DOI:
10.1039/C6NJ02671K
(Paper)
New J. Chem., 2017,
41, 347-357
Identification and development of pyrazolo[4,3-c]pyridine carboxamides as Mycobacterium tuberculosis pantothenate synthetase inhibitors†
Received
(in Montpellier, France)
26th August 2016
, Accepted 9th November 2016
First published on 10th November 2016
Abstract
The purpose of this study was to prepare various 1-((1-(substituted)-1H-1,2,3-triazol-4-yl)methyl)-N,3-diphenyl-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxamides using click chemistry. The synthesized compounds were characterized using various analytical techniques like NMR spectroscopy, mass spectrometry, and elemental analysis and screened for in vitro antitubercular activity against Mycobacterium tuberculosis (MTB) H37Rv strain and two ‘wild’ strains Spec. 210 and Spec. 192 using a classical test-tube method of successive dilution in Youmans' modification of the Proskauer and Beck liquid medium and were evaluated for a MTB pantothenate synthetase (PS) inhibition study as well. The final analogues exhibited minimum inhibitory concentration ranging from 24.72 to >200 μM. Among the compounds, 1-((1-(4-methoxyphenyl)-1H-1,2,3-triazol-4-yl)methyl)-N,3-diphenyl-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxamide (7d) was found to be the most active compound with IC50 1.01 ± 0.32 μM against MTB PS; it inhibited MTB with MIC 24.72 μM and it was non-cytotoxic at 50 μM in the RAW 267 cell line. Further, 7d was docked into the crystal structure of MTB PS to investigate its binding interaction pattern.
Introduction
Tuberculosis (TB) is contagious and airborne. It has a major impact on society due to the emergence of multi-drug resistant strains of Mycobacterium tuberculosis (MTB). In addition, multidrug-resistant Mycobacterium tuberculosis (MDR-TB) and especially extensively drug resistant TB (XDR-TB) are now increasing in incidence in many areas around the world.1 Around 9.6 million people fell ill with TB in 2014, including 1.2 million cases among people living with HIV. Globally in 2014, an estimated 0.48 million developed MDR-TB and there were an estimated 0.19 million deaths from MDR-TB.1 Therefore, the development of new therapeutic drugs targeting MDR-TB and XDR-TB is important due to the lack of effective drug treatments.2
The treatment of TB infections is generally carried out through the use of a combination of drugs including isoniazid (INH), rifampicin (RMP), pyrazinamide, ethambutol (ETB) and streptomycin.3–6
In order to overcome the issue of resistance there is a challenging need to develop new drugs using novel targets of MTB.
The biosynthetic pathway of pantothenate involves four steps catalyzed by panB, panC, panD, and panE genes.7 The last step of pantothenate biosynthesis, viz., the ATP-dependent condensation of D-pantoate and β-alanine to form pantothenate, is catalyzed by PanC. Pantothenate is essential for several processes such as cell signaling, fatty acid metabolism, and synthesis of non-ribosomal peptides and polyketides.8 The biosynthesis of pantothenate is essential for the growth of MTB. The pantothenate biosynthesis pathway is a latent drug target and hence is important for determining the growth and virulence of MTB, and PS is a very good target for developing new therapeutics to treat TB.9,10
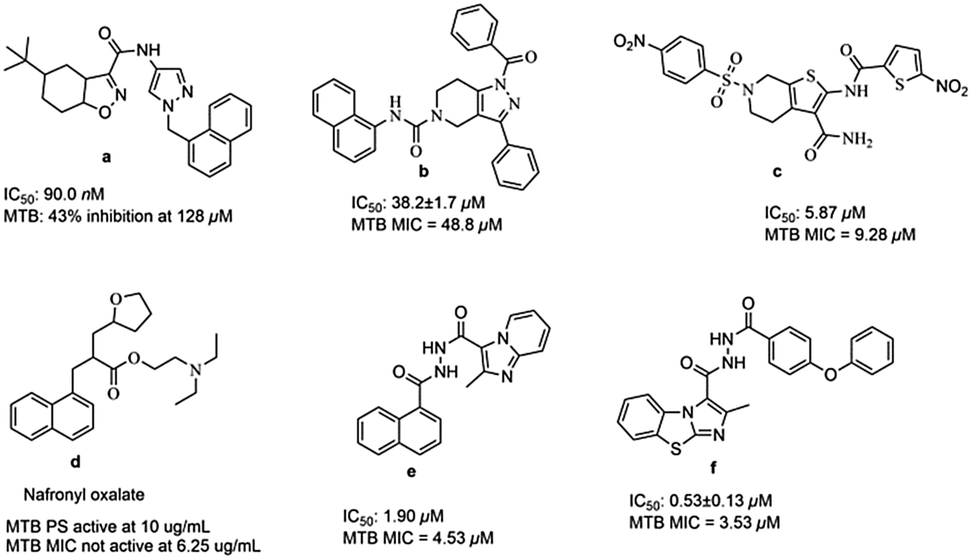
Till now many MTB PS inhibitors have been reported (Fig. 1), which include 5-tert-butyl-N-pyrazol-4-yl-4,5,6,7-tetrahydrobenzo[d]isoxazole-3-carboxamide derivatives (a),8 3-phenyl-4,5,6,7-tetrahydro-1H-pyrazolo[4,3-c]pyridine derivatives (b),11 2,6-disubstituted 4,5,6,7-tetrahydrothieno[2,3-c]pyridine-3-carboxamide derivatives (c),12 nafronyl oxalate (d),13N′-(1-naphthoyl)-2-methylimidazo[1,2-a]pyridine-3-carbohydrazide (e),14 and imidazo[2,1-b]thiazole and benzo[d]imidazo[2,1-b]thiazole derivatives (f).15
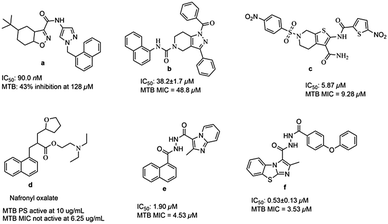 |
| | Fig. 1 Structures of literature reported MTB PS inhibitors. | |
Pyrazole derivatives are known to contain a broad spectrum of pharmacological properties such as antifungal,16 antidiabetic,17 antitumor,18 and antibacterial properties.19,20 Pyrazoles play an essential role in biologically active compounds and therefore signify an interesting template for medicinal chemistry.21 Mamolo et al. reported 5-aryl-1-isonicotinoyl-3-(pyridin-2-yl)-4,5-dihydro-1H-pyrazole derivatives which exhibited an interesting in vitro antimycobacterial activity against MTB, with minimum inhibitory concentration (MIC) values ranging from 8 to 16 μg mL−1.22 Ravindra et al. reported 3-(4-chlorophenyl)-4-substituted pyrazole derivatives with MIC values ranging from 0.35 to 3.15 μg mL−1 against MTB H37Rv.23 Chovatia et al. reported 1-acetyl-3,5-diphenyl-4,5-dihydro-(1H)-pyrazole derivatives which were screened against MTB H37Rv.24 A series of N-phenyl-3-(4-fluorophenyl)-4-substituted pyrazole derivatives were reported to exhibit significant antimycobacterial activity with IC50 values ranging from 0.47 to 118.0 μM against MTB H37Rv.25
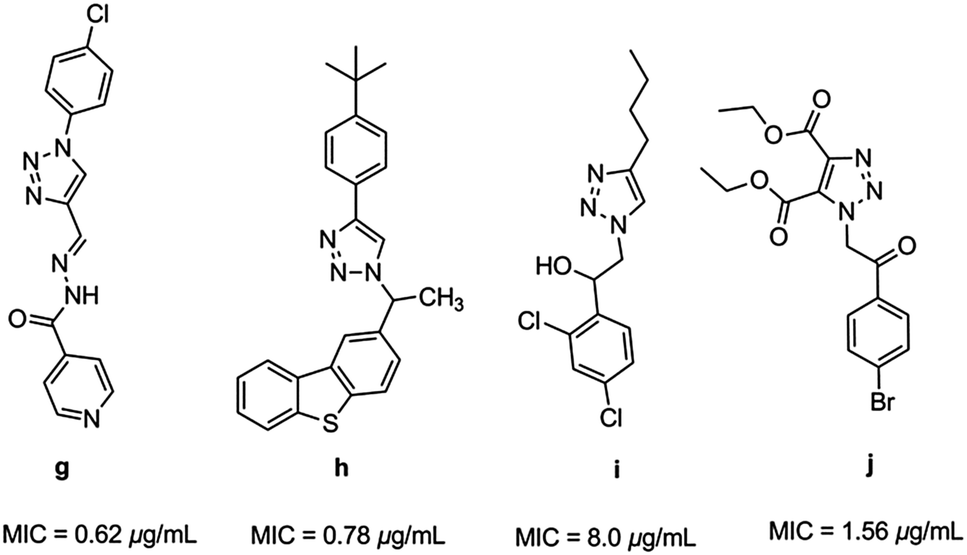
Triazoles have several medicinal applications.26 It is quite obvious that the favorable properties of the 1,2,3-triazole ring like moderate dipole character, hydrogen bonding capability, rigidity and stability under in vivo conditions are responsible for their enhanced biological activities.27,28 For instance, a variety of 1H-1,2,3-triazole compounds have been known to exhibit antitubercular activity (g–j) (Fig. 2).29–33
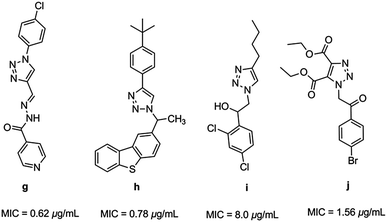 |
| | Fig. 2 Heterocyclic hybrids of [1,2,3] triazole with antitubercular activity. | |
In the present work, we have designed and developed novel MTB PS inhibitors by the molecular hybridization approach using the two known antimycobacterial agents.
Design and chemistry
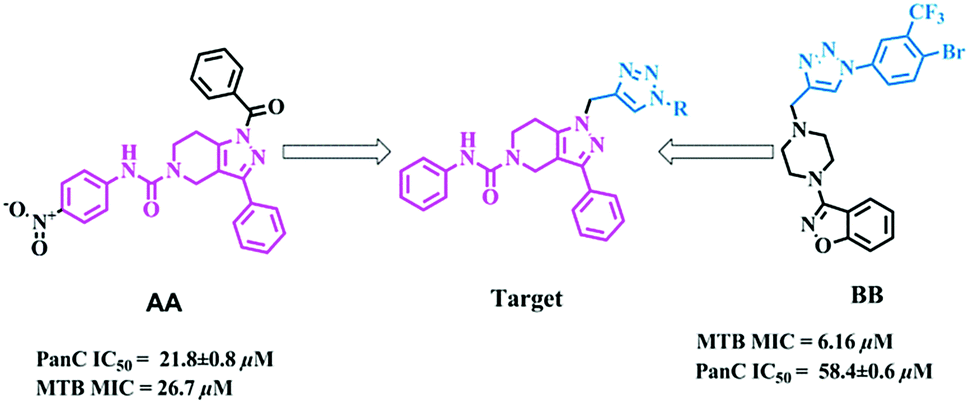
The molecular hybridization approach involves rational design of new ligands or the recognition of pharmacophoric sub-units in the molecular structure of two or more known bioactive derivatives which, through the adequate combination of these subunits, lead to the design of new hybrid architectures that maintain preselected characteristics of the original templates. In this work, we designed novel MTB inhibitors by hybridizing reported MTB PS inhibitors, 1-benzoyl-N-(4-nitrophenyl)-3-phenyl-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxamide (AA)12 and 3-(4-((1-(4-bromo-3-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-4-yl)methyl)piperazin-1-yl)benzo[d]isoxazole (BB),34 anticipating a new lead in the development of novel MTB PS inhibitors with potential MTB MIC (Fig. 3). BB is our most active compound from our previous work and when screened for MTB PS it exhibited panC IC50 58.4 μM.
