
Biosurfactant functionalized single-walled carbon nanotubes to promote laccase bioelectrocatalysis†
Masato
Tominaga
*a,
Aiko
Sasaki
b,
Masayuki
Tsushida
c and
Makoto
Togami
b
aGraduate School of Science and Engineering, Saga University, Saga 840-8502, Japan. E-mail: masato@cc.saga-u.ac.jp
bGraduate School of Science and Technology, Kumamoto University, Kumamoto 860-8555, Japan
cFaculty of Engineering, Kumamoto University, Kumamoto 860-8555, Japan
First published on 17th November 2016
Abstract
Fast oxygen (O2) reduction at high positive potential is essential to obtain effective green energy conversion systems. Here, in an attempt to develop a desirable O2 reduction biocathode for fuel cells using laccase (Lac), we modify the surface of single-walled carbon nanotubes (SWCNTs) with various biosurfactants to obtain fast direct electron transfer from the SWCNTs to Lac, resulting in O2 reduction starting from a potential close to the redox equilibrium potential of the oxygen/water couple. We found that pyranoside- and sugar-type surfactants behaved as effective modifier layers of SWCNTs to facilitate Lac bioelectrocatalysis. In particular, SWCNTs modified with the pyranoside-type surfactant n-octyl-β-D-glucopyranoside and the sugar-type surfactant n-decanoyl-N-methyl-D-glucamine exhibited electron transfer rates between the type-1 Cu site of Lac and the modified electrodes of 4000 and 2500 s−1, respectively. The number of modifier layers adsorbed onto SWCNTs strongly influenced its effect on Lac bioelectrocatalysis.
Introduction
Oxygen (O2) reduction catalysts that promote faster reaction at lower energy are required for both industrial and biological systems. Cytochrome c oxidase is an important O2 reduction catalyst in the respiratory system of biological systems, producing an electrochemical potential difference induced by the proton concentration difference between the inner and outer membranes of mitochondria with high energy conversion efficiency.1 From the viewpoint of industrial systems, the development of low-cost catalysts with high activity for O2 reduction is important for fuel cell technology.2 Multicopper oxidases such as laccase (Lac) and bilirubin oxidase have been widely studied as biocatalysts in fuel cells for O2 reduction.3–17 In nature, multicopper oxidases catalytically oxidize many types of organic compounds using O2 as an electron acceptor. Multicopper oxidases act as an efficient O2 reduction biocatalyst for cathodes in fuel cells when the type-1 (T1) Cu site of a multicopper oxidase can accept electrons from the electrode.18,19 Such enzymes have numerous advantages. First, these enzymes are able to fully reduce O2 to water by four-electron reduction without releasing reactive species such as H2O2 and superoxide radicals as intermediates.4–12,19 Second, these enzymes are able to reduce O2 with a very high turnover frequency of >103 s−1 compared with that of platinum (Pt)-based catalysts (approximately 15 s−1 per Pt atom).7 Third, the O2 reduction potential of multicopper oxidase is at a high potential close to the equilibrium redox potential of the O2/H2O couple.2,14,20,21 The O2 reduction mechanism can be described as follows. The T1 Cu site is reduced by electron transfer from a reduced O2 molecule. Intramolecular electron transfer then occurs between the T1 Cu site and the T3 Cu site over a distance of ca. 1.3 nm,10 and O2 reduction occurs at the T2/T3 Cu site, where type-2/3 Cu (T2/T3 Cu) from two sites forms a trinuclear copper cluster.Direct-type electron transfer systems have mainly been investigated for biocathode electrocatalysts. Such systems do not use small compounds as electron shuttles between the T1 site and the electrode. Much effort has been devoted to the design of electrode surfaces.3,5–12,14,17,19–41 Direct-type electron transfer systems require a short distance between the T1 site of the immobilized enzyme and the electrode surface, which can be achieved by optimizing the enzyme molecular orientation so that the T1 Cu site faces the electrode surface. Thus, such a surface design has been achieved. For example, in studies on the binding affinity of Lac, bioelectrocatalytic currents from direct electron transfer systems have been observed at gold (Au) electrodes modified with aromatic compounds such as 4-aminothiophenol-, 2-aminophenol- and neocuproine-modified graphite electrodes, and 1-aminophyrene- and naphthalene-functionalized multi-walled carbon nanotubes.25,27,29,32 In particular, because of the affinity of the hydrophobic substrate binding pocket of the T1 Cu site of Lac, graphite electrodes modified with anthracene or anthraquinone displayed high current density and bioelectrocatalytic activity with long-term stability.26,28,35 Furthermore, a very fast heterogeneous electron transfer rate (>400 s−1) was obtained at functionalized Au nanoparticles with a diameter of ca. 5 nm, and the nanoparticle size affected the electron transfer rate.23,24,30
Recently, we reported that an interface between Lac and single-walled carbon nanotubes (SWCNTs) displaying very fast electron transfer was achieved by modification with the steroid biosurfactant sodium cholate (SC).36 Furthermore, the onset potential for the reduction reaction of O2 was very close to the equilibrium redox potential of the O2/H2O couple. Here we explore how the modification of the SWCNT electrode can influence the electron transfer of Lac with high O2 reduction potential. We focus on biosurfactant modifiers, such as pyranoside-, sugar-, and sugar-steroid-type surfactants, of SWCNT electrodes.
