a PEMEC mainly compose of ohmic loss, activation loss, and mass transport loss. Using an equivalent electrical circuit (EEC) to model the EIS curves can separate each of these losses. Therefore, an electrical circuit model was developed based on the generalized Randles equivalent circuit to investigate the effect of each loss.59–61 The EEC model is comprised by all the possible impact elements, including inductor (L), resistor of ohmic loss (Rohm), constant phase element (CPE), resistor of activation (R2) and Warburg diffusion element (Wd), which is shown is Fig. 6. ROhm mainly represents the ohmic losses of the whole PEMEC which is caused by membrane, CL, LGDLs, BP and all interfacial resistances between each component.62 The inductance of all the conductors in the PEMEC is assumed as L. R2 represents the activation losses, which are mainly related to the kinetics of the reactions at both anode and cathode. CPE is a flexible element, which represents a combination of resistor, capacitor and inductor. The surface reactivity, surface roughness, electrode porosity and surface inhomogeneity could be effectively affected by the CPE.61,63 The resistance of the Warburg diffusion element (Wd) is expressed by diffusion resistance (Rd) and it is used to judge the effect of diffusion in the PEMEC.61,64Fig. 6 shows an example of EIS fitting of a LGDL. Dots are for test results, while solid line represents EEC model fitting curve. The EIS fitting parameters of all LGDLs in this study have been listed in Table 3.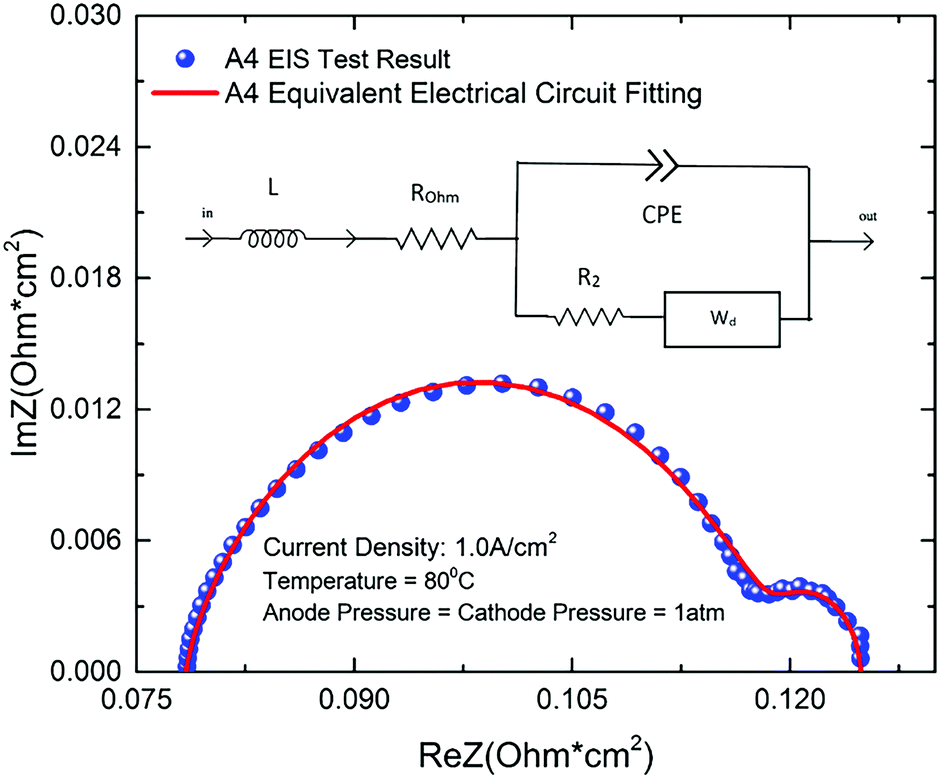 | ||
| Fig. 6 EIS results of sample A4 and its equivalent circuit fitting. | ||
| Sample | L Ind [H] | R Ohm [Ω cm2] | CPE_Q [F s(a−1)] | R 2 [Ω cm2] | R d [Ω cm2] | ERROR [%] |
|---|---|---|---|---|---|---|
| A1 | 1.23 × 10−8 | 0.0779 | 3.48 | 0.0406 | 0.006 | 0.31 |
| A2 | 1.19 × 10−8 | 0.0804 | 3.31 | 0.0410 | 0.005 | 0.29 |
| A3 | 1.28 × 10−8 | 0.0822 | 3.36 | 0.0411 | 0.004 | 0.23 |
| A4 | 1.24 × 10−8 | 0.0825 | 3.61 | 0.0418 | 0.004 | 0.31 |
| A5 | 1.21 × 10−8 | 0.0824 | 3.47 | 0.0420 | 0.004 | 0.20 |
| B3 | 1.24 × 10−8 | 0.0665 | 3.88 | 0.0363 | 0.003 | 0.45 |
| B4 | 1.20 × 10−8 | 0.0688 | 4.04 | 0.0365 | 0.003 | 0.48 |
| B5 | 1.28 × 10−8 | 0.0696 | 3.64 | 0.0381 | 0.002 | 0.33 |
From the Table 3, it can be seen that all the fitting error can be controlled within 0.5%, which means the fitting curve have a good coincidence with the experimental data and its feasibility is confirmed for the PEMECs with thin titanium LGDLs. For each electrical circuit element, the inductor parameters are very small and remain almost unchanged with different LGDLs. With the increase of porosity, the ohmic losses, activation losses and diffusion losses are all decreased, which lead to good performance of LGDLs. With the increase of pore size, the trend of the parameters is not obvious, which indicates that the effect of pore size is limited compared to porosity.