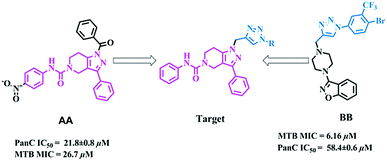 |
| | Fig. 3 Design strategy of the title compounds. | |
Results and discussion
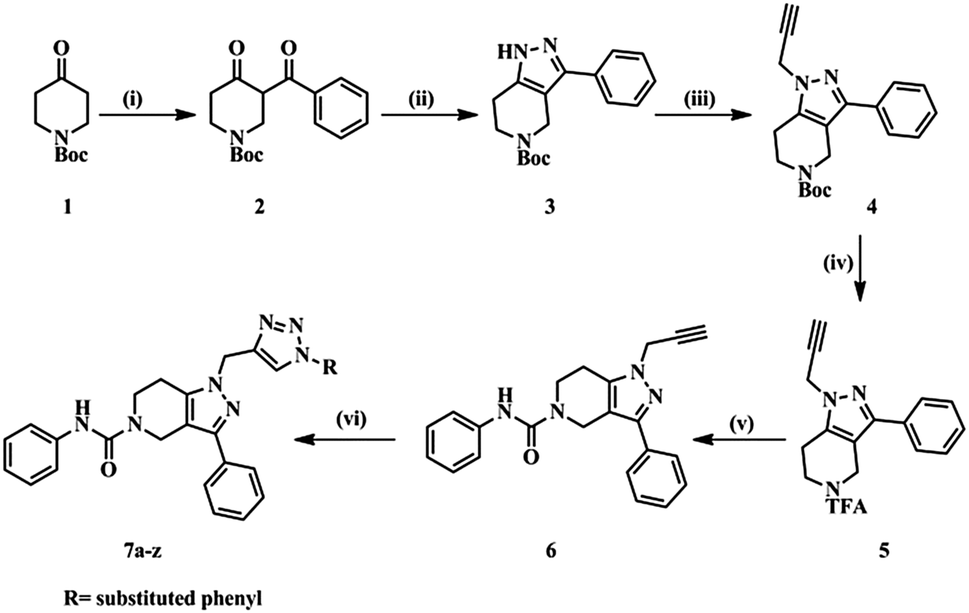
The designed molecules were synthesized in six steps (Scheme 1). Initially we prepared a 1,3-dicarbonyl intermediate (2) using 1-Boc-4-piperidone (1), morpholine, p-toluenesulfonic acid (catalytic) and benzoyl chloride (Stork-enamine reaction conditions). Treatment of 2 with hydrazine hydrate yielded a pyrazole ring (3).35 Compound 3 on reacting with propargyl bromide in the presence of CS2CO3 formed an N-alkyl product (4). Compound 4 was then deprotected using trifluoroacetic acid to yield compound 5. With weak base TEA, the more nucleophilic amine of the aliphatic ring reacted with phenylisocyanate yielding a urea derivative (6). The free acetylene group was converted to various 1H-1,2,3-triazoles using different aromatic azides via the click chemistry method.33 The purity of the compounds synthesized was checked by LC–MS and elemental analyses. The structures of the compounds were confirmed by spectral data. In 1H NMR and 13C NMR, the signals of the respective protons and carbons were verified on the basis of their chemical shifts, multiplicities, and coupling constants. The results of elemental analysis were within ±0.05 of the theoretical values.
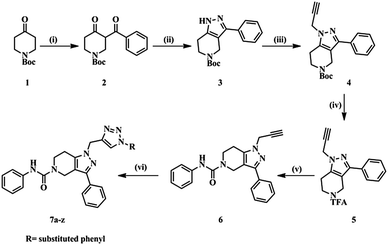 |
| | Scheme 1 Synthetic protocol of title compounds. | |
Reagents and conditions
(i) (a) Morpholine (1.0 eq.), PTSA (0.01 eq.), toluene, 100 °C, 16 h; (b) benzoyl chloride (1.1 eq.), TEA (2.0 eq.), DCM, 0 °C – rt, 4 h. (ii) N2H4 (1.0 eq.), EtOH, 0 °C – rt, 4 h. (iii) Propargyl bromide (80% in toluene) (1.2 eq.), Cs2CO3 (1.5 eq.), DMF, rt, 16 h. (iv) CF3COOH, DCM, rt, 16 h. (v) Phenyl isocyanate (1.3 eq.), TEA (3.0 eq.), DMF, rt, 4 h. (vi) Substituted aromatic azides, CuSO4·5H2O (10 mol%), sodium ascorbate (10 mol%), H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :t-BuOH (1:2), rt, 4 h.
:t-BuOH (1:2), rt, 4 h.
In vitro MTB screening
All the synthesized compounds were tested for their capacity to inhibit the growth of MTB. In the assay, three different M. tuberculosis strains were used. One of them was the reference strain M. tuberculosis H37Rv ATTC 25618 and the others were ‘wild’ strains isolated from tuberculosis patients.34,36 MTB strain spec. 210 was resistant to p-aminosalicylic acid (PAS), INH, ETB and RMP and another strain (Spec. 192) was fully sensitive to the administered tuberculostatics.37 In this study three different strains were used for screening as we wanted to know the kind of activity the synthesized compounds showed against the reference strain as well as against the strains isolated from TB patients. In this study the influence of the compound on the growth of mycobacteria at a certain concentration of 3.1, 6.2, 12.5, 25, 50 and 100 μg mL−1 was evaluated. INH was used as the reference drug. The in vitro antimycobacterial activity results of title compounds are presented in Table 1 in terms of MIC values (μM) and these values ranged from 24.72 to >200 μM.
Table 1 Results of antimycobacterial screening of title compounds
| Entry |
R |
MIC (μM) against MTB H37Rv |
MIC (μM) against MTB Spec. 192 |
MIC (μM) against MTB Spec. 210 |
|
7a
|
Phenyl |
105.14 |
105.14 |
105.14 |
|
7b
|
4-Methylphenyl |
25.53 |
25.53 |
51.06 |
|
7c
|
4-Ethylphenyl |
49.64 |
49.64 |
49.64 |
|
7d
|
4-Methoxyphenyl |
24.72 |
24.72 |
24.72 |
|
7e
|
4-Fluorophenyl |
>200 |
>200 |
>200 |
|
7f
|
4-Chlorophenyl |
196.08 |
196.08 |
196.08 |
|
7g
|
4-Bromophenyl |
90.18 |
90.18 |
90.18 |
|
7h
|
4-Iodophenyl |
166.26 |
166.26 |
166.26 |
|
7i
|
4-Nitrophenyl |
192.10 |
192.10 |
192.10 |
|
7j
|
4-Trifluoromethylphenyl |
91.98 |
91.98 |
91.98 |
|
7k
|
2-Chlorophenyl |
196.08 |
196.08 |
196.08 |
|
7l
|
2-Fluorophenyl |
>200 |
>200 |
>200 |
|
7m
|
2-Bromophenyl |
180.36 |
180.36 |
180.36 |
|
7n
|
2-Iodophenyl |
166.26 |
166.26 |
166.26 |
|
7o
|
2-Nitrophenyl |
192.10 |
192.10 |
192.10 |
|
7p
|
3-Chlorophenyl |
98.04 |
98.04 |
98.04 |
|
7q
|
3-Methoxyphenyl |
98.89 |
98.89 |
98.89 |
|
7r
|
2,4-Dichlorophenyl |
183.67 |
183.67 |
183.67 |
|
7s
|
3,4-Dichlorophenyl |
183.67 |
183.67 |
183.67 |
|
7t
|
3,5-Dichlorophenyl |
91.83 |
91.83 |
91.83 |
|
7u
|
3-Chloro-4-fluorophenyl |
189.40 |
189.40 |
189.40 |
|
7v
|
3,4-Difluorophenyl |
195.49 |
195.49 |
195.49 |
|
7w
|
3,4-Dimethoxyphenyl |
186.70 |
186.70 |
186.70 |
|
7x
|
4-Bromo-3 trifluoromethylphenyl |
160.65 |
160.65 |
160.65 |
|
7y
|
3,4-Dimethylphenyl |
198.57 |
198.57 |
198.57 |
|
7z
|
Benzo[d][1,3]dioxole |
192.47 |
192.47 |
192.47 |
| INH |
— |
≤22.60 |
≤22.60 |
≤91.14 |
Among the twenty six compounds screened, eight compounds (7b, 7c, 7d, 7g, 7j, 7p, 7q and 7t) showed activity against MTB with MIC <100 μM. Three compounds (7b, 7c, and 7d) inhibited MTB with MIC <50 μM. Compound 7d (1-((1-(4-methoxyphenyl)-1H-1,2,3-triazol-4-yl)methyl)-N,3-diphenyl-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)carboxamide) was found to be the most active compound with in vitro MIC 24.72 μM. In addition, compounds 7b, 7c and 7d with MIC <50 μM were further subjected to toxicity studies in a normal cell line to analyze the selectivity profile.
Amongst the synthesized derivatives, electron donating group containing compounds showed significant anti-TB activity. Structure–activity relationship studies are explained based on the activity of compound 7a. Structural changes at 4th position alter the activity. Compound 7a inhibited the growth of MTB H37Rv strain by 99% at 105.14 μM. In this series, the introduction of electron donating groups at the 4th position of the phenyl ring increased the activity. The introduction of an electron donating ethyl group (7c) increased the activity by two fold with MIC 49.64 μM. The presence of an electron donating methyl group (7b) increased the activity by four fold with MIC 25.53 μM. With the introduction of a methoxy group (7d) the activity increased by four fold (MIC 24.72 μM); with the same methoxy group at meta position of the phenyl ring (7q, MIC 98.89 μM) the activity remained unaltered compared to compound 7a. The presence of electron withdrawing groups, viz., F, Cl, Br and I, resulted in a decrease in activity compared to compounds 7b, 7c, and 7d. The presence of electron withdrawing groups at ortho or meta position also decreased the activity. The introduction of an electron donating methoxy group at meta and para positions (7w, MIC 186.70 μM) resulted in a decrease in activity by two fold compared to compound 7a. In conclusion, the presence of an electron donating methoxy group at the para position of the phenyl ring enhanced the activity. Amongst the synthesized derivatives, 7d emerged as the most active compound.
Pantothenate synthetase enzyme inhibition studies
PS enzyme inhibition studies were carried out on synthesized compounds by estimating the amount of NAD+ produced.7,15 The NAD+ produced can be examined spectrophotometrically at 340 nm. In the initial screening at 50 μM, all compounds exhibited more than 50% inhibition against MTB PS and their IC50 values were further determined. Most of the compounds showed good IC50 values ranging from 0.91 ± 0.32 to 8.97 ± 0.05 μM (Table 2). Seven compounds (7b, 7d, 7h, 7p, 7r, 7s and 7v) inhibited MTB PS with IC50 <2.00 μM. Compounds 7d and 7s emerged as the most active compounds with IC50 1.01 ± 0.32 and 0.91 ± 0.32 μM respectively.
Table 2 Docking scores and MTB PS assay
| Compound |
In silico
|
In vitro |
| Entry |
XP GScore |
Glide energy |
MTB PanC IC50 μM |
| Co-ligand |
−8.32 |
−78.66 |
— |
|
7a
|
−7.19 |
−71.33 |
2.13 ± 0.12 |
|
7b
|
−5.69 |
−58.61 |
1.54 ± 0.22 |
|
7c
|
−6.44 |
−64.89 |
2.56 ± 0.04 |
|
7d
|
−8.19 |
−66.25 |
1.01 ± 0.32 |
|
7e
|
−6.61 |
−67.18 |
3.41 ± 0.08 |
|
7f
|
−3.25 |
−69.57 |
6.36 ± 0.12 |
|
7g
|
−5.24 |
−65.40 |
2.46 ± 0.07 |
|
7h
|
−7.61 |
−73.33 |
1.04 ± 0.55 |
|
7i
|
−3.80 |
−62.50 |
8.97 ± 0.05 |
|
7j
|
−4.70 |
−58.79 |
5.04 ± 0.54 |
|
7k
|
−4.62 |
−65.89 |
2.78 ± 0.07 |
|
7l
|
−4.46 |
−61.60 |
5.93 ± 0.64 |
|
7m
|
−6.31 |
−71.76 |
3.54 ± 0.55 |
|
7n
|
−5.24 |
−60.42 |
3.92 ± 0.19 |
|
7o
|
−6.33 |
−72.18 |
4.94 ± 0.03 |
|
7p
|
−7.69 |
−70.79 |
1.14 ± 0.19 |
|
7q
|
−4.25 |
−65.23 |
7.78 ± 0.56 |
|
7r
|
−6.07 |
−67.82 |
1.29 ± 0.14 |
|
7s
|
−7.83 |
−69.26 |
0.91 ± 0.32 |
|
7t
|
−5.99 |
−63.35 |
4.76 ± 0.02 |
|
7u
|
−5.37 |
−63.08 |
6.43 ± 0.12 |
|
7v
|
−7.37 |
−64.55 |
1.56 ± 0.11 |
|
7w
|
−4.96 |
−64.16 |
8.21 ± 0.03 |
|
7x
|
−4.44 |
−57.95 |
6.54 ± 0.21 |
|
7y
|
−6.60 |
−56.87 |
6.91 ± 0.07 |
|
7z
|
−4.84 |
−73.86 |
8.39 ± 0.14 |
Docking study
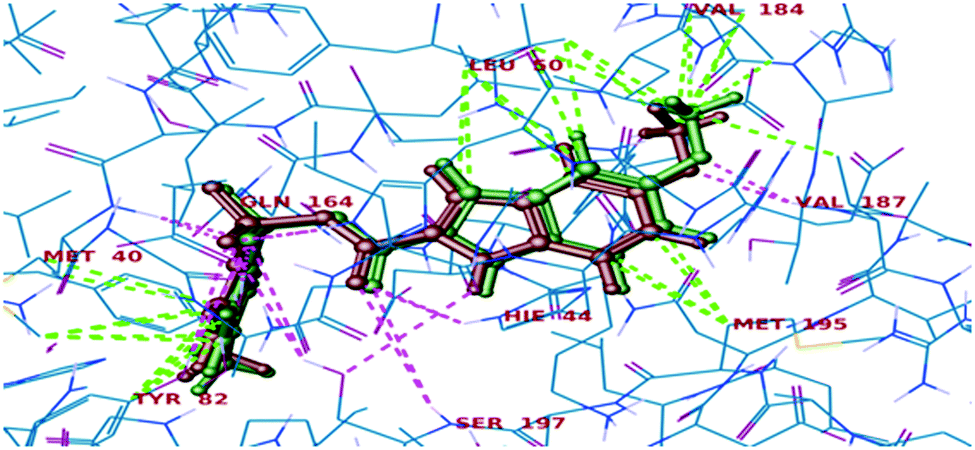
All the final compounds were docked into the crystal structure of MTB PS protein (PDB ID: 3IUB) to know the exact binding pattern with the receptor. Validation of the docking protocol revealed that the RMSD value obtained by comparing the experimental binding mode of the co-crystallized ligand (as in the X-ray structure) and its re-docked pose (Fig. 4) was 0.76, which suggested that the docking procedure could be relied on for further docking studies.