Experimental
Fungal Lac from Trametes sp. (Enzyme Commission Number EC 1.10.3.2) was obtained from Amano Enzyme (Nagoya, Japan) and purified according to a previous report.33 The purity of Lac was confirmed by SDS-PAGE, which gave a single band at approximately 63 kDa. The Lac concentration was determined from its molar absorption coefficient of 5700 (mol dm−3)−1 cm−1 at 614 nm.34 The pyranoside-type modifiers n-octyl-β-D-glucopyranoside (OGP) and n-decyl-β-D-maltopyranoside (DMP) were used, and n-decanoyl-N-methyl-D-glucamine (MEGA10) was used as a sugar-type modifier. Sugar-steroid-type modifiers N,N-bis(3-D-gluconamidopropyl)cholamide (BIGCHAP) and N,N-bis(3-D-gluconamidopropyl)deoxycholamide (deoxyBIGCHAP) were also investigated. In addition, 3-[(3-cholamidopropyl)dimethylammonio]propanesulfonate (CHAPS) and 3-[(3-cholamidopropyl)dimethylammonio]-2-hydroxypropanesulfonate (CHAPSO) were used as zwitterion-steroid-type modifiers. All modifiers were obtained from Dojindo Laboratories (Kumamoto, Japan), and their structures are shown in Fig. 1. The pyranoside-type modifiers OGP and DMP and the sugar-type modifier MEGA10 have alkyl chains. BIGCHAP and deoxyBIGCHAPS have two large glucamine moieties that bind to the steroid nucleus. Under the present experimental conditions, both CHAPS and CHAPSO have a negatively charged sulfonic acid and a positively charged ammonium ion bound to the steroid nucleus.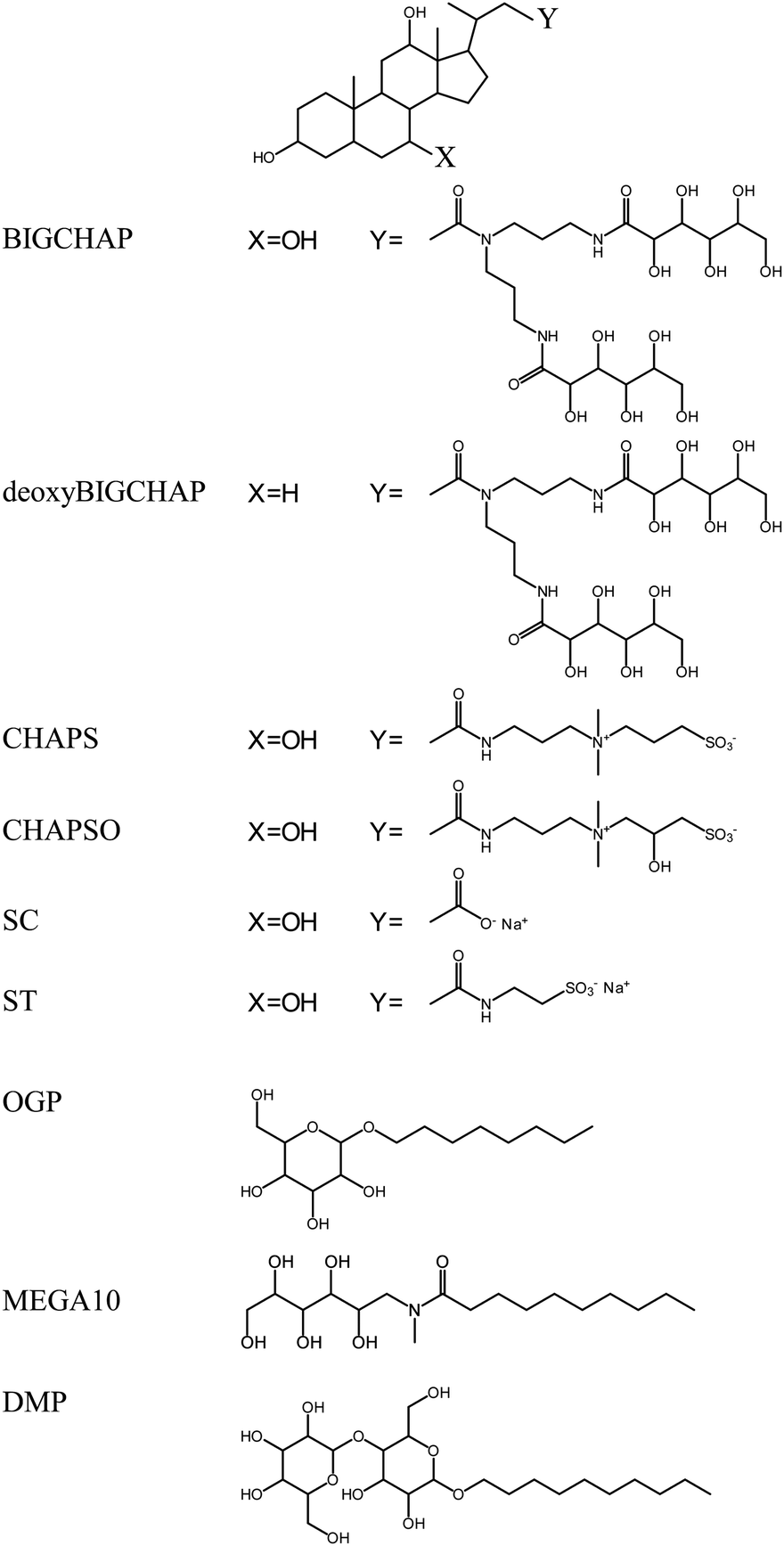 | ||
| Fig. 1 Chemical structures of the pyranoside-, sugar-, sugar-steroid- and zwitterion-steroid-type modifiers. | ||
SWCNTs were synthesized on the surface of an Au wire using chemical vapor deposition as described in our previous reports.36,37,42,43 The Au wire surface was completely covered with SWCNTs. Field-emission scanning electron microscopy (FE-SEM) measurements indicated that the thickness of the SWCNT layer was ca. 20 μm. FE-SEM and transmission electron microscopy (TEM) observations showed that pieces of SWCNTs were bundled with each other, resulting in tubes with diameters of 5–50 nm. In the TEM images of the surface edge of the SWCNT layer, individual tube structures with an estimated wall thickness of 0.30–0.35 nm were observed, indicating that the structures were SWCNTs. Raman spectroscopic measurements revealed that the ratio of the