Table 4 shows the fitting parameters of EIS with LGDL A4 at different temperatures at a current density of 1.0 A cm−2. It can be found that the inductor changed at different temperatures. With the increase of temperature, the ohmic losses and diffusion losses are decreased while the activation losses are increased. It should be noted that the ohmic losses dominate the losses which leads to better performance at high temperature.
| Temperature [°C] | L Ind [H] | R Ohm [Ω cm2] | CPE_Q [F s(a−1)] | R 2 [Ω cm2] | R d [Ω cm2] | ERROR [%] |
|---|---|---|---|---|---|---|
| 20 | 2.73 × 10−8 | 0.135 | 3.23 | 0.0390 | 0.014 | 0.22 |
| 40 | 2.15 × 10−8 | 0.110 | 3.33 | 0.0391 | 0.008 | 0.26 |
| 60 | 1.73 × 10−8 | 0.0905 | 3.46 | 0.0399 | 0.007 | 0.44 |
| 80 | 1.24 × 10−8 | 0.0822 | 3.61 | 0.0418 | 0.004 | 0.31 |
In order to better evaluate the impacts of pore morphologies on the performance of the PEMEC and to optimize the design of LGDLs, the fundamental understanding of the porescale electrochemical reactions in operating PEMECs was highly desirable. The electrochemical reaction sites are located at the center part of a PEMEC behind the LGDL and other components. Additionally, conventional LGDLs are typically made of titanium fibers or foam, and their random pore morphologies and interconnected structures hinder optical accessibility to the PEMEC center. The development of thin titanium LGDLs with well-tunable pore morphologies and straight pore structures makes direct visualization of porescale reactions possible. By taking advantage of the innovative design of transparent electrochemical PEMECs, and developing high-speed and microscale characterization systems, in situ rapid and microscale electrochemical reactions and associated multiphase transport with microsecond time resolution were revealed. Fig. 7(A) shows the visualization results of the rapid micro-scale reactions occurred in a typical pore on the anode side, which is a single frame of a video (Supplementary 1, ESI†). The field of view focused on the LGDL pore with a diameter of 791 μm located at the middle of the channel, and it can be seen that all the catalyst layer in the pore are exposed to the liquid water. Based on the conventional triple-phase boundary reaction theory, the electrochemical reactions should occur on the whole surface of catalyst layer of the pore. However, the micro oxygen bubbles only nucleate only along the rim of the pore.