 |
| | Fig. 4 Superimposed view of the co-crystallized ligand (green) with its X-ray pose (red) in 3IUB. | |
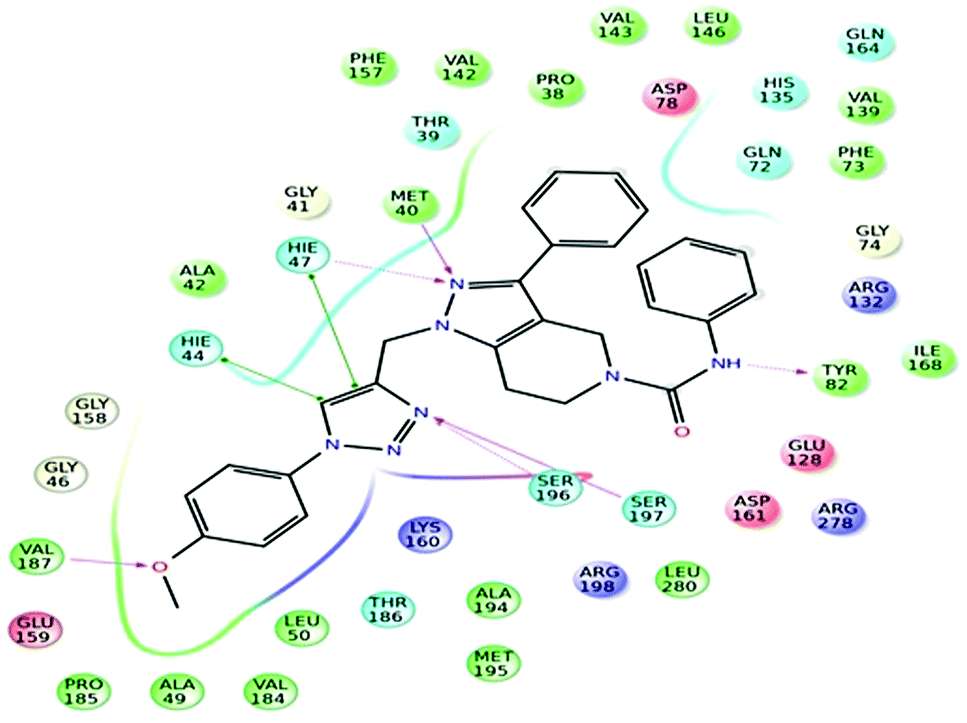
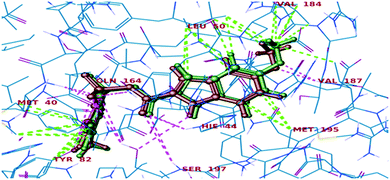
Further, in the docking studies, molecules exhibited good binding energy in the range of −3.25 to −8.19 kcal mol−1 and exhibited good fitness with the MTB PS protein. Several compounds displayed hydrogen bonding interactions with amino acid residues HIE47, HIE44, Met40, Ser196, Tyr82 and Ser197. 1-((1-(4-Methoxyphenyl)-1H-1,2,3-triazol-4-yl)methyl)-N,3-diphenyl-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)carboxamide (7d), one of the most active ligands, with IC50 1.01 ± 0.32 μM showed a docking score of −8.19 kcal mol−1. The active site in the hydrophobic pocket is within the vicinity of Leu146, Val187, Val142, Met195, Ala42, Leu50, Pro38, Ile168 and Met40 and some polar amino acid residues HIE47, Ser196, Ser197 and Thr82. The ligand also exhibited hydrogen bonding interactions with Met40, Ser197, and Val187 residues.15 The binding pattern of 7d with MTB PS is shown in Fig. 5.
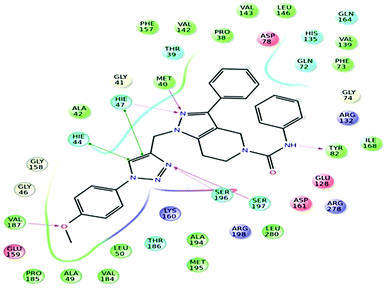 |
| | Fig. 5 Docked pose of compound 7d inside the 3IUB, showing a two-dimensional interactive diagram. | |
In vitro cytotoxicity studies
The active compounds 7b, 7c and 7d were evaluated by Promega Cell Titer 96 non-radioactive cell proliferation assay to analyze the selectivity profile against mouse macrophage (RAW264.7) cell lines.38 The IC50 values and selectivity index (SI) values are tabulated in Table 3. The most active compound (7d) showed an SI value of 13.76. The results imply that the compounds are suitable for further investigation against TB.
Table 3 Cytotoxicity results of the active compounds
| Entry |
MIC (μM) in MTB H37Rv |
IC50 approximationb |
SI values IC50/MICa |
|
Selectivity index.
Units in μM.
|
|
7b
|
25.53 |
335.19 |
13.12 |
|
7c
|
49.64 |
370.12 |
7.45 |
|
7d
|
24.72 |
340.15 |
13.76 |
Single crystal X-ray crystallographic structure of compound 7g
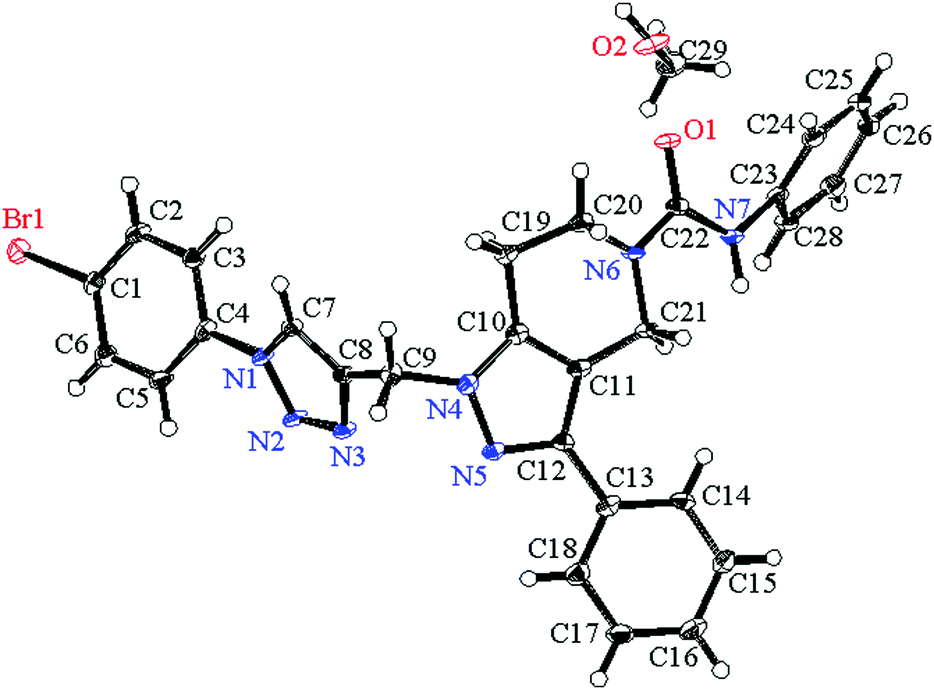
Suitable crystals of compound 7g for X-ray crystallographic study were grown from methanol solution. The single crystal X-ray diffraction measurement of the molecule (C28H24N7OBr·CH3OH) was performed on a Rigaku XtaLAB P200 diffractometer using graphite monochromatized Mo-Kα radiation (λ = 0.71073 Å) on 0.1 mm × 0.1 mm × 0.1 mm a pale yellow crystal. Data were collected and processed using CrysAlisPro (Rigaku Oxford Diffraction). The data were collected at a temperature of −180 ± 1 °C to a maximum 2θ value of 58.2°. Of the 19673 reflections collected, 6164 were unique (Rint = 0.0791) and equivalent reflections were merged. The diffraction data were refined and the structure was solved using the Crystal Structure 4.2.2 software program. The structure was solved by direct methods and expanded using Fourier techniques. The non-hydrogen atoms were refined anisotropically. Hydrogen atoms were refined using the riding model. The compound crystallized into a triclinic crystal system with P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. In a single unit cell four partially occupying molecules along with two methanol solvent molecules of crystallization are observed with Z = 2. The basic crystallographic data are shown in Table 4. The molecular structure of the compound with methanol solvent of crystallization is given as an ORTEP diagram in Fig. 6. The part of the molecule containing the p-bromophenyl ring is directly attached to triazole ring nitrogen. These two rings are not coplanar; the phenyl ring plane is deviated from the triazole plane by around 30°. The torsional angle between these two rings with selected bonds C5–C4–N1–N2 = 30.90 and C3–C4–N1–C7 = 31.71 degrees. The deviation is comparatively less in the other part of the molecule. The pyrazole ring plane with respect to the attached phenyl ring plane deviated by around 7.5°, and the corresponding dihedral angles for the selected four bonds are N5–C12–C13–C18 = 7.54 and C11–C12–C13–C14 = 7.34 degrees. Crystallographic data for compound 7g are deposited in the Cambridge Crystallographic Data Center and the corresponding deposition number is CCDC 1500428.
space group. In a single unit cell four partially occupying molecules along with two methanol solvent molecules of crystallization are observed with Z = 2. The basic crystallographic data are shown in Table 4. The molecular structure of the compound with methanol solvent of crystallization is given as an ORTEP diagram in Fig. 6. The part of the molecule containing the p-bromophenyl ring is directly attached to triazole ring nitrogen. These two rings are not coplanar; the phenyl ring plane is deviated from the triazole plane by around 30°. The torsional angle between these two rings with selected bonds C5–C4–N1–N2 = 30.90 and C3–C4–N1–C7 = 31.71 degrees. The deviation is comparatively less in the other part of the molecule. The pyrazole ring plane with respect to the attached phenyl ring plane deviated by around 7.5°, and the corresponding dihedral angles for the selected four bonds are N5–C12–C13–C18 = 7.54 and C11–C12–C13–C14 = 7.34 degrees. Crystallographic data for compound 7g are deposited in the Cambridge Crystallographic Data Center and the corresponding deposition number is CCDC 1500428.