intensities of the G-band to the D-band (IG/ID) was ca. 20 using a 514.5 nm (2.41 eV) excitation laser. This high IG/ID value indicated that the synthesized SWCNTs were highly crystalline.44 The radial breathing mode of the SWCNTs in Raman spectra indicated that the diameter distribution of the SWCNTs was 0.9–1.6 nm.45–47 Immediately after synthesis, the fresh SWCNT electrodes were immersed in 0.2% (w/v) modifier aqueous solutions for 24 h. After gentle rinsing, the modified SWCNT electrodes were immersed in 0.1 mol dm−3 acetate buffer solution (pH 5) containing 10 μmol dm−3 Lac for 30 min. The electrode surface was then rinsed gently with the buffer solution before performing electrochemical measurements. The modified Lac-immobilized SWCNTs are denoted as modifier-SWCNTs.
Voltammetric measurements were performed at 25 °C using a conventional three-electrode cell with Ag|AgCl|saturated KCl as the reference electrode and a Pt plate as the counter electrode. All potentials are reported with respect to Ag|AgCl|saturated KCl (+199 mV vs. NHE). A 1.0 mol dm−3 phosphate–citrate buffer solution (pH 3.0) was used as the electrolyte, and was purged with high-purity argon before the measurements. The buffer solution was stirred with a magnetic stirrer during measurements to obtain a steady-state sigmoidal shape (with plateau current) in the voltammograms of Lac adsorbed on the electrodes. Rotating disk electrodes were not used in this study because the SWCNT layers tended to peel off the Au surface. The cell temperature was controlled at 25 °C by a thermostated incubator.
Results and discussion
BIGCHAP, deoxyBIGCHAP, CHAPS and CHAPSO, all of which have a steroid nucleus, did not show good results as modifiers for Lac-immobilized SWCNTs to obtain bioelectrocatalytic O2 reduction activity because of the direct electron transfer reaction of immobilized Lac with SWCNT electrodes (Fig. 2). The steady-state catalytic currents at these modifier-SWCNTs electrodes were smaller than that of an unmodified SWCNT electrode. In particular, BIGCHAP and deoxyBIGCHAP inhibited the electron transfer of Lac with SWCNTs because their catalytic currents were much smaller than that of the unmodified SWCNT electrode. We previously reported that at SC- and sodium taurocholate (ST)-modified SWCNT electrodes, large catalytic current