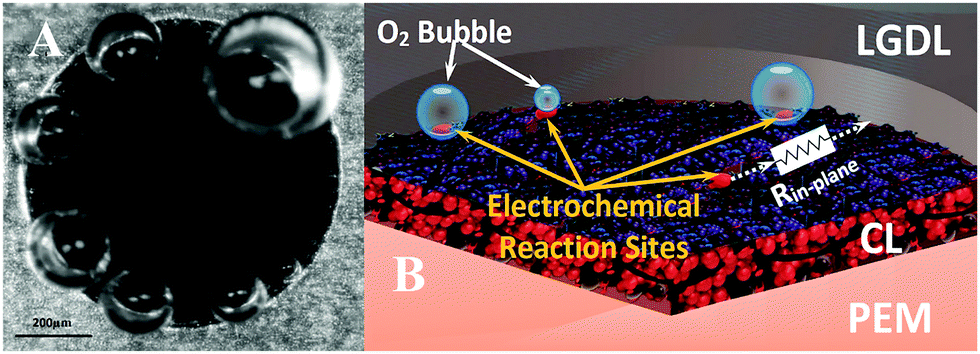 | ||
| Fig. 7 Visualization and schematic of the electrochemical reaction at 2.0 A cm−2 at pore scale (A) screenshot of visualization video shows the electrochemical reaction phenomenon in one pore. (B) Schematic of electrochemical reaction occurred at pore scale. | ||
From Fig. 7 and the supplementary video (ESI†), the rapid microbubble dynamics, including its nucleation, growth and detachment, can be observed clearly. Even under the current density of 2.0 A cm−2, the bubble detaches rapidly within the pore in a few milliseconds, and its detachment diameter is much smaller than the pore sizes. In our previous study, the bubble nucleation sites have been verified to be same at electrochemical reaction sites.65 Therefore, all bubbles nucleate along the rim of each pore, which also indicate the electrochemical reactions only occur at the triple-phase boundary (TPB) sites achieved at the rim zone of the pore. The CL sites that don't satisfy TPB conditions will not have electrochemical reaction, and the bubble will not nucleate and grow. As shown is Fig. 7(B), there is an in-plane resistance between the sites and LGDL which are expressed as Rin-plane. The Rin-plane closes to zero when the sites are located at the rim of the pore. Although the catalyst is expected to transport electrons, the Rin-plane of the IrRuOx catalyst layer has been found to be more than 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times larger than the thin titanium LGDL. The large in-plane ohmic losses in catalyst layers prevent the electrochemical reactions from occurring in the middle region of pores and act as an open circuit.
000 times larger than the thin titanium LGDL. The large in-plane ohmic losses in catalyst layers prevent the electrochemical reactions from occurring in the middle region of pores and act as an open circuit.
A Hitachi HF3300 scanning transmission electron microscope (STEM) equipped with a Bruker energy-dispersive X-ray spectrometer (EDS) and both high-angle annular dark-field (HAADF) and secondary electron (SE) detectors was used to image the morphology and composition of the different catalyst layers. Cross-sectional specimens were prepared by diamond-knife ultramicrotomy with a target thickness of 50–100 nm. Fig. 8 shows the cross-section and top-view morphology of the CCMs which were examined by SEM and STEM, and it can be seen that both the anode and cathode catalyst particles (IrRuOx and Pt Black, respectively) form non-uniformly distributed agglomerates on the surface membrane.66 The anode and cathode catalyst layers are roughly 20 μm and 15 μm thick, respectively, and the particle size of cathode PtB is smaller than anode. The membrane is observed above the anode, marked by the high fluorine signal in Fig. 8F. Fluorine was also present throughout the PtB cathode and is important for proton conduction throughout the electrode. The microstructures of the IrRuOx particles at the anode vary widely, which may cause different physical properties at a different point on the CL. At the anode electrode, water is split into electrons, protons and atomic oxygen at reaction sites. The TPB exists where water, good electron and proton conductivity and catalytically-active sites all meet. It can be assumed that not all of the sites on the CL yield electrochemical reactions; these are limited to sites where a TPB exist, which are distributed randomly on the CL.
 | ||
| Fig. 8 SEM and STEM images of catalyst coated membrane (A) SEM image of cross-section of catalysts coated membrane with IrRuOx anode at top side and PtB cathode at bottom side. (B) Top view images of catalyst particles for IrRuOx. (C) Top view images of catalyst particles for PtB. (D) STEM image of IrRuOx at anode with complementary EDS spectrum images for (E) IrRu and (F) F. (G) STEM image of PtB at cathode with complementary EDS spectrum images for (H) Pt and (I) F. | ||
From the above phenomena and conclusions, some assumptions are made to establish a model that could investigate the performance of PEMEC using titanium thin/well-tunable LGDLs: (a) there are n reaction sites on each length l on the rim of the pore; (b) δRm is the local resistance of the reaction site m, all reaction sites are in parallel with each other (the difference of resistance between each sites probably due to the non-uniformly distributed anode catalysts which can be seen in Fig. 8); (c) At is the active reaction area of the PEMEC which is 5 cm2 in this study; (d) D and ε are the pore diameter and porosity of the thin LGDL, respectively; (e) I is the current of the PEMEC. So the total reaction sites N can be calculated by eqn (2).