Table 4 Crystal data and structure refinement for 7g
| Empirical formula |
C28H24BrN7O·CH3OH |
| Formula weight |
586.49 |
| Crystal color, habit |
Light yellow |
| Crystal dimensions |
0.1 mm × 0.1 mm × 0.1 mm |
| Crystal system |
Triclinic |
| Lattice type |
Primitive |
| Lattice parameters |
a = 7.8845(3) Å |
|
b = 11.9643(4) Å |
|
c = 14.9423(4) Å |
|
α = 102.268(2) Å |
|
β = 101.916(2) Å |
|
γ = 95.003(3) Å |
|
δ = 1334.95(8) Å3 |
| Space group |
P (#2) |
|
Z value |
2 |
|
D
calc
|
1.459 g cm−3 |
|
F
000
|
604.00 |
|
μ(MoKα) |
15.855 cm−1 |
| Radiation |
Mo-Kα (λ = 0.71073 Å) |
| Radiation monochromator |
Graphite |
| Voltage, current |
50 kV, 40 mA |
| Temperature |
−180.0 °C |
| Maximum 2θ |
58.2° |
| Number of measured reflections |
19673° |
| Number of unique reflections |
6164 (Rint = 0.0791) |
| Number of parameters |
380 |
| Goodness-of-fit on F2 |
1.00 |
| Δρmax,mix (e− Å−3) |
1.89, −0.78 |
| Reflection/parameter ratio |
16.22 |
| Residuals: R1 (I > 2.00σ(I)) |
0.0479 |
| Residuals: R (all reflections) |
0.0538 |
| Residuals: wR2 (all reflections) |
0.1931 |
| Crystal refinement |
Crystal structure 4.2.2 |
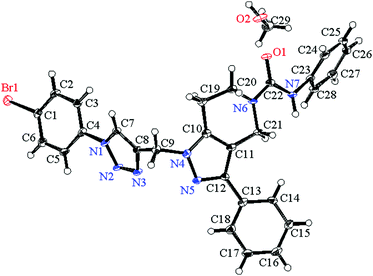 |
| | Fig. 6 ORTEP diagram showing the X-ray crystal structure of compound 7g with a methanol solvent of crystallization. | |
Conclusions
In this work, we designed novel 1-((1-(substituted)-1H-1,2,3-triazol-4-yl)methyl)-N,3-diphenyl-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxamide derivatives by a molecular hybridization approach using reported MTB PS inhibitors and substituted 1H-1,2,3-triazole antitubercular compounds. Twenty six compounds were synthesized and well characterized. Of these compounds 7d showed better MTB PS inhibition and MTB MIC than one of the lead compounds AA. Thus, this 1-(substituted)-1H-1,2,3-triazole scaffold could be further optimized to develop MTB PS specific agents. In conclusion, it has been shown that the potency and low cytotoxicity of the title compounds make them suitable leads for synthesizing new compounds with better anti-tubercular activity.
Experimental
Materials and methods
The chemicals and solvents were procured from a commercial source. The solvents and reagents were of LR grade and if necessary were purified before use. Thin-layer chromatography (TLC) was carried out on aluminium-supported silica gel plates (Merck 60 F254) with visualization of components by UV light (254 nm). Column chromatography was carried out on silica gel (Merck, 100–200 mesh). 1H NMR and 13C NMR spectra were recorded at 400 MHz and 101 MHz respectively using a Bruker AV 400 spectrometer (Bruker CO., Switzerland) in CDCl3 and DMSO-d6 solution with tetramethylsilane as the internal standard, and chemical shift values (δ) are given in ppm. Melting points were determined on an electro thermal melting point apparatus (Stuart-SMP30) in open capillary tubes and are uncorrected. Elemental analyses were performed using an Elementar Analysensysteme GmbH vario MICRO cube CHN Analyzer. Mass spectra (ESI-MS) were recorded on a Shimadzu MS/ESI mass spectrometer. Purity of all tested compounds was determined by LC-MS/MS on a Shimadzu system and was greater than 95%.
Chemistry
tert-Butyl 3-benzoyl-4-oxopiperidine-1-carboxylate (2).
In a two necked 100 mL round-bottom flask equipped with a Dean-stark trap, a reflux condenser and an internal thermocouple, compound 1 (5.0 g, 23.15 mmol), toluene (50 mL), morpholine (2.1 mL, 23.15 mmol), and p-toluenesulfonic acid (catalytic) were added sequentially. The reaction mixture was refluxed under a N2 atmosphere for 16 h. The solvent was evaporated and the crude reaction mixture was dissolved in DCM (40 mL) and then triethylamine (5.35 mL, 37.65 mmol) was added at 0 °C under a N2 atmosphere followed by the addition of benzoyl chloride (2.9 mL, 25.1 mmol) over 10 min. The ice bath was then removed and the reaction solution was stirred at room temperature for 4 h. On completion of the reaction, as indicated by TLC, the reaction was quenched with NaHCO3 solution and extracted with DCM. The organic layers were collected, washed with saturated brine solution, dried over anhydrous Na2SO4 and concentrated in vacuo. The resultant crude product was purified by column chromatography [ethyl acetate/hexane (10–15%)] to get the 1,3-dicarbonyl compound 2 (7.0 g, 92%) as a colorless liquid. ESI-MS found 304 (M + H)+.
tert-Butyl 3-phenyl-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxylate (3).
A stirred solution of tert-butyl 3-benzoyl-4-oxopiperidine-1-carboxylate (2) (7.6 g, 25.05 mmol) in ethanol was cooled to 0 °C and hydrazine hydrate (0.8 mL, 25.05 mmol) was added and stirred for 4 h. On completion of the reaction, as indicated by TLC, the reaction mixture was concentrated under reduced pressure and the crude residue was purified by column chromatography to get compound 3 (6.9 g, 92%) as an off-white solid. ESI-MS found 300 (M + H)+. 1H NMR (400 MHz, CDCl3) δ 9.48 (s, 1H), 7.58–7.19 (m, 5H), 4.82 (s, 2H), 3.91 (t, J = 7.6 Hz, 2H), 2.83 (t, J = 7.8 Hz, 2H), 1.34 (s, 9H); 13C NMR (100 MHz, CDCl3) 163.54, 146.99, 143.56, 136.64, 132.75, 129.31, 127.39, 117.45, 81.67, 43.91, 36.89, 31.49, 27.79; anal. calcd for C17H21N3O2: (%) C, 68.20; H, 7.07; N, 14.04, found: C, 68.24; H, 7.09; N, 14.13.
tert-Butyl-3-phenyl-1-(prop-2-yn-1-yl)-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxylate (4).
A solution of tert-butyl 3-phenyl-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxylate (3) (5.0 g, 16.70 mmol) in DMF was cooled to 0 °C and Cs2CO3 (8.16 g, 25.05 mmol) and propargyl bromide (80% in toluene) (1.64 mL, 21.17 mmol) were added and allowed to reach room temperature and stirred for 16 h. On completion of the reaction, as indicated by TLC, the reaction was quenched with cold water and extracted with diethyl ether. The organic layers were collected, washed with saturated brine solution, dried over anhydrous Na2SO4 and concentrated in vacuo. The resultant crude product was purified by column chromatography [ethyl acetate/hexane (20–30%)] to get compound 4 (4.6 g, 83%) as a semisolid. ESI-MS found 338.15 (M + H)+. 1H NMR (400 MHz, CDCl3) δ 7.60–7.23 (m, 5H), 4.80 (s, 2H), 4.56 (s, 2H), 3.92 (t, J = 7.4 Hz, 2H), 2.81 (t, J = 7.6 Hz, 2H), 2.72 (s, 2H), 1.31 (s, 9H); 13C NMR (100 MHz, CDCl3) 163.57, 147.64, 143.65, 136.76, 132.87, 129.38, 127.40, 117.51, 81.7, 77.96, 69.05, 43.91, 42.07, 36.77, 31.59, 27.90; anal. calcd for C20H23N3O2: (%) C, 69.79; H, 6.96; N, 13.20, found: C, 69.80; H, 7.01; N, 13.23.
3-Phenyl-1-(prop-2-yn-1-yl)-4,5,6,7-tetrahydro-1H-pyrazolo[4,3-c]pyridine (5).
A solution of tert-butyl 3-phenyl-1-(prop-2-yn-1-yl)-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxylate (4) (4.0 g, 11.85 mmol) in CH2Cl2 was cooled to 0 °C and CF3COOH (4.5 mL, 59.27 mmol) was added dropwise and stirred at room temperature for 16 h. The reaction mixture was concentrated under reduced pressure and the crude residue was washed with hexane and diethyl ether to get compound 5 (2.6 g, 92%) as an off-white solid. ESI-MS showed 238.10 (M + H)+.
N,3-Diphenyl-1-(prop-2-yn-1-yl)-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxamide (6).
Phenylisocyanate (1.3 mL, 10.95 mmol) was added to a stirred solution of compound 5 (2.0 g, 8.42 mmol) and Et3N (3.5 mL, 25.26 mmol) in DMF at 0 °C under a N2 atmosphere, and stirred at room temperature for 4 h. On completion of the reaction, as indicated by TLC, the reaction was quenched with cold water and then the solid product was filtered and washed with water and hexane to get the key intermediate 6 (2.7 g, 90%) as an off-white solid. ESI-MS showed 357.15 (M + H)+. 1H NMR (400 MHz, DMSO-d6) δ 8.74 (s, 1H), 7.88 (d, J = 8.6 Hz, 2H), 7.78 (d, J = 8.8 Hz, 2H), 7.67 (d, J = 7.6 Hz, 2H), 7.72–7.64 (m, 2H), 7.28–7.18 (m, 2H), 5.47 (s, 2H), 4.71 (s, 2H), 3.86 (t, J = 7.6 Hz, 2H), 2.90 (t, J = 7.5 Hz, 2H), 2.69 (s, 2H); 13C NMR (100 MHz, CDCl3) 164.58, 155.57, 143.57, 139.51, 136.69, 132.85, 129.38, 128.92, 128.12, 127.40, 121.16, 117.52, 81.73, 77.94, 69.0, 44.01, 42.12, 23.94; anal. calcd for C22H20N4O: (%) C, 74.14; H, 5.66; N, 15.72, found: C, 74.16; H, 5.67; N, 15.73.
N,3-Diphenyl-1-((1-phenyl-1H-1,2,3-triazol-4-yl)methyl)-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxamide (7a–z).
A solution of N,3-diphenyl-1-(prop-2-yn-1-yl)-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxamide (6) (0.20 g, 1.0 equiv.) was reacted with substituted phenyl azides (1.2 equiv.) in the presence of sodium ascorbate (0.01 equiv.), CuSO4·5H2O (0.02 equiv.) and t-BuOH:H2O (2:1) at rt for 4 h. On completion of the reaction, as indicated by TLC, the reaction was quenched with cold water and extracted with DCM. The DCM layers were collected, washed with saturated brine solution, dried over anhydrous Na2SO4 and concentrated in vacuo. The resultant crude product was purified by column chromatography [MeOH/DCM (1–3%)] to yield the title compounds 7a–z.
N,3-Diphenyl-1-((1-phenyl-1H-1,2,3-triazol-4-yl)methyl)-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxamide (7a).
Off-white solid (83%); m.p. 221–223 °C; IR (KBr) νmax/cm−1 3490, 3021, 2843, 1650, 1410, 1340, 1060. 1H NMR (400 MHz, DMSO-d6) δ 8.88 (s, 1H), 8.75 (s, 1H), 7.94–7.86 (m, 2H), 7.69–7.66 (m, 2H), 7.58 (dd, J = 8.6, 7.1 Hz, 2H), 7.50–7.42 (m, 5H), 7.38–7.28 (m, 1H), 7.28–7.15 (m, 2H), 6.94 (t, J = 7.2, 1.2 Hz, 1H), 5.49 (s, 2H), 4.72 (s, 2H), 3.83 (t, J = 5.8 Hz), 2.98 (t, J = 5.8 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 155.67, 145.55, 144.46, 140.85, 139.15, 136.98, 133.97, 130.32, 129.20, 129.14, 128.74, 127.78, 126.51, 122.38, 122.39, 120.61, 120.48, 112.00, 44.40, 42.17, 41.16, 40.59, 22.33. EI-MS m/z 476.20 (M + H)+; anal. calcd for C28H25N7O: (%) C, 70.72; H, 5.30; N, 20.62; found: C, 70.74; H, 5.31; N, 20.63.
N,3-Diphenyl-1-((1-(p-tolyl)-1H-1,2,3-triazol-4-yl)methyl)-1,4,6,7-tetrahydro-5H-pyrazolo[4,3-c]pyridine-5-carboxamide (7b).