was obtained compared with that of unmodified SWCNTs.36 Both SC and ST have a steroid nucleus like BIGCHAP, deoxyBIGCHAP, CHAPS and CHAPSO, differing in their Y side chain that binds to the steroid nucleus (Fig. 1). These results clearly indicate that the Y side chain plays an important role in direct electron transfer between Lac and SWCNTs. The large size of the side chain would not be suitable for a modifier to promote Lac bioelectrocatalysis.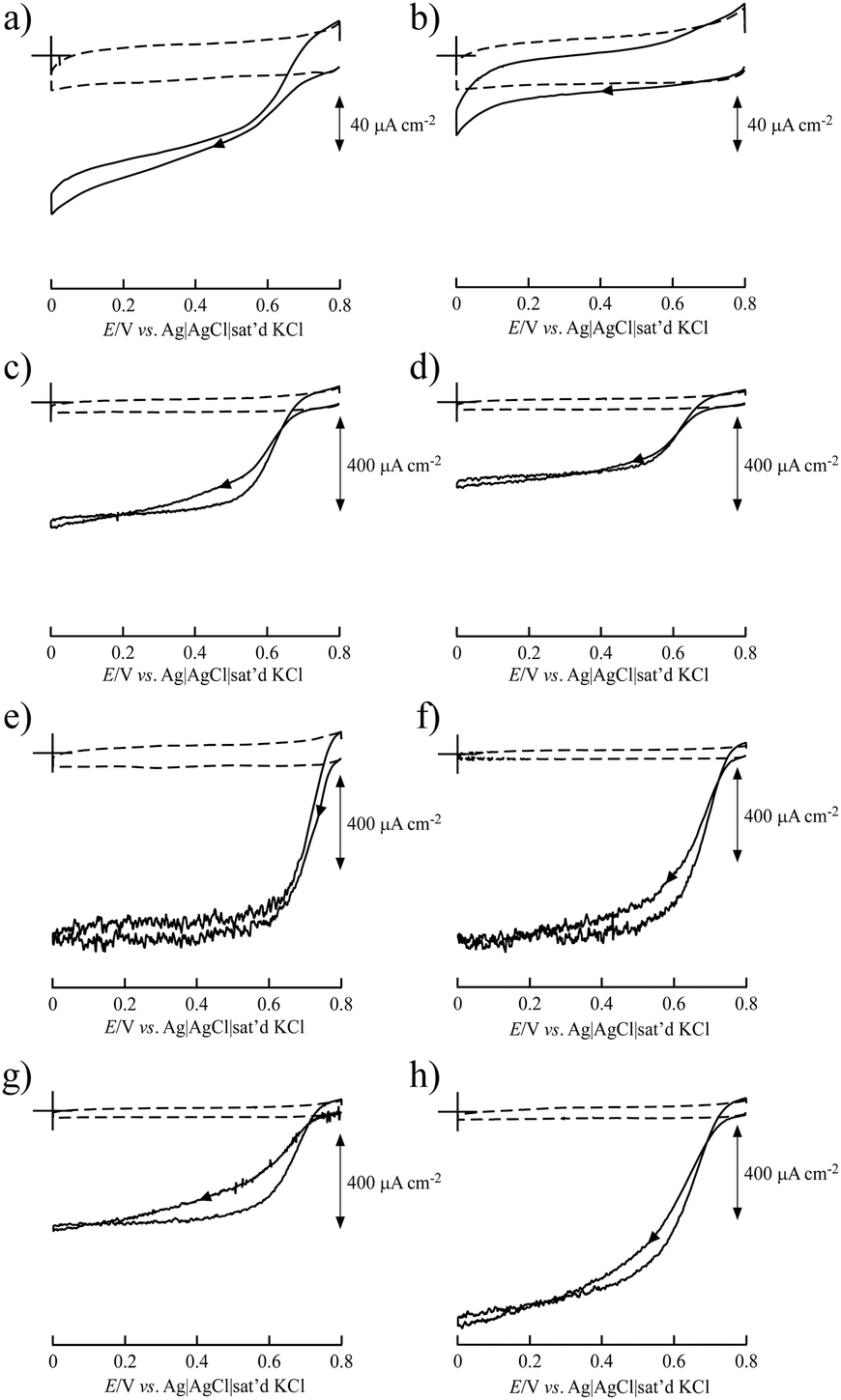 | ||
| Fig. 2 Cyclic voltammograms of the bioelectrocatalytic current of O2 reduction by Lac immobilized on (a) BIGCHAP–SWCNTs, (b) deoxyBIGCHAP–SWCNTs, (c) CHAPS–SWCNTs, (d) CHAPSO–SWCNTs, (e) OGP–SWCNTs, (f) MEGA10–SWCNTs, (g) DMP–SWCNTs and (h) unmodified SWCNTs in the presence of O2 (solid lines) and Ar gas (dashed lines). The potential sweep rate was 10 mV s−1. | ||
The OGP–SWCNT and MEGA10–SWCNT electrodes showed O2 reduction in the presence of Lac from a higher positive potential than that of an unmodified SWCNT electrode. In particular, the onset potential of the OGP–SWCNT electrode was 0.794 V, which was almost the same as that obtained using the SC–SWCNT electrode.36 The onset potential of the MEGA10–SWCNT electrode was slightly lower (0.777 V). An interesting result was obtained for the DMP–SWCNT electrode, which displayed an onset potential of 0.741 V, ca. 50 mV lower than that of the OGP–SWCNT electrode, even though its catalytic current was almost half that of the OGP–SWCNT electrode. OGP has a glucopyranoside group, and DMP has a maltopyranoside group. Maltopyranoside is a dimer of glucopyranoside groups. The difference in Lac bioelectrocatalysis using OGP–SWCNT and DMP–SWCNT electrodes would be caused by the amount of modifiers adsorbed on the SWCNTs. The surface coverage, which is the number of monolayers of a modifier when it is adsorbed homogeneously on a SWCNT surface, was evaluated to be 4.4 (±2.2) for OGP and 30 (±30) for DMP. The estimated values have a large associated error, but still clearly indicate that a larger amount of DMP than OGP was adsorbed onto the SWCNTs. Such a large amount of DMP on the SWCNT surface could inhibit electron transfer between Lac and SWCNTs. The reason for a larger amount of DMP is unclear. An aggregated structure of DMP on the SWCNT surface may be suggested.