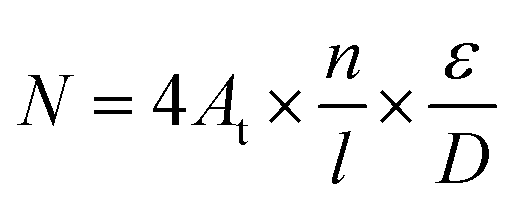 | (2) |
| Vm = Im × δRm (m = 1, 2, 3,…,N) | (3) |
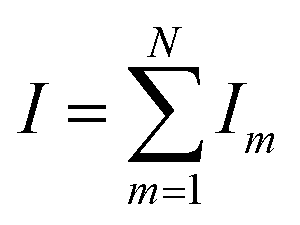 | (4) |
So, it can be concluded that as the total reaction sites increases, the cell voltage of the PEMEC will decrease. It can also be found that with larger pore size, a large amount of catalyst sites located away from the rim will not behave normally due to the large in-plane resistance, which will result in worse performance and catalyst underutilization. By increasing the porosity or decreasing the pore size, the number of reaction sites can be increased and more catalysts are active. Meanwhile, the total ohmic losses can be decreased due to more parallel resistances which exist in the equivalent circuit, which will lead to better performance. On the other hand, the increase of the total reaction sites N means more active catalyst, which will impact the kinetics and decrease the activation loss. In the Butler–Volmer model of kinetics, the activation potential is related to many factors, such as reaction mechanism, catalyst morphology, operating parameters, species concentrations and so on.55 The larger porosity LGDLs with more reaction sites will lead to a larger roughness factor which causes higher effective exchange current density and result in a smaller activation overpotential. It can be concluded that the smaller ohmic loss and activation loss are the two main reasons why larger porosity thin LGDLs can achieve better performance.
4 Conclusion
In this study, a set of thin and planar titanium LGDLs with well-tunable pore morphologies are developed to promote PEMEC performance, and to precisely investigate the impacts of the LGDL pore size and porosity. The thin LGDLs have exhibited exceptional performance. At a current density of 2.0 A cm−2 with a porosity of 0.7 and a pore size of 400 μm, the required voltage reaches 1.661 V, the lowest value that has been publicly reported so far. The PEMEC has a better performance with a larger porosity under a fixed pore size. It can also be found that the PEMEC performance decreases gradually with the increase of pore size from 100 to 800 μm, but pore-size impacts are not as significant as porosity. Additionally, operating temperatures have a large impact on the PEMEC performance. The PEMEC performance is significantly improved when the temperature increased from 20 °C to 80 °C. For better understanding the performance mechanisms, EIS evaluations are conducted and comprehensive equivalent electrical circuits, including CPE and Warburg diffusion element, are established to quantify the ohmic, activation and transport losses, respectively. The thin/well-tunable titanium-based LGDLs remarkably reduce mass transport, ohmic and activation losses. By taking advantage of the straight-through pores of the novel LGDLs and transparent PEMECs, the direct visualizations of the electrochemical reactions were captured with a high-speed and micro-scale visualization system to interpret the effects of pore size and porosity on the PEMEC performance. The observation reveals that the oxygen bubble only generated and nucleated at the rim of the pores and the PEMEC performance is closely related to the number of the reaction sites. Larger porosity and smaller pore size will increase the reaction sites and enhance the PEMEC performance. Furthermore, due to the thin feature of the novel LGDL, not only the thickness/volume/weight of the PEMEC stack can be reduced greatly, but also the materials used for LGDLs can be decreased which helps to reduce the cost. The precisely controlled pore parameters are extremely valuable to advance numerical modeling of electrochemical reactions and associated multiphase flow as well. Because all titanium thin LGDLs in this study have a better performance than the conventional ones (like titanium felt), they are expected to have many potential applications in energy and environmental engineering. More work will be performed to investigate the other parameters that may have affect the PEMEC performance.Nomenclature