Light yellow solid (87%); m.p. 204–206 °C; (KBr) νmax/cm−1 3455, 3025, 2867, 1645, 1420, 1348, 1062. 1H NMR (400 MHz, DMSO-d6) δ 8.81 (s, 1H), 8.75 (s, 1H), 7.77 (d, J = 8.5 Hz, 2H), 7.67 (d, J = 7.2 Hz, 2H), 7.48–7.42 (m, 4H), 7.40–7.30 (m, 3H), 7.26–7.20 (m, 2H), 6.94 (t, J = 6.8 Hz, 1H), 5.47 (s, 2H), 4.71 (s, 2H), 3.83 (t, J = 5.6 Hz, 2H), 2.97 (t, J = 5.3 Hz, 2H), 2.36 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 155.68, 145.54, 144.42, 140.91, 139.16, 138.77, 134.74, 133.95, 130.71, 129.21, 128.82, 127.78, 126.48, 122.38, 122.24, 120.49, 120.47, 111.99, 44.46, 42.18, 41.66, 22.29, 21.06. EI-MS m/z 490.23 (M + H)+; anal. calcd for C28H25N7O: (%) C, 71.15; H, 5.56; N, 20.03; found: C, 71.16; H, 5.58; N, 20.06.
1-((1-(4-Ethylphenyl)-1H-1,2,3-triazol-4-yl)methyl)-N,3-diphenyl-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxamide (7c).
Off-white solid (89%); m.p. 128–130 °C; IR (KBr) νmax/cm−1 3478, 3031, 2913, 1635, 1422, 1341, 1056. 1H NMR (400 MHz, DMSO-d6) δ 8.82 (s, 1H), 8.75 (s, 1H), 7.78 (d, J = 8.4 Hz, 2H), 7.68 (d, J = 7.3 Hz, 2H), 7.48–7.42 (m, 4H), 7.40–7.30 (m, 3H), 7.26–7.20 (m, 2H), 6.94 (t, J = 6.8 Hz, 1H), 5.47 (s, 2H), 4.71 (s, 2H), 3.83 (t, J = 5.6 Hz, 2H), 2.97 (t, J = 5.4 Hz, 2H), 2.76 (m, 2H), 1.36 (t, 3H). 13C NMR (101 MHz, DMSO-d6) δ 155.17, 145.09, 144.67, 144.21, 143.62, 140.38, 138.66, 133.47, 129.93, 128.66, 128.22, 127.25, 126.04, 121.89, 121.84, 121.77, 119.95, 114.78, 48.12, 44.19, 41.67, 28.23, 21.85, 14.45. EI-MS m/z 504.25 (M + H)+; anal. calcd for C30H29N7O: (%) C, 71.55; H, 5.80; N, 19.47; found: C, 71.56; H, 5.82; N, 19.48.
1-((1-(4-Methoxyphenyl)-1H-1,2,3-triazol-4-yl)methyl)-N,3-diphenyl-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxamide (7d).
Brown solid (90%); m.p. 115–117 °C; IR (KBr) νmax/cm−1 3442, 3027, 2832, 1645, 1424, 1365, 1034, 1020. 1H NMR (400 MHz, DMSO-d6) δ 8.76 (s, 1H), 8.73 (s, 1H), 7.81–7.78 (m, 2H), 7.68–7.64 (m, 2H), 7.47–7.41 (m, 4H), 7.35–7.31 (m, 1H), 7.25–7.21 (m, 2H), 7.11 (d, J = 9.1 Hz, 2H), 6.95 (d, J = 7.4 Hz, 1H), 5.46 (s, 2H), 4.70 (s, 2H), 3.83 (s, 3H), 3.82 (t, J = 7.1 Hz, 2H), 2.97 (t, J = 5.4 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.47, 153.18, 145.02, 143.72, 140.36, 138.63, 133.49, 129.91, 128.64, 128.24, 127.27, 126.01, 121.88, 121.86, 121.77, 119.98, 114.78, 111.48, 55.51, 48.12, 44.93, 41.67, 22.13. EI-MS m/z 506.20 (M + H)+; anal. calcd for C29H27N7O2: (%) C, 68.89; H, 5.38; N, 19.39; found: C, 68.91; H, 5.39; N, 19.41.
1-((1-(4-Fluorophenyl)-1H-1,2,3-triazol-4-yl)methyl)-N,3-diphenyl-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxamide (7e).
Off-white solid (82%); m.p. 158–160 °C; IR (KBr) νmax/cm−1 3490, 3021, 2843, 1655, 1410, 1340, 1120, 1060. 1H NMR (400 MHz, DMSO-d6) δ 8.85 (s, 1H), 8.74 (s, 1H), 8.00–7.90 (m, 2H), 7.71–7.63 (m, 2H), 7.50–7.29 (m, 7H), 7.28–7.17 (m, 2H), 6.94 (tt, J = 7.3, 1.2 Hz, 1H), 5.48 (s, 2H), 4.71 (s, 2H), 3.82 (t, J = 5.7 Hz, 2H), 2.97 (t, J = 5.4 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 161.01, 155.21, 145.02, 143.72, 140.36, 138.63, 133.49, 129.91, 128.64, 128.24, 127.27, 126.01, 121.88, 121.86, 121.77, 119.98, 114.78, 111.48, 50.19, 48.93, 41.87, 24.23. EI-MS m/z 494.20 (M + H)+; anal. calcd for C28H24FN7O: (%) C, 68.14; H, 4.90; N, 19.87; found: C, 68.16; H, 4.91; N, 19.89.
1-((1-(4-Chlorophenyl)-1H-1,2,3-triazol-4-yl)methyl)-N,3-diphenyl-1,4,6,7-tetrahydro-5H-pyrazolo[4,3-c]pyridine-5-carboxamide (7f).
White solid (89%); m.p. 145–147 °C; (KBr) νmax/cm−1 3428, 3027, 2834, 1647, 1412, 1343, 1043, 615. 1H NMR (400 MHz, DMSO-d6) δ 8.82 (s, 1H), 8.73 (s, 1H), 7.80 (d, J = 9.2 Hz, 2H), 7.66 (d, J = 8.5 Hz, 2H), 7.43 (t, J = 7.6 Hz, 4H), 7.37 (t, J = 7.5 Hz, 1H), 7.27–7.21 (m, 2H), 7.18 (d, J = 9.5 Hz, 2H), 6.91 (t, J = 7.8 Hz, 1H), 5.46 (s, 2H), 4.74 (s, 2H), 3.83 (t, J = 5.7 Hz, 2H), 2.96 (t, J = 5.2 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 155.28, 145.12, 143.92, 141.36, 138.63, 134.12, 133.39, 129.91, 128.64, 128.24, 127.27, 126.11, 121.88, 121.89, 121.77, 119.88, 114.78, 111.48, 52.19, 48.83, 41.67, 24.83. EI-MS m/z 510.20 (M + H)+; anal. calcd for C28H24ClN7O: (%) C, 65.94; H, 4.74; N, 19.23; found: C, 65.16; H, 4.75; N, 19.89.
1-((1-(4-Bromophenyl)-1H-1,2,3-triazol-4-yl)methyl)-N,3-diphenyl-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxamide (7g).
Pale yellow solid (87%); m.p. 150–152 °C; IR (KBr) νmax/cm−1 3492, 3022, 2913, 1657, 1422, 1332, 1055, 680. 1H NMR (400 MHz, DMSO-d6) δ 8.90 (s, 1H), 8.75 (s, 1H), 7.88 (d, J = 8.6 Hz, 2H), 7.78 (d, J = 8.8 Hz, 2H), 7.67 (d, J = 7.6 Hz, 2H), 7.45 (dt, J = 7.8, 3.6 Hz, 4H), 7.33 (t, J = 7.4 Hz, 1H), 7.23 (t, J = 7.8 Hz, 2H), 6.94 (t, J = 7.4 Hz, 1H), 5.48 (s, 2H), 4.71 (s, 2H), 3.82 (t, J = 5.6 Hz, 2H), 2.96 (t, J = 6.0 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 155.66, 145.58, 144.65, 140.84, 139.15, 136.17, 133.95, 133.19, 129.14, 128.74, 127.79, 126.45, 122.51, 122.43, 122.38, 121.92, 120.42, 112.05, 44.33, 42.24, 41.24, 22.22. EI-MS m/z 554.12 (M + H)+; 556.10 (M + H)+2; anal. calcd for C28H24BrN7O: (%) C, 60.66; H, 4.36; N, 17.68; found: C, 60.68; H, 4.37; N, 17.69.
1-((1-(4-Iodophenyl)-1H-1,2,3-triazol-4-yl)methyl)-N,3-diphenyl-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxamide (7h).
Light brown solid (80%); m.p. 147–149 °C; IR (KBr) νmax/cm−1 3485, 30311, 2905, 1658, 1402, 1043, 560. 1H NMR (400 MHz, DMSO-d6) δ 8.85 (s, 1H), 8.76 (s, 1H), 7.99 (d, J = 9.2 Hz, 2H), 7.76 (d, J = 8.5 Hz, 2H), 7.46 (t, J = 7.8 Hz, 4H), 7.35 (t, J = 7.5 Hz, 1H), 7.27–7.21 (m, 2H), 7.12 (d, J = 9.3 Hz, 2H), 6.92 (t, J = 7.8 Hz, 1H), 5.47 (s, 2H), 4.69 (s, 2H), 3.83 (t, J = 5.8 Hz, 2H), 2.97 (t, J = 5.4 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 155.48, 145.02, 144.23, 143.72, 140.36, 138.63, 129.91, 128.64, 128.24, 127.27, 126.01, 121.88, 121.86, 121.77, 119.98, 116.78, 115.48, 95.15, 45.23, 42.13, 41.67, 23.93. EI-MS m/z 602.12 (M + H)+; anal. calcd for C28H24IN7O: (%) C, 55.92; H, 4.03; N, 16.30; found: C, 55.94; H, 4.31; N, 16.32.
1-((1-(4-Nitrophenyl)-1H-1,2,3-triazol-4-yl)methyl)-N,3-diphenyl-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxamide (7i).
Light yellow solid (81%); m.p. 144–146 °C; IR (KBr) νmax/cm−1 3505, 3031, 2912, 1675, 1532, 1372, 1027. 1H NMR (400 MHz, DMSO-d6) δ 9.08 (s, 1H), 8.74 (s, 1H), 8.48–8.40 (m, 2H), 8.28–8.19 (m, 2H), 7.71–7.63 (m, 2H), 7.50–7.39 (m, 4H), 7.40–7.27 (m, 1H), 7.28–7.18 (m, 2H), 6.94 (tt, J = 7.3, 1.2 Hz, 1H), 5.51 (s, 2H), 4.71 (s, 2H), 3.83 (t, J = 5.7 Hz, 2H), 2.97 (t, J = 5.4 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 155.65, 147.17, 145.65, 145.10, 141.20, 140.83, 139.19, 133.92, 129.13, 128.74, 127.81, 126.50, 125.96, 122.89, 122.38, 121.14, 120.47, 112.03, 44.29, 42.15, 41.13, 22.28. EI-MS m/z 521.19 (M + H)+; anal. calcd for C28H24N8O3: (%) C, 64.61; H, 4.65; N, 21.53; found: C, 64.63; H, 4.66; N, 21.55.
N,3-Diphenyl-1-((1-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-4-yl)methyl)-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxamide (7j).
White solid (76%); m.p. 246–248 °C; IR (KBr) νmax/cm−1 3505, 3029, 2903, 1650, 1402, 1357, 1279, 1045. 1H NMR (400 MHz, DMSO-d6) δ 9.03 (s, 1H), 8.75 (s, 1H), 8.17 (d, J = 8.4 Hz, 2H), 7.97 (d, J = 8.5 Hz, 2H), 7.69–7.64 (m, 2H), 7.47–7.42 (m, 4H), 7.37–7.29 (m, 1H), 7.27–7.20 (m, 2H), 6.96–6.92 (m, 1H), 5.51 (s, 2H), 4.72 (s, 2H), 3.83 (t, J = 5.7 Hz, 2H), 2.97 (t, J = 6.7 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 155.66, 145.62, 144.86, 140.84, 139.75, 139.18, 133.94, 129.14, 128.73, 127.80, 127.60, 127.60, 126.51, 122.91, 122.71, 122.38, 121.06, 120.47, 112.02, 44.33, 42.16, 41.14, 22.30. EI-MS m/z 544.19 (M + H)+; anal. calcd for C29H24F3N7O3: (%) C, 64.08; H, 4.45; N, 18.04; found: C, 64.10; H, 4.46; N, 18.05.
1-((1-(2-Chlorophenyl)-1H-1,2,3-triazol-4-yl)methyl)-N,3-diphenyl-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxamide (7k).
Off-white solid (88%); m.p. 137–139 °C; IR (KBr) νmax/cm−1 3523, 3032, 2923, 1675, 1445, 1052, 602. 1H NMR (400 MHz, DMSO-d6) δ 8.87 (s, 1H), 8.75 (s, 1H), 7.81 (d, J = 9.2 Hz, 2H), 7.69 (d, J = 8.5 Hz, 2H), 7.40 (t, J = 7.6 Hz, 5H), 7.37 (t, J = 7.5 Hz, 1H), 7.27–7.21 (m, 1H), 7.19 (d, J = 9.5 Hz, 2H), 6.99 (t, J = 7.8 Hz, 1H), 5.42 (s, 2H), 4.64 (s, 2H), 3.84 (t, J = 5.7 Hz, 2H), 2.96 (t, J = 5.2 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 155.28, 145.22, 142.92, 141.36, 138.53, 136.23, 134.12, 133.39, 129.91, 128.64, 128.24, 127.27, 126.11, 121.88, 121.89, 121.77, 120.21, 119.88, 114.78, 111.48, 52.19, 48.83, 41.67, 24.83. EI-MS m/z 510.15 (M + H)+; EI-MS m/z 510.15 (M + H)+; anal. calcd for C28H24ClN7O: (%) C, 65.94; H, 4.74; N, 19.23; found: C, 65.16; H, 4.75; N, 19.89.
1-((1-(2-Fluorophenyl)-1H-1,2,3-triazol-4-yl)methyl)-N,3-diphenyl-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxamide (7l).
White solid (91%); m.p. 156–158 °C; IR (KBr) νmax/cm−1 3576, 3031, 2925, 1665, 1421, 1330, 1050. 1H NMR (400 MHz, DMSO-d6) δ 8.76 (s, 1H), 8.68 (s, 1H), 7.83 (t, J = 7.8 Hz, 1H), 7.68 (d, J = 7.6 Hz, 2H), 7.64–7.51 (m, 2H), 7.51–7.38 (m, 5H), 7.33 (t, J = 7.4 Hz, 1H), 7.24 (t, J = 7.7 Hz, 2H), 6.95 (t, J = 7.4 Hz, 1H), 5.51 (s, 2H), 4.72 (s, 2H), 3.83 (t, J = 6.1 Hz, 2H), 2.98 (t, J = 6.3 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 155.67, 153.06, 145.56, 143.95, 140.85, 139.19, 133.96, 131.82, 129.16, 128.75, 127.80, 126.51, 125.99, 125.74, 125.13, 122.38, 120.48, 117.66, 117.47, 112.00, 44.20, 42.16, 41.16, 22.34. EI-MS m/z 494.20 (M + H)+; anal. calcd for C28H24FN7O: (%) C, 68.14; H, 4.90; N, 19.87; found: C, 68.16; H, 4.91; N, 19.89.
1-((1-(2-Bromophenyl)-1H-1,2,3-triazol-4-yl)methyl)-N,3-diphenyl-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxamide (7m).
White solid (90%); m.p. 119–121 °C; IR (KBr) νmax/cm−1 3490, 3021, 2843, 1650, 1410, 1340, 570. 1H NMR (400 MHz, DMSO-d6) δ 8.75 (s, 1H), 8.71 (s, 1H), 7.81(d, J = 9.1 Hz, 2H), 7.67 (d, J = 8.4 Hz, 2H), 7.44 (t, J = 7.9 Hz, 5H), 7.33 (t, J = 7.4 Hz, 1H), 7.26–7.20 (m, 1H), 7.11 (d, J = 9.1 Hz, 2H), 6.94 (t, J = 7.8 Hz, 1H), 5.46 (s, 2H), 4.73 (s, 2H), 3.82 (t, J = 5.6 Hz, 2H), 2.97 (t, J = 5.4 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 155.21, 145.02, 144.23, 143.72, 140.36, 139.21, 138.63, 134.24, 133.49, 133.67, 129.91, 128.64, 128.24, 127.27, 126.01, 121.88, 121.86, 121.77, 120.08, 118.12, 48.12, 44.73, 41.67, 22.83. EI-MS m/z 554.12 (M + H)+, 556.10 (M + H)+2; anal. calcd for C28H24BrN7O: (%) C, 60.66; H, 4.36; N, 17.68; found: C, 60.68; H, 4.37; N, 17.69.
1-((1-(2-Iodophenyl)-1H-1,2,3-triazol-4-yl)methyl)-N,3-diphenyl-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxamide (7n).
White solid (87%); m.p. 188–120 °C; (KBr) νmax/cm−1 3512, 3025, 2843, 1650, 1408, 1344, 500. 1H NMR (400 MHz, DMSO-d6) δ 8.79 (s, 1H), 8.72 (s, 1H), 7.99 (d, J = 9.2 Hz, 2H), 7.76 (d, J = 8.5 Hz, 2H), 7.46 (t, J = 7.8 Hz, 5H), 7.35 (t, J = 7.5 Hz, 2H), 7.19 (d, J = 9.3 Hz, 2H), 6.98 (t, J = 7.8 Hz, 1H), 5.47 (s, 2H), 4.70 (s, 2H), 3.82 (t, J = 5.6 Hz, 2H), 2.97 (t, J = 5.4 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 155.46, 145.02, 144.23, 143.72, 140.36, 138.63, 130.23, 129.91, 128.64, 128.24, 127.61, 127.27, 126.01, 121.88, 121.86, 121.77, 119.98, 116.78, 115.48, 95.15, 48.23, 44.83, 41.67, 22.93. EI-MS m/z 602.12 (M + H)+; anal. calcd for C28H24IN7O: (%) C, 55.92; H, 4.03; N, 16.30; found: C, 55.94; H, 4.31; N, 16.32.
1-((1-(2-Nitrophenyl)-1H-1,2,3-triazol-4-yl)methyl)-N,3-diphenyl-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4h)-carboxamide (7o).
Off-white solid (86%); m.p. 118–120 °C; IR (KBr) νmax/cm−1 3505, 3027, 2846, 1655, 1525, 1410, 1360, 1025. 1H NMR (400 MHz, DMSO-d6) δ 9.07 (s, 1H), 8.75 (s, 1H), 8.49–8.41 (m, 2H), 8.28–8.20 (m, 2H), 7.73–7.62 (m, 2H), 7.50–7.39 (m, 4H), 7.40–7.27 (m, 1H), 7.28–7.18 (m, 2H), 6.94 (tt, J = 7.3, 1.2 Hz, 1H), 5.47 (s, 2H), 4.69 (s, 2H), 3.82 (t, J = 5.6 Hz, 2H), 2.97 (t, J = 5.4 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 155.18, 147.87, 147.15, 145.12, 143.72, 141.36, 138.63, 135.23, 134.56, 133.49, 129.91, 128.64, 128.24, 127.27, 126.01, 121.88, 121.86, 121.77, 120.98, 112.03, 44.29, 42.15, 41.13, 22.28. EI-MS m/z 521.19 (M + H)+; anal. calcd for C28H24N8O3: (%) C, 64.61; H, 4.65; N, 21.53; found: C, 64.63; H, 4.66; N, 21.55.
1-((1-(3-Chlorophenyl)-1H-1,2,3-triazol-4-yl)methyl)-N,3-diphenyl-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxamide (7p).
White solid (86%); m.p. 147–149 °C; IR (KBr) νmax/cm−1 3497, 3029, 2847, 1650, 1444, 1306, 1032, 753. 1H NMR (400 MHz, DMSO-d6) δ 8.80 (s, 1H), 8.75 (s, 1H), 7.89 (s, 1H), 7.81 (d, J = 9.2 Hz, 2H), 7.69 (d, J = 8.5 Hz, 2H), 7.40 (t, J = 7.6 Hz, 4H), 7.37 (t, J = 7.5 Hz, 1H), 7.27–7.21 (m, 1H), 7.19 (d, J = 9.5 Hz, 2H), 6.93 (t, J = 7.8 Hz, 1H), 5.42 (s, 2H), 4.64 (s, 2H), 3.84 (t, J = 5.7 Hz, 2H), 2.96 (t, J = 5.2 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 155.38, 145.42, 142.82, 141.39, 138.55, 136.23, 134.32, 133.30, 129.96, 128.74, 128.29, 127.20, 126.51, 121.98, 121.80, 121.75, 121.34, 120.31, 119.78, 118.78, 45.29, 42.80, 41.66, 22.73. EI-MS m/z 510.15 (M + H)+; anal. calcd for C28H24ClN7O: (%) C, 65.94; H, 4.74; N, 19.23; found: C, 65.95; H, 4.76; N, 19.24.
1-((1-(3-Methoxyphenyl)-1H-1,2,3-triazol-4-yl)methyl)-N,3-diphenyl-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxamide (7q).
Brown solid (79%); m.p. 135–136 °C; IR (KBr) νmax/cm−1 3502, 3023, 2875, 1657, 1454, 1350, 1090. 1H NMR (400 MHz, DMSO-d6) δ 8.78 (s, 1H), 8.75 (s, 1H), 7.80 (d, J = 9.1 Hz, 2H), 7.67 (d, J = 8.4 Hz, 2H), 7.47–7.41 (m, 4H), 7.34 (t, J = 7.4 Hz, 1H), 7.26–7.20 (m, 2H), 7.11 (d, J = 9.1 Hz, 2H), 6.94 (t, J = 7.8 Hz, 1H), 5.48 (s, 2H), 4.71 (s, 2H), 3.83 (t, J = 5.7 Hz, 2H), 2.96 (t, J = 5.5 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 156.25, 154.08, 145.22, 143.87, 141.36, 138.63, 133.49, 129.91, 128.64, 128.24, 127.27, 126.01, 125.32, 121.88, 121.76, 121.77, 120.14, 119.98, 114.78, 111.42, 53.51, 46.12, 44.73, 41.87, 22.21. EI-MS m/z 506.20 (M + H)+; anal. calcd for C29H27N7O2: (%) C, 68.89; H, 5.38; N, 19.39; found: C, 68.91; H, 5.39; N, 19.41.
1-((1-(2,4-Dichlorophenyl)-1H-1,2,3-triazol-4-yl)methyl)-N,3-diphenyl-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxamide (7r).
White solid (82%); m.p. 139–141 °C; IR (KBr) νmax/cm−1 3505, 3029, 2840, 1654, 1415, 1342, 1070, 740. 1H NMR (400 MHz, DMSO-d6) δ 8.77 (s, 1H), 8.73 (s, 1H), 7.80 (d, J = 9.2 Hz, 2H), 7.66 (d, J = 8.5 Hz, 2H), 7.69 (s, 1H), 7.62 (d, J = 9.5 Hz, 2H), 7.50 (t, J = 7.6 Hz, 2H), 7.41 (t, J = 7.5 Hz, 3H), 6.91 (t, J = 7.8 Hz, 1H), 5.45 (s, 2H), 4.69 (s, 2H), 3.84 (t, J = 5.7 Hz, 2H), 2.96 (t, J = 5.2 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 155.31, 145.53, 144.16, 142.72, 141.48, 139.56, 136.29, 135.74, 133.40, 129.99, 129.24, 128.44, 127.25, 126.21, 123.58, 123.49, 121.57, 120.21, 119.88, 114.78, 46.21, 44.63, 41.67, 22.23. EI-MS m/z 544.13 (M + H)+; anal. calcd for C28H23Cl2N7O: (%) C, 61.77; H, 4.26; N, 18.01; found: C, 61.79; H, 4.27; N, 18.03.
1-((1-(3,4-Dichlorophenyl)-1H-1,2,3-triazol-4-yl)methyl)-N,3-diphenyl-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxamide (7s).
Off-white solid (83%); m.p. 228–230 °C; IR (KBr) νmax/cm−1 3503, 3029, 2842, 1654, 1410, 1345, 1066, 745. 1H NMR (400 MHz, DMSO-d6) δ 8.78 (s, 1H), 8.74 (s, 1H), 7.81 (d, J = 9.2 Hz, 2H), 7.77 (s, 1H), 7.66 (d, J = 8.6 Hz, 2H), 7.62 (d, J = 9.4 Hz, 2H), 7.54 (t, J = 7.5 Hz, 2H), 7.44 (t, J = 7.4 Hz, 3H), 7.01 (t, J = 7.9 Hz, 1H), 5.43 (s, 2H), 4.63 (s, 2H), 3.82 (t, J = 5.7 Hz, 2H), 2.98 (t, J = 5.4 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 155.18, 146.41, 143.80, 141.43, 138.26, 136.49, 134.31, 133.41, 129.90, 128.52, 128.41, 127.43, 125.32, 121.78, 121.59, 121.47, 120.20, 119.82, 115.78, 111.78, 46.65, 46.24, 40.97, 22.73. EI-MS m/z 544.20 (M + H)+; anal. calcd for C28H23Cl2N7O: (%) C, 61.77; H, 4.26; N, 18.01; found: C, 61.79; H, 4.27; N, 18.03.
1-((1-(3,5-Dichlorophenyl)-1H-1,2,3-triazol-4-yl)methyl)-N,3-diphenyl-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxamide (7t).
White solid (85%); m.p. 139–141 °C; IR (KBr) νmax/cm−1 3505, 3027, 2846, 1650, 1411, 1345, 1030, 752. 1H NMR (400 MHz, DMSO-d6) δ 8.98 (s, 1H), 8.74 (s, 1H), 8.08 (d, J = 1.8 Hz, 2H), 7.78–7.64 (m, 3H), 7.45 (t, J = 7.4 Hz, 4H), 7.38–7.16 (m, 3H), 6.94 (t, J = 7.3 Hz, 1H), 5.49 (s, 2H), 4.71 (s, 2H), 3.83 (t, J = 5.8 Hz, 2H), 2.95 (t, J = 5.9 Hz, 2H13C NMR (101 MHz, DMSO-d6) δ 155.64, 145.60, 144.79, 140.84, 139.15, 138.57, 135.63, 133.93, 129.13, 128.73, 127.80, 126.50, 122.83, 122.38, 120.47, 119.25, 112.02, 44.35, 42.16, 41.12, 22.26. EI-MS m/z 544.13 (M + H)+; anal. calcd for C28H23Cl2N7O: (%) C, 61.77; H, 4.26; N, 18.01; found: C, 61.79; H, 4.27; N, 18.03.
1-((1-(3-Chloro-4-fluorophenyl)-1H-1,2,3-triazol-4-yl) methyl)-N,3-diphenyl-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxamide (7u).
White solid (77%); m.p. 240–242 °C; IR (KBr) νmax/cm−1 3505, 3022, 2840, 1654, 1410, 1345, 1178, 1020, 780. 1H NMR (400 MHz, DMSO-d6) δ 8.90 (s, 1H), 8.73 (s, 1H), 8.23 (dd, J = 6.4, 2.7 Hz, 1H), 8.01–7.92 (m, 1H), 7.71–7.62 (m, 3H), 7.49–7.39 (m, 4H), 7.38–7.28 (m, 1H), 7.28–7.18 (m, 2H), 6.98–6.89 (m, 1H), 5.48 (s, 2H), 4.70 (s, 2H), 3.82 (t, J = 5.8 Hz, 2H), 2.95 (t, J = 5.8 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 155.28, 145.09, 144.14, 140.34, 138.63, 133.46, 128.62, 128.22, 127.27, 126.00, 122.44, 122.30, 121.87, 120.95, 120.77, 120.58, 119.99, 118.13, 117.90, 111.51, 43.88, 41.67, 40.64, 21.79. EI-MS m/z 528.17 (M + H)+; anal. calcd for C28H23ClFN7O: (%) C, 63.70; H, 4.39; N, 18.57; found: C, 63.72; H, 4.40; N, 18.59.
1-((1-(3,4-Difluorophenyl)-1H-1,2,3-triazol-4-yl)methyl)-N,3-diphenyl-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxamide (7v).
Off-white solid (80%); m.p. 226–228 °C; IR (KBr) νmax/cm−1 3501, 3021, 2846, 1659, 1412, 1335, 1130, 1060. 1H NMR (400 MHz, DMSO-d6) δ 8.91 (s, 1H), 8.73 (s, 1H), 8.24 (dd, J = 6.5, 2.6 Hz, 1H), 8.01–7.91 (m, 1H), 7.70–7.62 (m, 3H), 7.46–7.40 (m, 4H), 7.35 (t, J = 7.4 Hz, 1H), 7.20 (t, J = 7.9 Hz, 2H), 7.04 (t, J = 7.3 Hz, 1H), 5.48 (s, 2H), 4.70 (s, 2H), 3.82 (t, J = 5.6 Hz, 2H), 2.95 (t, J = 5.4 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 155.21, 149.84, 149.21, 145.02, 143.72, 141.78, 140.36, 138.63, 133.49, 130.12, 129.91, 128.64, 128.24, 127.27, 126.01, 124.12, 121.88, 121.86, 121.77, 119.98, 45.22, 42.13, 40.81, 23.53. EI-MS m/z 528.17 (M + H)+; anal. calcd for C28H23F2N7O: (%) C, 65.74; H, 4.53; N, 19.17; found: C, 65.76; H, 4.54; N, 19.19.
1-((1-(3,4-Dimethoxyphenyl)-1H-1,2,3-triazol-4-yl)methyl)-N,3-diphenyl-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxamide (7w).
Brown solid (88%); m.p. 207–209 °C; IR (KBr) νmax/cm−1 3500, 3020, 2845, 1654, 1410, 1345, 1080. 1H NMR (400 MHz, DMSO-d6) δ 8.78 (s, 1H), 8.72 (s, 1H), 7.82 (d, J = 9.1 Hz, 2H), 7.67 (d, J = 8.4 Hz, 2H), 7.44 (t, J = 7.9 Hz, 4H), 7.33 (t, J = 7.4 Hz, 1H), 7.26 (m, 1H), 7.11 (d, J = 9.1 Hz, 2H), 6.94 (s, 1H), 5.46 (s, 2H), 4.72 (s, 2H), 3.84 (s, 6H), 3.82 (d, J = 7.2 Hz, 2H), 2.97 (t, J = 5.4 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.31, 153.21, 148.74, 139.76, 136.99, 135.44, 129.89, 129.54, 128.44, 127.45, 126.81, 123.98, 123.69, 121.77, 120.81, 119.88, 118.78, 114.25, 109.13, 101.21, 55.86, 46.90, 44.63, 40.32, 22.09. EI-MS m/z 536.20 (M + H)+; anal. calcd for C30H29N7O3: (%) C, 67.27; H, 5.47; N, 18.31; found: C, 67.29; H, 5.48; N, 18.33.
1-((1-(4-Bromo-3-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-4-yl)methyl)-N,3-diphenyl-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxamide (7x).
White solid (79%); m.p. 221–223 °C; IR (KBr) νmax/cm−1 3500, 3022, 2854, 1660, 1405, 1343, 1119, 1070. 1H NMR (400 MHz, DMSO-d6) δ 8.91 (s, 1H), 8.73 (s, 1H), 8.24 (dd, J = 6.5, 2.6 Hz, 1H), 8.01–7.82 (m, 1H), 7.69–7.53 (m, 3H), 7.46–7.41 (m, 4H), 7.36 (t, J = 7.4 Hz, 1H), 7.21 (t, J = 7.9 Hz, 2H), 7.04 (t, J = 7.3 Hz, 1H), 5.48 (s, 2H), 4.70 (s, 2H)), 3.82 (t, J = 5.6 Hz, 2H), 2.95 (t, J = 5.4 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 155.18, 145.02, 143.72, 140.36, 139.10, 138.63, 136.89, 136.12, 133.49, 130.23, 129.91, 128.64, 128.24, 127.27, 126.01, 121.88, 121.86, 121.77, 120.98, 120.01, 114.78, 48.01, 46.32, 44.83, 22.19. EI-MS m/z 622.20, 624.15 (M + 1, M + 2)+; anal. calcd for C30H23BrF3N7O: (%) C, 55.96; H, 3.72; N, 15.75; found: C, 55.98; H, 3.74; N, 15.76.
1-((1-(3,4-Dimethylphenyl)-1H-1,2,3-triazol-4-yl)methyl)-N,3-diphenyl-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxamide (7y).
Off-white solid (84%); m.p. 221–223 °C; IR (KBr) νmax/cm−1 3505, 3022, 2844, 1658, 1412, 1345, 1076. 1H NMR (400 MHz, DMSO-d6) δ 8.82 (s, 1H), 8.79 (s, 1H), 7.78 (d, J = 8.6 Hz, 2H), 7.66 (d, J = 7.3 Hz, 2H), 7.47–7.42 (m, 3H), 7.44 (s, 1H), 7.41–7.31 (m, 2H), 7.27–7.20 (m, 2H), 6.93 (t, J = 6.9 Hz, 1H), 5.47 (s, 2H), 4.72 (s, 2H), 3.82 (t, J = 5.7 Hz, 2H), 2.97 (t, J = 5.5 Hz, 2H), 2.36 (s, 6H). 13C NMR (101 MHz, DMSO-d6) δ 159.25, 155.18, 145.02, 143.72, 140.36, 138.63, 136.89, 136.12, 133.49, 129.91, 128.64, 128.24, 127.27, 126.01, 121.88, 121.86, 121.77, 119.98, 114.78, 111.48, 45.51, 44.02, 41.57, 22.23, 20.92. EI-MS m/z 504.25 (M + H)+; anal. calcd for C30H29N7O: (%) C, 71.55; H, 5.80; N, 19.47; found: C, 71.56; H, 5.82; N, 19.49.
1-((1-(Benzo[d][1,3]dioxol-5-yl)-1H-1,2,3-triazol-4-yl)methyl)-N,3-diphenyl-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxamide (7z).
Brown solid (78%); m.p. 168–170 °C; IR (KBr) νmax/cm−1 3504, 3025, 2840, 1655, 1416, 1345, 1080. 1H NMR (400 MHz, DMSO-d6) δ 8.77 (s, 1H), 8.72 (s, 1H), 7.81 (d, J = 9.1 Hz, 2H), 7.67 (d, J = 8.4 Hz, 2H), 7.44 (t, J = 7.9 Hz, 4H), 7.33 (t, J = 7.4 Hz, 1H), 7.26 (m, 1H), 7.11 (d, J = 9.1 Hz, 2H), 6.94 (s, 1H), 5.94 (s, 2H), 5.46 (s, 2H), 4.70 (s, 2H), 3.82 (d, J = 7.1 Hz, 2H), 2.97 (t, J = 5.4 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.31, 148.74, 147.53, 139.76, 136.99, 135.44, 133.49, 129.89, 129.54, 128.44, 127.45, 126.81, 123.98, 123.69, 121.77, 120.81, 119.88, 114.78, 109.98, 101.21, 52.21, 48.63, 41.67, 24.73. EI-MS m/z 520.20 (M + H)+; anal. calcd for C29H25N7O3: (%) C, 67.05; H, 4.85; N, 18.87; found: C, 67.06; H, 4.87; N, 18.89.
Biological activity
MTB PS screening.
The MTB panC gene (Rv3602c) encoding the pantothenate synthetase was cloned and introduced into BL21 (DE3) cells and the protein was expressed.7,15 For the assay, to each well of a 96-well plate, 60 μL of PS reaction mixture containing 0.4 mM NADH, 10 mM MgCl2, 5 mM β-alanine, 5 mM pantoic acid, 1 mM potassium phosphoenolpyruvate,10 mM ATP, and 20 μL of enzyme mixture consisting of 18 units per mL each of chicken muscle myokinase, rabbit muscle lactate dehydrogenase and rabbit muscle pyruvate kinase, diluted in 100 mM HEPES buffer, were added. The reaction mixture and the enzyme mixture were added to the plate to a final volume of 100 μL with 100 mM HEPES buffer (pH 7.8). The concentration of the enzyme was determined based on the range-finding experiments by varying the concentration of enzymes. Compound solutions were then added to the plates (from 50 μM to lower concentration) and the reaction was started with the addition of 10 μL of 4.32 pM MTB PS, diluted in buffer. The test plate was immediately moved to a microplate reader and the reduction of NADH was quantified at 340 nm. The reaction elements except MTB PS were mixed in the well and the background reaction was calculated; MTB PS was then added and the reaction kinetics was monitored. Reactions were carried out at 37 °C in a heat controlled Perkin Elmer Victor X3 Spectrophotometer. % inhibition was calculated using the following formula: 100 × [(1 − compound rate − background rate)/(full reaction rate − background rate)].7,15
In vitro MTB screening.
The antimycobacterial activities of compounds 7a–z were evaluated against the MTB H37Rv strain and two “wild” strains extracted from tuberculosis patients: one strain is Spec. 210 resistant to PAS, INH, ETB and RMP and the other strain is Spec. 192 fully sensitive to the administered anti-TB agents. In vitro anti-TB activity was studied by a classical test-tube method of successive dilution in Youmans' modification of the Proskauer and Beck liquid medium containing 10% bovine serum.36,37 Bacterial suspensions were prepared from 14 day old cultures of gradually growing strains. Solutions of compounds in DMSO were tested. Stock solutions contained 10 mg of compounds in 1 mL. Dilutions (in geometric progression) were prepared in Youmans' medium.37 The medium without compounds and containing INH as reference drug was used for comparison. Incubation was performed at 37 °C. The MIC values were determined as MIC inhibiting the growth of tested TB strains in relation to the probe with no tested compound. The influence of the compound on the growth of bacteria at concentrations of 3.12, 6.25, 12.5, 25, 50 and 100 μg mL−1 was evaluated.
In vitro cytotoxicity screening.
Compounds 7b, 7c and 7d were further tested for toxicity in a RAW 264.7 cell line at the concentration of 50 μM. After 72 h of exposure, viability was evaluated on the basis of cellular translation of MTT into a formazan product by the Promega Cell Titer 96 nonradioactive cell proliferation assay.38,39
Docking study.
Docking studies of the title compounds (7a–z) were performed using Glide 5.9 (Extra Precision) running on maestro version 9.4, in order to investigate their in silico binding affinity as well as their binding pattern with the enzyme pantothenate synthetase.40 The enzyme used for the docking study was retrieved from the RCSB Protein Data Bank (PDB ID: 3IUB) in complex with the co-crystallised ligand (indole-2-carboxamide derivative). The selected protein consists of two chains of asymmetric units (A and B), both consisting of co-crystallized ligand. In the current study, unit A was separated and used further for docking studies. The protein preparation wizard of the Schrödinger suite was used for the preparation of selected protein. The protein was pre-processed separately by deleting the substrate co-factor as well as the crystallographically observed water molecules (water without H bonds), followed by optimization of hydrogen bonds. After assigning charge and protonation position, finally energy was minimized with a root mean square deviation (RMSD) value of 0.30 Å using the optimized potentials for liquid simulations-2005 (OPLS-2005) force field.41 Finally the energy minimized protein and the co-crystallized ligand were used to build energy grids using the default value of protein atom scaling (1.0 Å) within a cubic box of 14 Å dimensions, centered on the centroid of the X-ray ligand pose.42 The structures of 7a–z were drawn using ChemSketch and converted to 3D structures with the help of the 3D optimization tool. Using the LigPrep 2.6 module, the drawn ligands were geometry optimized; partial atomic charges were computed using the OPLS-2005 force field.43 Finally, the prepared ligands were docked with the prepared protein using the Glide 5.9 module, in extra precision mode (XP). The leading docked pose (with the lowest Glide score value) found from Glide was analyzed. The RMSD value was calculated by comparing the experimental binding mode of the co-crystallized ligand (as in the X-ray pose) and its re-docked pose to ensure accuracy and reliability of the docking procedure.
Acknowledgements
The financial assistance provided by the Department of Science and Technology (DST) (F.No.SB/S1/OC-60/2013) and DST FIST grant (SR/FST/CSI-240/2012), Government of India, New Delhi, is gratefully acknowledged. A. S. thanks DST for providing a junior research fellowship and S. S. thanks CSIR for providing a junior research fellowship.
Notes and references
- World Health Organization (WHO) – Global Tuberculosis Control report 2015, http://www.who.int/tb/publications/global_report/gtbr15_main_text.pdf.
- P. M. S. Chauhan, N. Sunduru and M. Sharma, Future Med. Chem., 2010, 2, 1469 CrossRef CAS PubMed
 .
.
- D. L. Combs, R. J. O'Brien and L. J. Geiter, Ann. Intern. Med., 1990, 112, 397 CrossRef CAS PubMed .
- M. A. Stratton and M. T. Reed, Clin. Pharmacol., 1986, 5, 977 CAS .
- D. A. Mitchison, Tubercle, 1985, 66, 219 CrossRef CAS PubMed .
- M. Yan and S. Ma, ChemMedChem, 2012, 7, 2063 CrossRef CAS PubMed .
- R. Zheng and J. S. Blanchard, Biochemistry, 2001, 40, 12904 CrossRef CAS PubMed .
- S. Velaparthi, M. Brunsteiner, R. Uddin, B. Wan, S. G. Franzblau and P. A. Petukhov, J. Med. Chem., 2008, 51, 1999 CrossRef CAS PubMed .
- S. Wang and D. Eisenberg, Protein Sci., 2003, 12, 1097 CrossRef CAS PubMed .
- Y. Yang, P. Gao, Y. Liu, X. Ji, M. Gan, Y. Guan, X. Hao, Z. Li and C. Xiao, Bioorg. Med. Chem. Lett., 2011, 21, 3943 CrossRef CAS PubMed .
- G. Samala, P. B. Devi, R. Nallangi, J. P. Sridevi, P. Yogeeswari and D. Sriram, Eur. J. Med. Chem., 2013, 69, 356 CrossRef CAS PubMed .
- G. Samala, P. B. Devi, R. Nallangi, J. P. Sridevi, P. Yogeeswari and D. Sriram, Bioorg. Med. Chem., 2014, 22, 1938 CrossRef CAS PubMed .
- E. L. White, K. Southworth, L. Ross, S. Cooley, R. B. Gill, M. I. Sosa, A. Manouvakhova, L. Rasmussen, C. Goulding, D. Eisenberg and T. M. Fletcher, J. Biomol. Screening, 2007, 12, 100 CrossRef CAS PubMed .
- G. Samala, R. Nallangi, P. B. Devi, S. Saxena, R. Yadav, J. P. Sridevi, P. Yogeeswari and D. Sriram, Bioorg. Med. Chem., 2014, 22, 4223 CrossRef CAS PubMed .
- G. Samala, P. B. Devi, S. Saxena, N. Meda, P. Yogeeswari and D. Sriram, Bioorg. Med. Chem., 2016, 24, 1298 CrossRef CAS PubMed .
- P. P. Deohate, J. P. Deohate and B. N. Berad, Asian J. Chem., 2004, 16, 255 CAS .
- K. L. Kees, J. J. Fitzgerald, K. E. Steiner, J. F. Mattes, B. Mihan, T. Tosi, D. Moondoro and M. L. McCaleb, J. Med. Chem., 1996, 39, 3920 CrossRef CAS PubMed .
-
(a) P. Diana, A. Carbone, P. Barraja, A. Martorana, O. Gia, L. DallaVia and G. Cirrincione, Bioorg. Med. Chem. Lett., 2007, 17, 6134 CrossRef CAS PubMed ;
(b) H. J. Park, K. Lee, S. J. Park, B. Ahn, J. C. Lee, H. Y. Cho and K. I. Lee, Bioorg. Med. Chem. Lett., 2005, 15, 3307 CrossRef CAS PubMed .
- R. Aggarwal, S. P. Singh, V. Kumar and P. Tyagi, Bioorg. Med. Chem., 2006, 14, 1785 CrossRef CAS PubMed .
-
(a) N. Harikrishan, A. M. Isloor, K. Ananda, A. Obaid and H. K. Fun, New J. Chem., 2016, 40, 73 RSC ;
(b) N. K. Piyush, P. S. Shailesh and K. R. Dipak, New J. Chem., 2014, 38, 2902 RSC ;
(c) R. Manikannan, R. Venkatesan, S. Muthusubramanian, P. Yogeeswari and D. Sriram, Bioorg. Med. Chem. Lett., 2010, 20, 6920 CrossRef CAS PubMed .
- D. Raffa, B. Maggio, M. V. Raimondi, S. Cascioferro, F. Plescia, G. Cancemi and G. Daidone, Eur. J. Med. Chem., 2015, 97, 732 CrossRef CAS PubMed .
- M. G. Mamolo, D. Zampieri, V. Falagiani, L. Vio and E. Banfi, Farmaco, 2001, 56, 593 CrossRef CAS PubMed .
- R. B. Pathak, P. T. Chovatia and H. H. Parekh, Bioorg. Med. Chem. Lett., 2012, 22, 5129 CrossRef CAS PubMed .
- P. T. Chovatia, J. D. Akabari, P. K. Kachhadia, P. D. Zalavadia and H. S. Joshi, J. Serb. Chem. Soc., 2007, 71, 713 CrossRef .
- R. C. Khunt, V. M. Khedkar, R. S. Chawda, N. A. Chauhan, A. R. Parikh and E. C. Coutinho, Bioorg. Med. Chem. Lett., 2012, 22, 666 CrossRef CAS PubMed .
- A. Suresh, N. Suresh, S. Misra, M. M. Krishna Kumar and K. V. G. Chandra Sekhar, ChemistrySelect, 2016, 1, 1705 CrossRef CAS .
-
(a) M. Bhat, G. K. Nagarjuna, R. Kayarmar, S. K. Peethamber and R. Mohammed Shafeeulla, RSC Adv., 2016, 6, 59375 RSC ;
(b) H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128 CrossRef CAS PubMed .
- D. Kumar, G. B. Khare, S. Kidwai, A. K. Tyagi, R. Singh and D. S. Rawat, Eur. J. Med. Chem., 2014, 81, 301 CrossRef CAS PubMed .
- S. R. Patpi, L. Pulipati, P. Yogeeswari, D. Sriram, N. Jain, B. Sridhar, R. Murthy, T. Anjana Devi, S. V. Kalivendi and S. Kantevari, J. Med. Chem., 2012, 55, 3911 CrossRef CAS PubMed .
- N. Boechat, V. F. Ferreira, S. B. Ferreira, M. L. G. Ferreira, F. C. da Silva, M. M. Bastos, M. S. Costa, M. S. Lourenço, A. C. Pinto, A. U. Krettli, A. C. Aguiar, B. M. Teixeira, N. V. da Silva, P. R. C. Martins, F. F. M. Bezerra, A. S. Camilo, G. P. da Silva and C. C. P. Costa, J. Med. Chem., 2011, 54, 5988 CrossRef CAS PubMed .
- S. Kim, S. Cho, T. Oh and P. Kim, Bioorg. Med. Chem. Lett., 2012, 22, 6844 CrossRef CAS PubMed .
- P. Shanmugavelan, S. Nagarajan, M. Sathishkumar, A. Ponnuswamy, P. Yogeeswari and D. Sriram, Bioorg. Med. Chem. Lett., 2011, 21, 7273 CrossRef CAS PubMed .
- H. N. Nagesh, K. Mahalakshmi Naidu, D. Harika Rao, J. P. Sridevi, D. Sriram, P. Yogeeswari and K. V. G. Chandra Sekhar, Bioorg. Med. Chem. Lett., 2013, 23, 6805 CrossRef CAS PubMed .
- K. Mahalakshmi Naidu, S. Srinivasarao, N. Agnieszka, A. Ewa, M. M. Krishna Kumar and K. V. G. Chandra Sekhar, Bioorg. Med. Chem. Lett., 2016, 26, 2245 CrossRef PubMed .
- X. M. Ye, A. W. Konradi, J. Smith, D. L. Aubele, A. W. Garofalo, J. Marugg, M. L. Neitzel, C. M. Semko, H. L. Sham, M. Sun, A. P. Truong, J. Wu, H. Zhang, E. Goldbach, J. M. Sauer, E. F. Brigham, M. Bova and G. S. Basi, Bioorg. Med. Chem. Lett., 2010, 20, 2195 CrossRef CAS PubMed .
- G. P. Youmans, Am. Rev. Tuberc., 1947, 56, 376 CAS .
- G. P. Youmans and A. S. Youmans, J. Bacteriol., 1949, 58, 247 CAS .
- P. B. Devi, G. Samala, J. P. Sridevi, S. Saxena, M. Alvala, E. G. Salina, D. Sriram and P. Yogeeswari, ChemMedChem, 2014, 9, 2538 CrossRef CAS PubMed .
- D. Gerlier and N. Thomasset, Immunol. Methods, 1986, 94, 57 CrossRef CAS PubMed .
- Glide, Schrödinger, LLC, New York, 2013, version 5.9.
- W. L. Jorgensen, D. S. Maxwell and R. J. Tirado, J. Am. Chem. Soc., 1996, 118, 11225 CrossRef CAS .
- S. Chander, A. Penta and S. Murugesan, J. Pharm. Res., 2014, 8, 552 CAS .
- Lig-Prep, Schrödinger, LLC, New York, 2013, version 2.6.
Footnote |
| † Electronic supplementary information (ESI) available: Photophysical data, copies of NMR spectra for products and single crystal X-ray data. CCDC 1500428. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6nj02671k |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a
*a