The evaluated surface coverages of BIGCHAP, deoxyBIGCHAP, CHAPS and CHAPSO were 2.2 (±0.4), 7.0 (±3.0), 2.4 (±0.9) and 1.8 (±0.5), respectively. These values are similar to that of SC (2.9 (±0.9)), indicating that the lower bioelectrocatalysis at BIGCHAP–SWCNT, deoxyBIGCHAP–SWCNT, CHAPS–SWCNT and CHAPSO–SWCNT electrodes was not caused by the large amount of modifier on the SWCNTs. The surface concentration of Lac (Γ) revealed the important role of the Y side chain of these biosurfactants with a steroid nucleus in bioelectrocatalysis. The estimated Γ values of BIGCHAP–SWCNTs, deoxyBIGCHAP–SWCNTs, CHAPS–SWCNTs and CHAPSO–SWCNTs were 5.0 (±2.6), 4.2 (±2.3), 10.5 (±1.8) and 11.0 (±2.8) pmol cm−2, respectively. These Γ values are almost the same as that of SC–SWCNTs (7.3 (±1.3) pmol cm−2). The Γ value when Lac adsorbs onto the SWCNT surface in a monolayer is expected to be ca. 5 pmol cm−2 according to the crystallographic structure of a fungal Lac from Trametes versicolor with dimensions of 6.5 × 5.5 × 4.5 nm.13 Therefore, the obtained Γ values for the modified SWCNTs indicate that there are one or two layers of the adsorbed Lac. Considering these results, the poor bioelectrocatalysis at BIGCHAP–SWCNT, deoxyBIGCHAP–SWCNT, CHAPS–SWCNT and CHAPSO–SWCNT electrodes would be due to two possible reasons as follows. First, at these modified SWCNT electrodes, the amount of electrochemically active Lac (Γa) that can participate in direct electron transfer with SWCNTs may be lower than that at the SC–SWCNT electrode, even though their Γ values are higher than those of SC–SWCNTs. Second, the heterogeneous electron transfer reaction rate (k°) of these modified SWCNTs would be lower than that of SC–SWCNTs. To further investigate these two possibilities, simulations were carried out.
Fig. 3 shows the simulated curve of the steady-state current density (js) for the adsorption model developed by the Kano–Ikeda group.48–50 The obtained onset potential for O2 reduction was similar to the previously reported value.14,17–38 This result suggests that the T1 Cu site is the primary electron acceptor from the electrode and shuttles electrons to the T2/T3 Cu site, where O2 is fully reduced to water (Fig. 4). The simulated analysis based on the above electron transfer mechanism was carried out. The simulated curve fitted well with the experimental bioelectrocatalytic current curves corrected for the background current when the individual values of the parameters determined (as shown in Table 1) were used. The determined k° values at BIGCHAP–SWCNTs, deoxyBIGCHAP–SWCNTs, CHAPS–SWCNTs and CHAPSO–SWCNTs were 1500, 300, 2000 and 2300 s−1, respectively, which are lower than that at SC–SWCNTs (4000 s−1). The Γa/Γ value is the ratio of the amount of electrochemically active Lac to the total amount of the adsorbed Lac at a modifier-SWCNT interface. The Γa/Γ values of BIGCHAP–SWCNTs, deoxyBIGCHAP–SWCNTs, CHAPS–SWCNTs and CHAPSO–SWCNTs were 0.07, 0.03, 0.20 and 0.13, respectively. These values are also smaller than that of SC–SWCNTs (0.52). Considering both the low k° and small Γa/Γ values of BIGCHAP–SWCNTs, deoxyBIGCHAP–SWCNTs, CHAPS–SWCNTs and CHAPSO–SWCNTs, the poorer bioelectrocatalysis at these modified SWCNTs compared with that at SC–SWCNTs could