| BP | Bipolar plate |
| CCM | Catalyst-coated membrane |
| CD | Current distributor |
| CL | Catalyst layer |
| CPE | Constant phase element |
| DI | De-ionized |
| EDS | Energy dispersive X-ray spectroscopy |
| EEC | Equivalent electrical circuit |
| EIS | Electrochemical impedance spectroscopy |
| F | Fluorine |
| HAADF | High angle annular dark field |
| Ir | Iridium |
| LGDL | Liquid/gas diffusion layer |
| MG | Modular galvanodynamic |
| ML | Microporous layers |
| MPD | Mean pore diameter |
| MPL | Micro porous layer |
| PEM | Proton exchange membrane |
| PEMEC | Proton exchange membrane electrolyzer cell |
| PEMFC | Proton exchange membrane fuel cell |
| Pt | Platinum |
| Ru | Ruthenium |
| SE | Secondary electron |
| SEM | Scanning electron microscopy |
| SOEC | Solid oxide electrolyzer cell |
| STEM | Scanning transmission electron microscopy |
| Ti | Titanium |
| TPB | Triple phase boundary |
Acknowledgements
The authors greatly appreciate the support from U.S. Department of Energy's National Energy Technology Laboratory under Award DE-FE0011585. This research was partially conducted at the Center for Nanophase Materials Sciences, which is a DOE Office of Science User Facility. The authors also wish to express their appreciation to Dr Bo Han, Dr Lee Leonard, Dr Jacqueline Anne Johnson, William Barnhill, Stuart Steen, Alexander Terekhov, Douglas Warnberg, Kate Lansford, and Andrew Mays for their help.References
- J. W. Ager, M. R. Shaner, K. A. Walczak, I. D. Sharp and S. Ardo, Energy Environ. Sci., 2015, 8, 2811–2824 CAS.
- W. T. Hong, M. Risch, K. A. Stoerzinger, A. Grimaud, J. Suntivich and Y. Shao-Horn, Energy Environ. Sci., 2015, 8, 1404–1427 CAS.
- J. A. Turner, Science, 1999, 285, 687–689 CrossRef CAS PubMed.
- W. Isherwood, J. R. Smith, S. M. Aceves, G. Berry, W. Clark, R. Johnson, D. Das, D. Goering and R. Seifert, Energy, 2000, 25, 1005–1020 CrossRef CAS.
- X. Chen, C. Li, M. Grätzel, R. Kostecki and S. S. Mao, Chem. Soc. Rev., 2012, 41, 7909–7937 RSC.
- C. A. Rodriguez, M. A. Modestino, D. Psaltis and C. Moser, Energy Environ. Sci., 2014, 7, 3828–3835 CAS.
- Y. Hou, M. R. Lohe, J. Zhang, S. Liu, X. Zhuang and X. Feng, Energy Environ. Sci., 2016, 9, 478–483 CAS.
- X. Lai, J. E. Halpert and D. Wang, Energy Environ. Sci., 2012, 5, 5604–5618 CAS.
- H. Tang, C. M. Hessel, J. Wang, N. Yang, R. Yu, H. Zhao and D. Wang, Chem. Soc. Rev., 2014, 43, 4281–4299 RSC.
- M. Debe, S. Hendricks, G. Vernstrom, M. Meyers, M. Brostrom, M. Stephens, Q. Chan, J. Willey, M. Hamden and C. K. Mittelsteadt, J. Electrochem. Soc., 2012, 159, K165–K176 CrossRef CAS.
- A. Javadekar, A. Jayakumar, R. J. Gorte, J. M. Vohs and D. Buttrey, J. Electrochem. Soc., 2012, 159, A386–A389 CrossRef CAS.
- D. Shapiro, J. Duffy, M. Kimble and M. Pien, Sol. Energy, 2005, 79, 544–550 CrossRef CAS.
- W. Smith, J. Power Sources, 2000, 86, 74–83 CrossRef CAS.
- H.-Y. Jung, S.-Y. Huang and B. N. Popov, J. Power Sources, 2010, 195, 1950–1956 CrossRef CAS.
- F. Barbir, Sol. Energy, 2005, 78, 661–669 CrossRef CAS.
- D.-H. Ha, B. Han, M. Risch, L. Giordano, K. P. Yao, P. Karayaylali and Y. Shao-Horn, Nano Energy, 2016 DOI:10.1016/j.nanoen.2016.04.034.
- J. Baxter, Z. Bian, G. Chen, D. Danielson, M. S. Dresselhaus, A. G. Fedorov, T. S. Fisher, C. W. Jones, E. Maginn and U. Kortshagen, Energy Environ. Sci., 2009, 2, 559–588 CAS.
- M. B. Gorensek and C. W. Forsberg, Int. J. Hydrogen Energy, 2009, 34, 4237–4242 CrossRef CAS.