be caused by the slightly unsuitable molecular orientation of Lac for the electron transfer reaction. Taking into account the high bioelectrocatalysis at SC–SWCNTs and ST–SWCNTs in addition to the above results for BIGCHAP–SWCNTs, deoxyBIGCHAP–SWCNTs, CHAPS–SWCNTs and CHAPSO–SWCNTs, the Y side chain has an important role in directing Lac molecular orientation, which influences direct electron transfer of Lac with SWCNTs. The results indicate that zwitterionic and bulky Y side chains do not direct Lac to adopt a suitable molecular orientation for electron transfer with SWCNTs. We tried to investigate the biosurfactant adsorption structure on the SWCNT interface by using TEM and SEM. However, the results were not clear. This indicates that the adsorption structure may not be uniform, because had it been uniform, we could have observed a clear image. It is known that Lac can be intelligently oriented onto electrode architectures whereby the moieties with structural similarities to the natural substrates of Lac are incorporated on specially designed electrode architectures. The Y side chain might have such an architectural role in Lac molecular orientation onto a SWCNT electrode.25,27,29,32,39,40
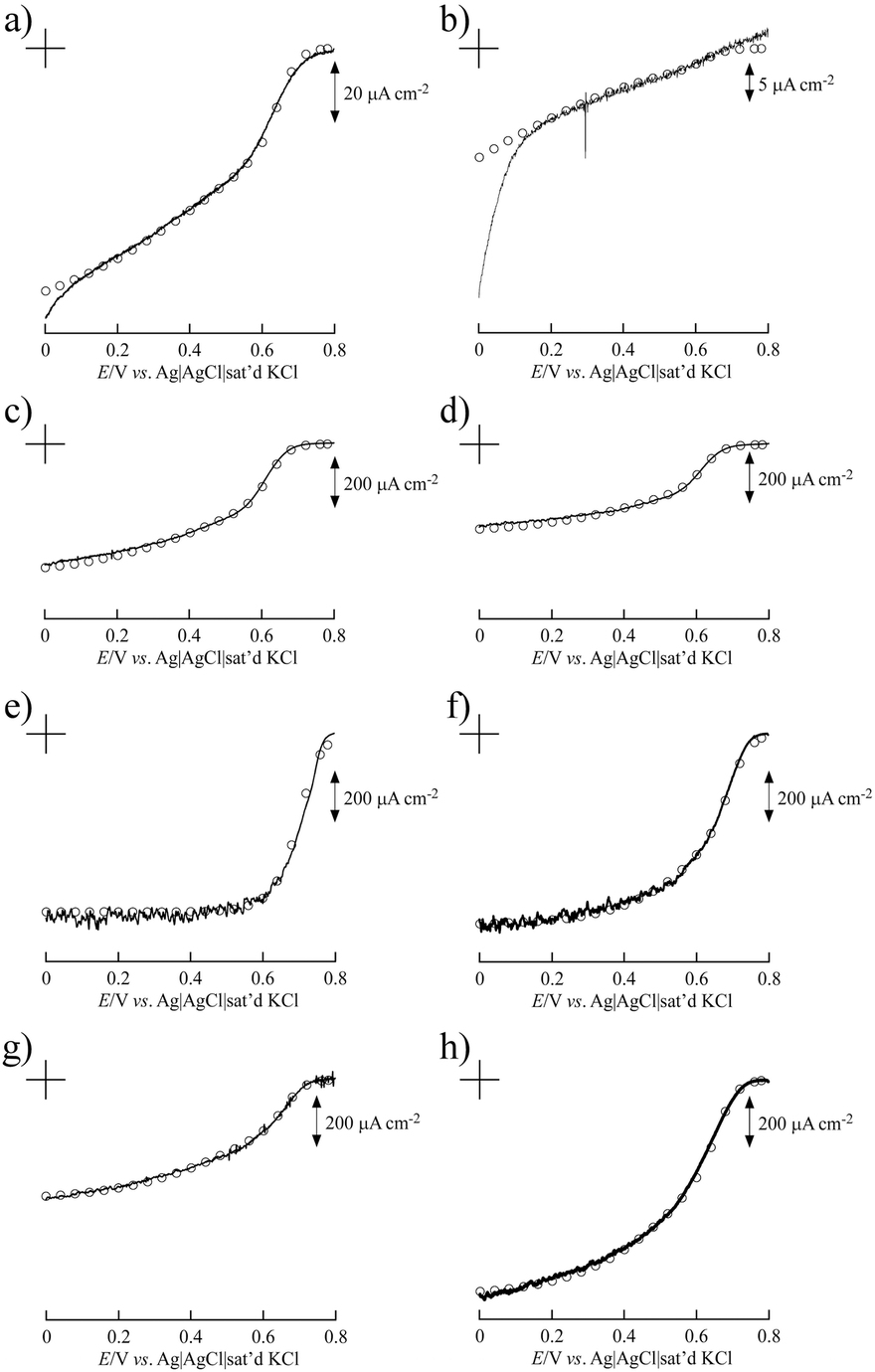 | ||
| Fig. 3 Background current-corrected steady-state voltammograms of the bioelectrocatalytic current of O2 reduction at Lac immobilized on (a) BIGCHAP–SWCNTs, (b) deoxyBIGCHAP–SWCNTs, (c) CHAPS–SWCNTs, (d) CHAPSO–SWCNTs, (e) OGP–SWCNTs, (f) MEGA10–SWCNTs, (g) DMP–SWCNTs and (h) unmodified SWCNTs. The open circles represent the simulated curves of steady-state current density using the parameters given in Table 1. | ||
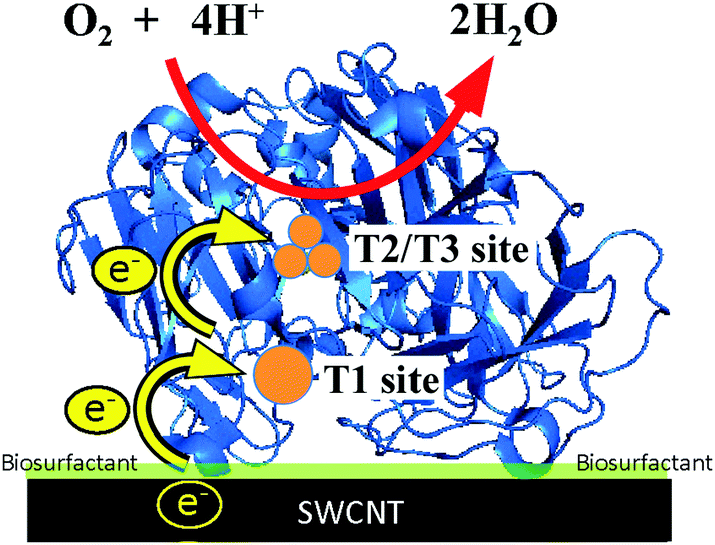 | ||
| Fig. 4 Schematic representation of the electron transfer pathway of a biosurfactant-modified SWCNT electrode. | ||
| Biosurfactant | Γ a/10−12 mol cm−2 | k°/s−1 | α | E°′/V vs. Ag|AgCl|sat'd KCl |
|---|---|---|---|---|
| BIGCHAP | 0.36 | 1500 | 0.10 | 0.625 |
| deoxyBIGCHAP | 0.11 | 300 | 0.10 | 0.575 |
| CHAPS | 2.1 | 2000 | 0.12 | 0.605 |
| CHAPSO | 1.4 | 2300 | 0.13 | 0.605 |
| OGP | 2.8 | 4000 | 0.50 | 0.715 |
| MEGA10 | 3.0 | 2500 | 0.20 | 0.685 |
| DMP | 1.9 | 1700 | 0.15 | 0.645 |
| None | 3.4 | 1900 | 0.17 | 0.645 |
The k° value at the OGP–SWCNT interface, 4000 s−1, was the highest of the samples, and the same as that at the SC–SWCNT interface although the Γa value of OGP–SWCNTs (2.8 pmol cm−2) was slightly smaller than that of SC–SWCNTs (4.2 pmol cm−2). In contrast, the k° value at DMP–SWCNTs (1700 s−1) was quite different from that of OGP–SWCNTs, indicating that the amount of modifier strongly affected k°. The k° value at MEGA10–SWCNTs (2500 s−1) indicated an improved electron transfer in this sample compared with that at an unmodified interface, but was slightly lower than that at SC–SWCNTs. These results reveal that pyranoside- and sugar-type modifiers such as OGP and MEGA10 have the ability to improve the electron transfer between Lac and SWCNTs. From the present investigation, at least the tendency of the modifier of pyranoside- and sugar-type surfactants to promote Lac bioelectrocatalysis would be clarified, although the effect of the modifier on the interface between Lac and SWCNTs is still unclear.