- M. Uzunoglu, O. Onar and M. Alam, Renewable Energy, 2009, 34, 509–520 CrossRef CAS.
- C.-J. Liu, U. Burghaus, F. Besenbacher and Z. L. Wang, ACS Nano, 2010, 4, 5517–5526 CrossRef CAS PubMed.
- H. Oh, J. Park, K. Min, E. Lee and J.-Y. Jyoung, Appl. Energy, 2015, 149, 186–193 CrossRef CAS.
- J. Qi, X. Lai, J. Wang, H. Tang, H. Ren, Y. Yang, Q. Jin, L. Zhang, R. Yu and G. Ma, Chem. Soc. Rev., 2015, 44, 6749–6773 RSC.
- B. Han, J. Mo, Z. Kang and F.-Y. Zhang, Electrochim. Acta, 2016, 188, 317–326 CrossRef CAS.
- B. Han, J. Mo, Z. Kang and F.-Y. Zhang, AIAA 2015-3915, 2015.
- B. Han, S. M. Steen, J. Mo and F.-Y. Zhang, Int. J. Hydrogen Energy, 2015, 40, 7006–7016 CrossRef CAS.
- J. Fall, D. Humphreys and S. Guo, J. Fuel Cell Sci. Technol., 2009, 6, 031003 CrossRef.
- G. Chen, C. C. Waraksa, H. Cho, D. D. Macdonald and T. E. Mallouka, J. Electrochem. Soc., 2003, 150, E423–E428 CrossRef CAS.
- J. O. M. Bockris, Materials, 2011, 4, 2073–2091 CrossRef.
- A. Nikiforov, I. Petrushina, E. Christensen, A. Tomás-García and N. Bjerrum, Int. J. Hydrogen Energy, 2011, 36, 111–119 CrossRef CAS.
- R. E. Fuentes, J. Farell and J. W. Weidner, Electrochem. Solid-State Lett., 2011, 14, E5–E7 CrossRef CAS.
- S. S. Dihrab, K. Sopian, M. Alghoul and M. Y. Sulaiman, Renewable Sustainable Energy Rev., 2009, 13, 1663–1668 CrossRef CAS.
- J. Mo, S. M. Steen, F.-Y. Zhang, T. J. Toops, M. P. Brady and J. B. Green, Int. J. Hydrogen Energy, 2015, 40, 12506–12511 CrossRef CAS.
- F.-Y. Zhang, S. G. Advani and A. K. Prasad, J. Power Sources, 2008, 176, 293–298 CrossRef CAS.
- T. Matsuura, M. Kato and M. Hori, J. Power Sources, 2006, 161, 74–78 CrossRef CAS.
- S. Arisetty, A. K. Prasad and S. G. Advani, J. Power Sources, 2007, 165, 49–57 CrossRef CAS.
- H. Tawfik, Y. Hung and D. Mahajan, J. Power Sources, 2007, 163, 755–767 CrossRef CAS.
- H. Wang and J. Turner, Fuel Cells, 2010, 10, 510–519 CrossRef CAS.
- S. Grigoriev, P. Millet, S. Volobuev and V. Fateev, Int. J. Hydrogen Energy, 2009, 34, 4968–4973 CrossRef CAS.
- P. Millet, D. Dragoe, S. Grigoriev, V. Fateev and C. Etievant, Int. J. Hydrogen Energy, 2009, 34, 4974–4982 CrossRef CAS.
- L. Ma, S. Sui and Y. Zhai, Int. J. Hydrogen Energy, 2009, 34, 678–684 CrossRef CAS.
- C. M. Hwang, M. Ishida, H. Ito, T. Maeda, A. Nakano, Y. Hasegawa, N. Yokoi, A. Kato and T. Yoshida, Int. J. Hydrogen Energy, 2011, 36, 1740–1753 CrossRef CAS.
- C. M. Hwang, M. Ishida, H. Ito, T. Maeda, A. Nakano, A. Kato and T. Yoshida, J. Power Sources, 2012, 202, 108–113 CrossRef CAS.
- H. Ito, T. Maeda, A. Nakano, C. M. Hwang, M. Ishida, A. Kato and T. Yoshida, Int. J. Hydrogen Energy, 2012, 37, 7418–7428 CrossRef CAS.
- H. Ito, T. Maeda, A. Nakano, A. Kato and T. Yoshida, Electrochim. Acta, 2013, 100, 242–248 CrossRef CAS.
- J. Mo, S. M. Steen, B. Han, Z. Kang, A. Terekhov, F.-Y. Zhang, S. T. Retterer and D. A. Cullen, AIAA 2015-3914, 2015.
- J. Mo, S. M. Steen, S. Retterer, D. A. Cullen, A. Terekhov and F.-Y. Zhang, ECS Trans., 2015, 66, 3–10 CrossRef CAS.
- F.-Y. Zhang, A. K. Prasad and S. G. Advani, J. Micromech. Microeng., 2006, 16, N23 CrossRef CAS.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS.
- A. T. Marshall, S. Sunde, M. Tsypkin and R. Tunold, Int. J. Hydrogen Energy, 2007, 32, 2320–2324 CrossRef CAS.
- J. Xu, G. Liu, J. Li and X. Wang, Electrochim. Acta, 2012, 59, 105–112 CrossRef CAS.
- J. Xu, R. Miao, T. Zhao, J. Wu and X. Wang, Electrochem. Commun., 2011, 13, 437–439 CrossRef CAS.
- J. Mo, Z. Kang, G. Yang, S. T. Retterer, D. A. Cullen, T. J. Toops, J. B. Green and F.-Y. Zhang, Appl. Energy, 2016, 177, 817–822 CrossRef CAS.
- S. Sun, Y. Xiao, D. Liang, Z. Shao, H. Yu, M. Hou and B. Yi, RSC Adv., 2015, 5, 14506–14513 RSC.
- N. Dale, M. Mann, H. Salehfar, A. Dhirde and T. Han, J. Fuel Cell Sci. Technol., 2010, 7, 031010 CrossRef.
- M. M. Mench, Fuel cell engines, John Wiley & Sons, 2008 Search PubMed.
- H. Su, V. Linkov and B. J. Bladergroen, Int. J. Hydrogen Energy, 2013, 38, 9601–9608 CrossRef CAS.
- J. M. Spurgeon and N. S. Lewis, Energy Environ. Sci., 2011, 4, 2993–2998 CAS.
- S. Grigoriev, V. Porembsky and V. Fateev, Int. J. Hydrogen Energy, 2006, 31, 171–175 CrossRef CAS.
- J. Mainka, G. Maranzana, J. Dillet, S. Didierjean and O. Lottin, J. Electrochem. Soc., 2010, 157, B1561–B1568 CrossRef CAS.
- D. Malevich, E. Halliop, B. A. Peppley, J. G. Pharoah and K. Karan, J. Electrochem. Soc., 2009, 156, B216–B224 CrossRef CAS.
- A. Ter Heijne, O. Schaetzle, S. Gimenez, F. Fabregat-Santiago, J. Bisquert, D. P. Strik, F. Barriere, C. J. Buisman and H. V. Hamelers, Energy Environ. Sci., 2011, 4, 5035–5043 CAS.
- S.-J. Seo, J.-J. Woo, S.-H. Yun, H.-J. Lee, J.-S. Park, T. Xu, T.-H. Yang, J. Lee and S.-H. Moon, Phys. Chem. Chem. Phys., 2010, 12, 15291–15300 RSC.
- J.-B. Jorcin, M. E. Orazem, N. Pébère and B. Tribollet, Electrochim. Acta, 2006, 51, 1473–1479 CrossRef CAS.
- K. M. Jeong, C. K. Lee and H. J. Sohn, J. Electrochem. Soc., 1992, 139, 1927–1931 CrossRef CAS.
- J. Mo, Z. Kang, S. T. Retterer, D. A. Cullen, T. J. Toops, J. B. Green, M. M. Mench and F.-Y. Zhang, Sci. Adv., 2016 DOI:10.1126/sciadv.1600690.
- J. Peron, Z. Shi and S. Holdcroft, Energy Environ. Sci., 2011, 4, 1575–1591 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ee02368a |
| ‡ This manuscript has been authored by UT-Battelle, LLC under Contract No. DE-AC05-00OR22725 with the U.S. Department of Energy. The United States Government retains and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, world-wide license to publish or reproduce the published form of this manuscript, or allow others to do so, for United States Government purposes. The Department of Energy will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://energy.gov/downloads/doe-public-access-plan). |
| § Present affiliation: National Renewable Energy Laboratory, USA. |
| This journal is © The Royal Society of Chemistry 2017 |