Conclusions
The results presented here show that the pyranoside- and sugar-type modifier layers on SWCNT electrodes can promote very fast heterogeneous direct electron transfer between the T1 Cu site of fungal Lac and SWCNTs. The many aggregated layers of the pyranoside-type modifier on SWCNTs, as observed for DMP–SWCNTs, inhibit this electron transfer reaction. Sugar-functionalized steroid modifiers such as BIGCHAP and deoxyBIGCHAP did not promote electron transfer between Lac and SWCNTs.Acknowledgements
This work was financially supported by the Grants-in-Aid for Scientific Research (24550159) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.Notes and references
- S. Yoshikawa and A. Shimada, Chem. Rev., 2015, 115, 1936 CrossRef CAS PubMed.
- I. Katsounaros, S. Cherevko, A. R. Zeradjanin and K. J. J. Mayrhofer, Angew. Chem., Int. Ed., 2014, 53, 102 CrossRef CAS PubMed.
- M. R. Tarasevich, A. I. Yaropolov, V. A. Bogdanovskaya and S. D. Varfolomeev, Bioelectrochem. Bioenerg., 1979, 6, 393 CrossRef CAS.
- E. I. Solomon, U. M. Sundaram and T. E. Machonkin, Chem. Rev., 1996, 96, 2563 CrossRef CAS PubMed.
- M. K. Jeon, C. H. Lee, G. I. Park and K. H. Kang, J. Power Sources, 2012, 216, 400 CrossRef CAS.
- S. C. Barton, J. Gallaway and P. Atanassov, Chem. Rev., 2004, 104, 4867 CrossRef CAS PubMed.
- J. A. Cracknell, K. A. Vincent and F. A. Armstrong, Chem. Rev., 2008, 108, 2439 CrossRef CAS PubMed.
- S. Shleev, J. Tkac, A. Christenson, T. Ruzgas, A. I. Yaropolov, J. W. Whittaker and L. Gorton, Biosens. Bioelectron., 2005, 20, 2517 CrossRef CAS PubMed.
- D. Leech, P. Kavanagh and W. Schuhmann, Electrochim. Acta, 2012, 84, 223 CrossRef CAS.
- D. L. Johnson, J. L. Thompson, S. M. Brinkmann, K. A. Schuller and L. L. Martin, Biochemistry, 2003, 42, 10229 CrossRef CAS PubMed.
- N. Mano and L. Edembe, Biosens. Bioelectron., 2013, 50, 478 CrossRef CAS PubMed.
- U. N. Dwivedi, P. Singh, V. P. Pandey and A. Kumar, J. Mol. Catal. B: Enzym., 2011, 68, 117 CrossRef CAS.
- K. Pionetek, M. Antorini and T. Choinowski, J. Biol. Chem., 2002, 277, 37663 CrossRef PubMed.
- S. Shleev, V. Andoralov, M. Falk, C. T. Reimann, T. Ruzgas, M. Srnec, U. Ryde and L. Rulíšek, Electroanalysis, 2012, 24, 1524 CrossRef CAS.
- A. Zebda, S. Cosnier, J.-P. Alcaraz, A. L. Goff, C. Gondran, F. Boucher, F. Giroud, K. Gorgy, H. Lamraoui and P. Cinquin, Sci. Rep., 2013, 3, 1 Search PubMed.
- A. Zebda, C. Gondran, A. L. Goff, M. Holzinger, P. Cinquin and S. Cosnier, Nat. Commun., 2011, 370, 1 Search PubMed.
- S. Shleev, G. Shumakovich, O. Morozova and A. Yaropolov, Fuel Cells, 2010, 10, 726 CrossRef CAS.
- O. V. Morozova, G. P. Shumakovich, M. A. Gorbacheva, S. V. Shleev and A. I. Yaropolov, Biochemistry, 2007, 72, 1136 CAS.
- L. Betancor, G. R. Johnson and H. R. Luckarift, ChemCatChem, 2013, 5, 46 CrossRef CAS.
- I. Matijošyte, I. W. C. E. Arends, R. A. Sheldon and S. Vries, Inorg. Chim. Acta, 2008, 361, 1202 CrossRef.
- M. Pita, D. Mate, D. Gonzalez-Perez, S. Shleev, V. M. Fernandez, M. Alcalde and A. L. D. Lacey, J. Am. Chem. Soc., 2014, 136, 5892 CrossRef CAS PubMed.
- S. Shleev, A. Jarosz-Wilkolazka, A. Khalunina, O. Morozova, A. Yaropolov, T. Ruzgas and L. Gorton, Bioelectrochemistry, 2005, 67, 115 CrossRef CAS PubMed.
- C. Gutiérrez-Sánchez, M. Pita, C. Vaz-Domínguez, S. Shleev and A. L. D. Lacey, J. Am. Chem. Soc., 2012, 134, 17212 CrossRef PubMed.
- D. Pankratov, R. Sundgerg, D. B. Suyatin, J. Sotres, A. Barrantes, T. Ruzgas, I. Maximov, L. Montelius and S. Shleev, RSC Adv., 2014, 4, 38164 RSC.
- M. Jönsson-Niedziolka, A. Kaminska and M. Opallo, Electrochim. Acta, 2010, 55, 8744 CrossRef.
- C. Blanford, R. S. Heath and F. A. Armstrong, Chem. Commun., 2007, 1710 RSC.
- R. P. Ramasamy, H. R. Luckarift, D. M. Ivnitski, P. B. Atanassov and G. R. Johnson, Chem. Commun., 2010, 46, 6045 RSC.
- S. Lörcher, P. Lopes, A. Kartashov and E. E. Ferapontova, ChemPhysChem, 2013, 14, 2112 CrossRef PubMed.
- M. Karaskiewicz, E. Nazaruk, K. Zelechowska, J. F. Biernat, J. Rogalski and R. Bilewicz, Electrochem. Commun., 2012, 22, 181 CrossRef.
- H. Qiu, C. Xu, X. Huang, Y. Ding, Y. Qu and P. Gao, J. Phys. Chem. C, 2009, 113, 2521 CAS.
- C. Vaz-Dominguez, S. Campuzano, O. Rüdiger, M. Pita, M. Gorbacheva, S. Shleev, V. M. Fernandez and A. L. D. Lacey, Biosens. Bioelectron., 2008, 24, 531 CrossRef CAS PubMed.
- H. L. Pang, J. Liu, D. Hu, X. H. Zang and J. H. Chen, Electrochim. Acta, 2010, 55, 6611 CrossRef CAS.
- Y. Kamitaka, S. Tsujimura, N. Setoyama, T. Kajino and K. Kano, Phys. Chem. Chem. Phys., 2007, 9, 1793 RSC.
- R. Santucci, T. Ferri, L. Morpurgo, I. Savini and L. Avigliano, Biochem. J., 1998, 332, 611 CrossRef CAS PubMed.
- N. Lalaoui, A. L. Goff, M. Holzinger, M. Mermoux and S. Cosnier, Chem. – Eur. J., 2015, 21, 3198 CrossRef CAS PubMed.
- M. Tominaga, A. Sasaki and M. Togami, Anal. Chem., 2015, 87, 5417 CrossRef CAS PubMed.
- M. Tominaga, M. Togami, M. Tsushida and D. Kawai, Anal. Chem., 2014, 86, 5053 CrossRef CAS PubMed.
- S. Shleev, A. Christenson, V. Serezhenkov, D. Burbaev, A. Yaropolov, L. Gorton and T. Ruzgas, Biochem. J., 2005, 385, 745 CrossRef CAS PubMed.
- M. T. Meredith, M. Minson, D. Hickey, K. Artyushkova, D. T. Glatzhofer and S. D. Minteer, ACS Catal., 2011, 1, 1683 CrossRef CAS.
- R. D. Milton, F. Giroud, A. E. Thumser, S. D. Minteer and R. C. T. Slade, Chem. Commun., 2014, 49, 94 RSC.
- A. Navaee and A. Salimi, J. Mater. Chem. A, 2015, 3, 7623 CAS.
- M. Tominaga, S. Sakamoto and H. Yamaguchi, J. Phys. Chem. C, 2012, 116, 9498 CAS.
- S. Sakamoto and M. Tominaga, Chem. – Asian J., 2013, 8, 2680 CrossRef CAS PubMed.
- F. Tuinstra and J. L. Koenig, J. Chem. Phys., 1970, 53, 1126 CrossRef CAS.
- R. Saito, G. Dresselhaus and M. S. Dresselhaus, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 13202 CrossRef.
- A. Jorio, R. Saito, J. H. Hafner, C. M. Lieber, M. Hunter, T. McClure, G. Dresselhaus and M. S. Dresselhaus, Phys. Rev. Lett., 2001, 86, 1118 CrossRef CAS PubMed.
- H. Kataura, Y. Kumazawa, Y. Maniwa, I. Umezu, S. Suzuki, Y. Ohtsuka and Y. Achiba, Synth. Met., 1999, 103, 2555 CrossRef CAS.
- S. Tsujimura, T. Nakagawa, K. Kano and T. Ikeda, Electrochemistry, 2004, 72, 437 CAS.
- M. Tominaga, M. Ohtani and I. Taniguchi, Phys. Chem. Chem. Phys., 2008, 10, 6928 RSC.
- S. Tsujimura, K. Kano and T. Ikeda, J. Electroanal. Chem., 2005, 576, 113 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nj02287a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |