
Metal catalyst free cyclization of 3-alkynyl substituted 2-(indol-3-yl)quinoxalines in TFA alone: a new synthesis of indolophenazines†
K. Shiva
Kumar
*a,
Boyapally
Bhaskar
a,
Meesa Siddi
Ramulu
a,
N. Praveen
Kumar
a,
Mohd Ashraf
Ashfaq
b and
Manojit
Pal
*b
aDepartment of Chemistry, Osmania University, Hyderabad-500 007, India. E-mail: shivakumarkota@yahoo.co.in; Tel: +91 40 27682337
bDr Reddy's Institute of Life Sciences, Hyderabad Central University, Campus, Gachibowli, Hyderabad-500 046, India. E-mail: manojitpal@rediffmail.com; Tel: +91 40 6657 1500
First published on 22nd November 2016
Abstract
TFA alone was found to be remarkably effective for the intramolecular hydroarylation (IMHA) of alkynes when employed as a solvent in the cyclization of 3-alkynyl substituted 2-(indol-3-yl)quinoxalines. This simple and metal free cyclization method afforded a range of indolophenazines as new and potential cytotoxic agents. The use of excess TFA was found to be crucial for the success of this reaction.
The phenazine framework (A, Fig. 1) has been found to be an integral part of many bioactive compounds.1–4 For example, indolophenazine (B, Fig. 1) has been identified as a novel inhibitor (IC50 ∼ 3 μM) of human NAD(P)H quinoneoxido reductase (NQO1) in vitro from a series of NCI (National Cancer Institute) compounds.5 The compound also showed good interactions with NQO1 in silico. Notably, NQO1 being over-expressed in many solid tumours compared to the surrounding normal tissue has attracted attention as a potential target for selective anti-cancer drug development. Therefore inhibitors of NQO1 are useful as potential anticancer agents.6 However, to date B is the only indolophenazine derivative that has been assessed for pharmacological activities and no further studies around this class of compounds have been reported perhaps due to the lack of easy and straightforward access to the required analogues. All these observations prompted us not only to evaluate the in vitro anticancer properties of a series of indolophenazine derivatives (C, Fig. 1) related to B possessing a similar substitution pattern but also develop a convenient synthetic method for their easy access for further study.
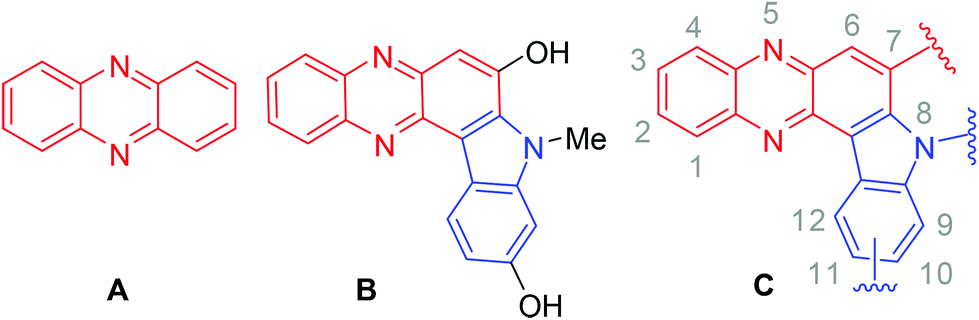 | ||
| Fig. 1 Phenazine (A), known indolophenazine (B) and its newly designed analogues. | ||
The synthesis of indolophenazine is not common in the literature. While acid catalyzed unusual Friedlander reactions leading to indolophenazine derivatives have been reported,7 only two compounds were prepared in low yields and the methodology did not appear to be a general and effective one for the preparation of other analogues. Recently, the transition-metal catalyzed intramolecular hydroarylation (IMHA) of alkyne has emerged as a powerful tool for the atom-economical construction of carbocyclic and heterocyclic structures.8,9 The use of this strategy for the synthesis of phenazine derivatives has also been reported10 (Scheme 1a), one of which involved a Pd-catalyzed intramolecular Fujiwara-hydroarylation10a and the other one involved ICl-promoted intramolecular cyclization.10b However, this strategy has not been explored for the synthesis of indolophenazine earlier. Moreover, either a catalyst e.g. Pd(OAc)2 (2 mol%) or a promoter e.g. ICl (stoichiometric amount) was necessary for the reaction to be successful in both the cases. In continuation of our longstanding interest in the synthesis of quinaxoline based bioactive molecules11 we now report our preliminary results on unexpected but straightforward TFA-mediated intramolecular cyclization of 2-(indol-3-yl)-3-(ethynyl)quinoxalines (1) leading to 7-substituted 8H-indolo[3,2-a]phenazines (2 or B) (Scheme 1b). The current methodology appeared to be attractive as it does not involve the use of any additional catalyst or reagent. We also present the effect of synthesized indolophenazines on the growth of cancer cell lines in vitro.
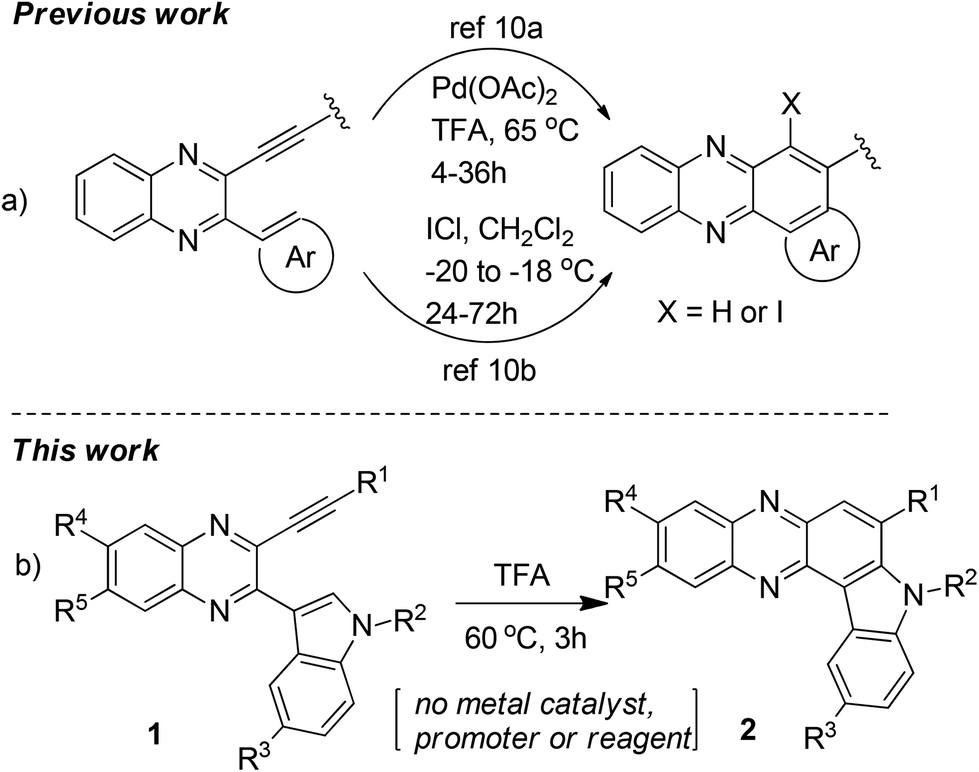 | ||
| Scheme 1 Approaches based on intramolecular hydroarylation (IMHA) of alkynes related to quinaxoline. | ||
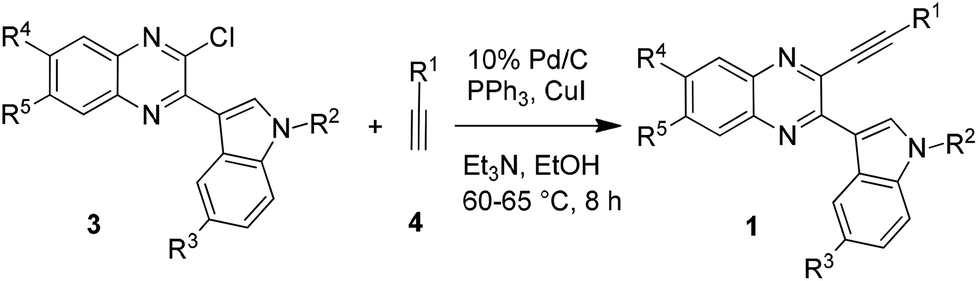
Having anticipated the IMHA of alkyne as a potential approach towards the synthesis of indolophenazines (2) we required the supply of the corresponding starting materials i.e. 3-ethynyl substituted 2-(indol-3-yl)quinoxalines (1) to test our proposed strategy. While the sequential Sonogashira/Suzuki coupling as used earlier10a could be adopted for the preparation of 1 in the present case too, the non-availability of the required variety of indole-3-boronic acids made this approach a bit challenging. We therefore adopted a slightly different strategy that allowed variation of the indole moiety of 1. Thus the starting compounds (1a–q) were prepared via a two step method i.e. the AlCl3 induced heteroarylation followed by Sonogashira type coupling.11a Accordingly, the reaction of 2,3-dichloroquinoxaline with various indoles in the presence of AlCl3 (see the ESI†) afforded 2-chloro-3-(indol-3-yl)quinoxaline derivatives (3) which on Pd/C-catalyzed coupling with a variety of terminal alkynes (4) yielded the desired compound (1) (Table 1). Notably, an attempt to prepare 1via an alternative approach involving the AlCl3 induced reaction of 3-alkynyl substituted 2-chloroquinoxaline with indole failed as an inseparable mixture of products was obtained in this case.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
|
|
||||
|---|---|---|---|---|
| Entry | 3; R2, R3, R4, R5 = | Alkyne 4; R1 = | Product 1; R2, R3, R1, R4, R5 = | %Yieldb |
| a All reactions were carried out by using 3 (1.0 equiv.), 4 (1.5 equiv.), 10% Pd/C (0.035), PPh3 (0.35 equiv.), CuI (0.06 equiv.) and Et3N (3.0 equiv.) in EtOH (5 mL) at 60–65 °C for 8 h under nitrogen. b Isolated yields. | ||||
| 1 | 3a; H, H, H, H | 4a; Ph | 1a; H, H, Ph, H, H | 85 |
| 2 | 3a | 4b; (CH2)3Me | 1b; H, H, (CH2)3Me, H, H | 87 |
| 3 | 3a | 4c; (CH2)4Me | 1c; H, H, (CH2)4Me, H, H | 74 |
| 4 | 3a | 4d; (CH2)5Me | 1d; H, H, (CH2)5Me, H, H | 81 |
| 5 | 3b; Me, H, H, H | 4a | 1e; Me, H, C6H5, H, H | 81 |
| 6 | 3b | 4b | 1f; Me, H, (CH2)3Me, H, H | 87 |
| 7 | 3b | 4c | 1g; Me, H, (CH2)4Me, H, H | 85 |
| 8 | 3b | 4d | 1h; Me, H, (CH2)5Me, H, H | 82 |
| 9 | 3c; CH2Me, H, H, H | 4a | 1i; CH2Me, H, C6H5, H, H | 84 |
| 10 | 3c | 4b | 1j; CH2Me, H, (CH2)3Me, H, H | 81 |
| 11 | 3c | 4c | 1k; CH2Me, H, (CH2)4Me, H, H | 82 |
| 12 | 3c | 4d | 1l; CH2Me, H, (CH2)5Me, H, H | 84 |
| 13 | 3d; CH2Ph, H, H, H | 4a | 1m; CH2Ph, H, Ph, H, H | 81 |
| 14 | 3d | 4b | 1n; CH2Ph, H, (CH2)3Me, H, H | 85 |
| 15 | 3d | 4c | 1o; CH2Ph, H, (CH2)4Me, H, H | 82 |
| 16 | 3d | 4d | 1p; CH2Ph, H, (CH2)5Me, H, H | 82 |
| 17 | 3e; H, Br, H, H | 4a | 1q; H, Br, Ph, H, H | 81 |
| 18 | 3f; CH2CH3, H, CH3, CH3 | 4a | 1r; CH2CH3, H, Ph, CH3, CH3 | 80 |
| 19 | 3f | 4d | 1s; CH2CH3, H, (CH2)5Me, CH3, CH3 | 78 |
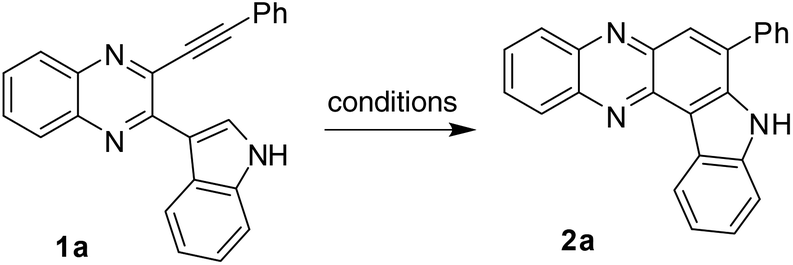
Next, to test the feasibility of the strategy based on IMHA of alkyne leading to 2 as well as to establish the optimized reaction conditions, compound 1a was chosen for this purpose. Accordingly, the reaction of 1a was examined under various conditions (Table 2). The reaction was initially performed using a transition metal catalyst (2 mol%) e.g. AgOTf or AgNO3 or Cu(OTf)2 in 1,2-dichloroethane (DCE) (entries 1–3, Table 2) when no formation of the desired product 2a was observed. The metal catalyst was then replaced by TFA and the reaction was performed in TFA–DCE (1:1) at 25 °C (entry 4, Table 2). The desired product 2a was obtained in this case albeit in a low yield. The yield increased significantly when the reaction temperature was increased to 60 °C (entry 5, Table 2) while the yield decreased with a decrease in volume of TFA used (entries 6 and 7, Table 2). A noteworthy increase in yield was observed when the reaction was performed in TFA alone (entry 8, Table 2). This was remarkable as the reaction proceeded in the absence of any metal catalyst or reagent and was completed within 3 h. Notably, TFA alone was found to be ineffective when applied to a similar system i.e. in the cyclization of 2-phenyl-3-(phenylethynyl)quinoxaline where the use of a Pd-catalyst was necessary.10a We were delighted with this observation and continued our study further. Since most of these reactions were performed at 60 °C the effect of a higher temperature and longer reaction time was examined. However, no further increase in product yield was observed (entries 9 and 10, Table 2). Notably, the use of stronger Lewis acids such as AlCl3 (1.1 equivalent) also afforded 2a in good yield (entry 11, Table 2). Once again the reduction in the AlCl3 quantity used decreased the product yield (entry 12, Table 2). This also prompted us to examine the use of one of the catalysts e.g. Cu(OTf)2 in a higher quantity. While the reaction was completed within 3 h in this case too it did not afford 3a (entry 13, Table 2) and a mixture of products was obtained. Nevertheless, we preferred TFA over AlCl3 to avoid the use of anhydrous atmosphere as well as moisture sensitive salts. Thus, performing the reaction in TFA alone at 60 °C for 3 h (entry 8, Table 2) was chosen as the best strategy for the preparation of 2a.
a
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst/solvent | Temp. (°C) | Time (h) | %Yieldb |
| a A mixture of 1a (0.034 mmol) and catalyst (2 mol%) in a solvent (2 mL) was stirred under open air. b Isolated yield. c Reaction was performed using 0.2 equiv. of catalyst. d 1.1 equiv. of reagent was used. e Reaction was performed under anhydrous conditions. f 0.6 equiv. of AlCl3 was used. | ||||
| 1 | AgOTf/DCE | 60 | 8 | 0c |
| 2 | AgNO3/DCE | 60 | 8 | 0c |
| 3 | Cu(OTf)2/DCE | 60 | 8 | 0c |
| 4 | TFA/DCE (1:1) |
25 | 8 | 30 |
| 5 | TFA/DCE (1:1) |
60 | 8 | 60 |
| 6 | TFA/DCE (1:2) |
60 | 8 | 40 |
| 7 | TFA/DCE (1:4) |
60 | 8 | 13 |
| 8 | TFA | 60 | 3 | 86 |
| 9 | TFA | 80 | 3 | 65 |
| 10 | TFA | 60 | 5 | 62 |
| 11 | AlCl3/DCE | 60 | 3 | 85d,e |
| 12 | AlCl3/DCE | 60 | 3 | 40e,f |
| 13 | Cu(OTf)2/DCE | 60 | 3 | 0d |
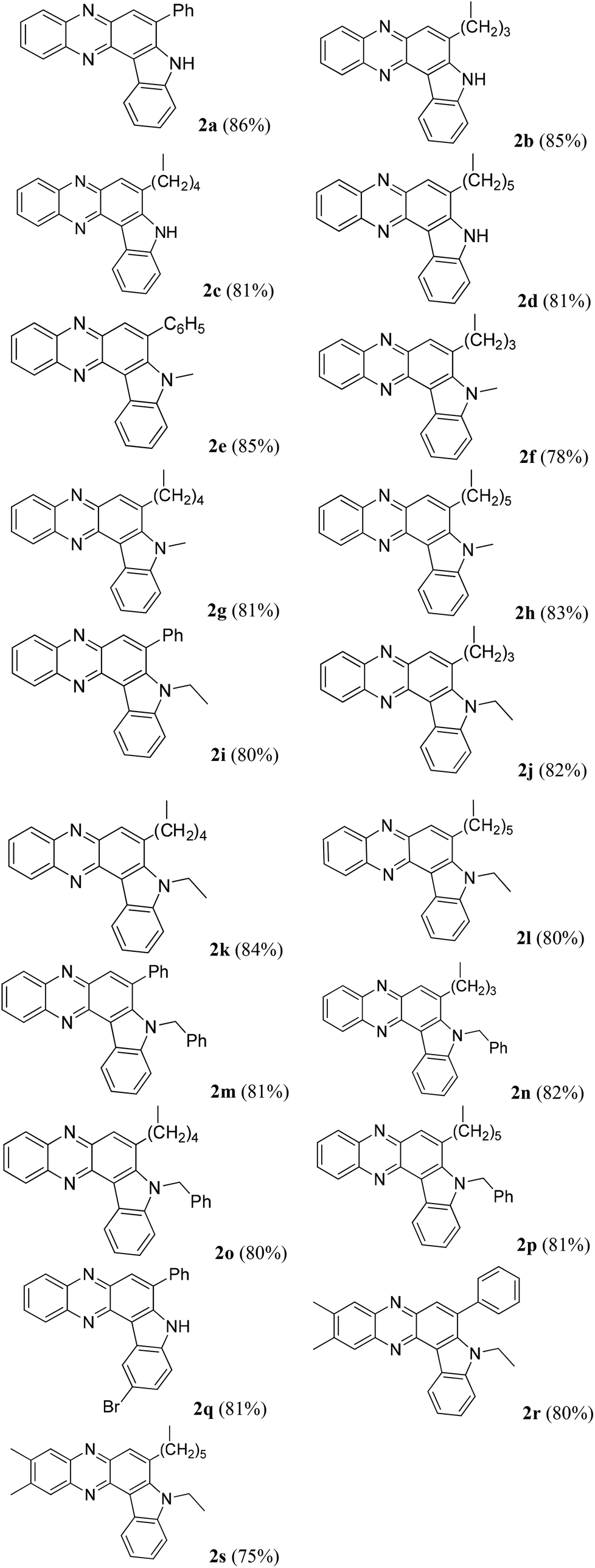
Having established a metal free process for the IMHA of alkynes leading to 2a we then focused on the application of this methodology for the synthesis of other analogues of 2a. It was also necessary to expand the scope and generality of this newly established methodology further. Thus a range of indolophenazine derivatives (2) were prepared from the starting alkyne (1) with diverse substitution patterns (Table 3). Indeed, the reaction proceeded well with R1 = aryl and aliphatic, R2 = H, Me, Et and benzyl and R3 = H and Br groups to give the desired product in good yields. Altogether 19 new indolophenazine derivatives were prepared and characterized by spectral (IR, NMR, MS) and HPLC data. This was supported by the molecular structures of 2h and 2l being confirmed by X-ray analysis (Fig. 2 and 3).12 We also examined the generality of the AlCl3 mediated method (entry 11, Table 2) which afforded the desired products in good yields (see Table S4 in the ESI†).
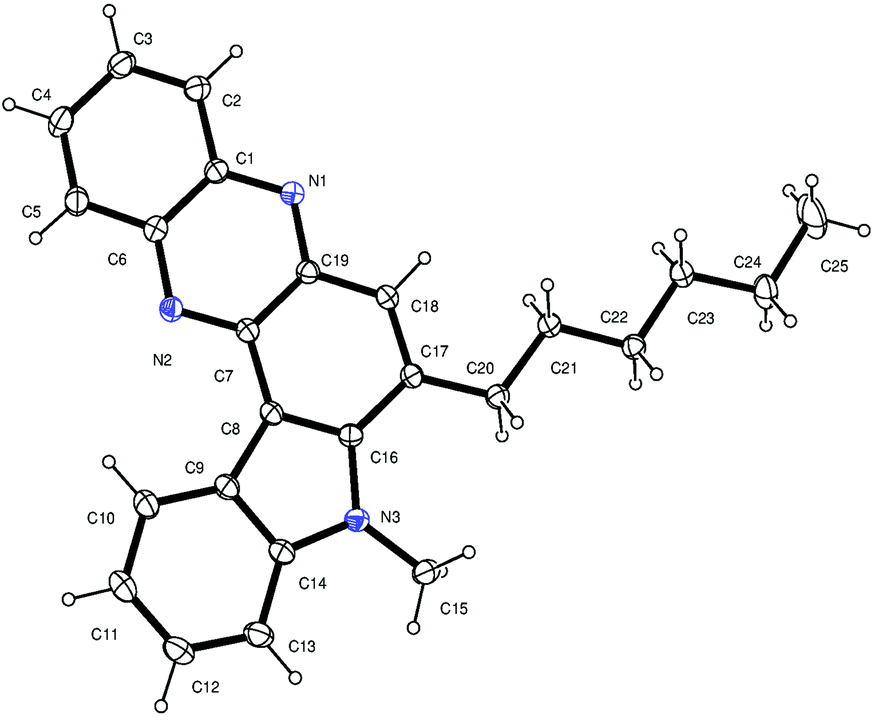 | ||
| Fig. 2 X-ray crystal structure of 2h (ORTEP diagram). Thermal ellipsoids are drawn at the 50% probability level. | ||
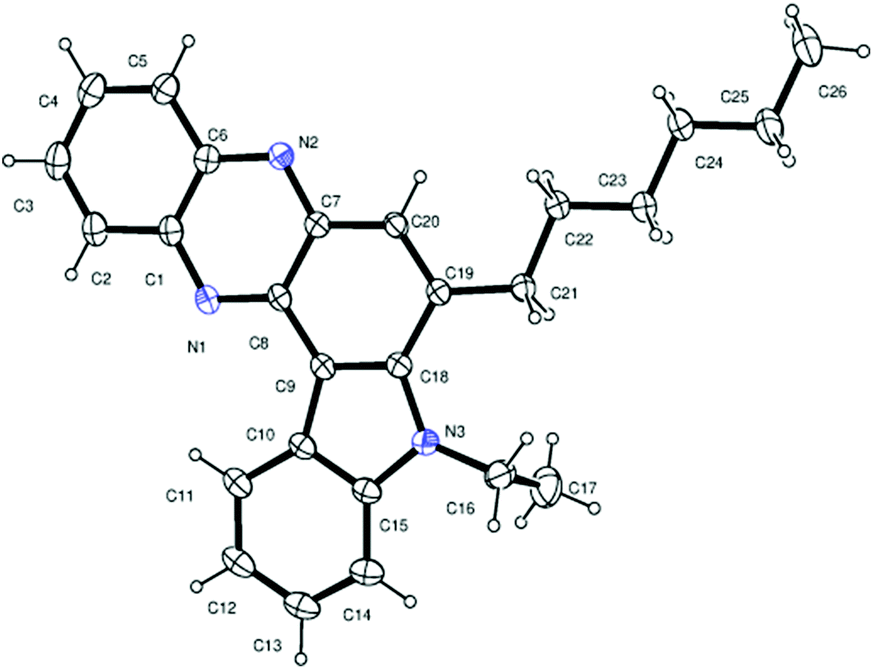 | ||
| Fig. 3 X-ray crystal structure of 2l (ORTEP diagram). Thermal ellipsoids are drawn at the 50% probability level. | ||
Based on the observation that the reaction proceeded well in the presence of a higher volume of TFA (entries 5–7 vs. 8, Table 2) a probable reaction mechanism is presented in Scheme 2.
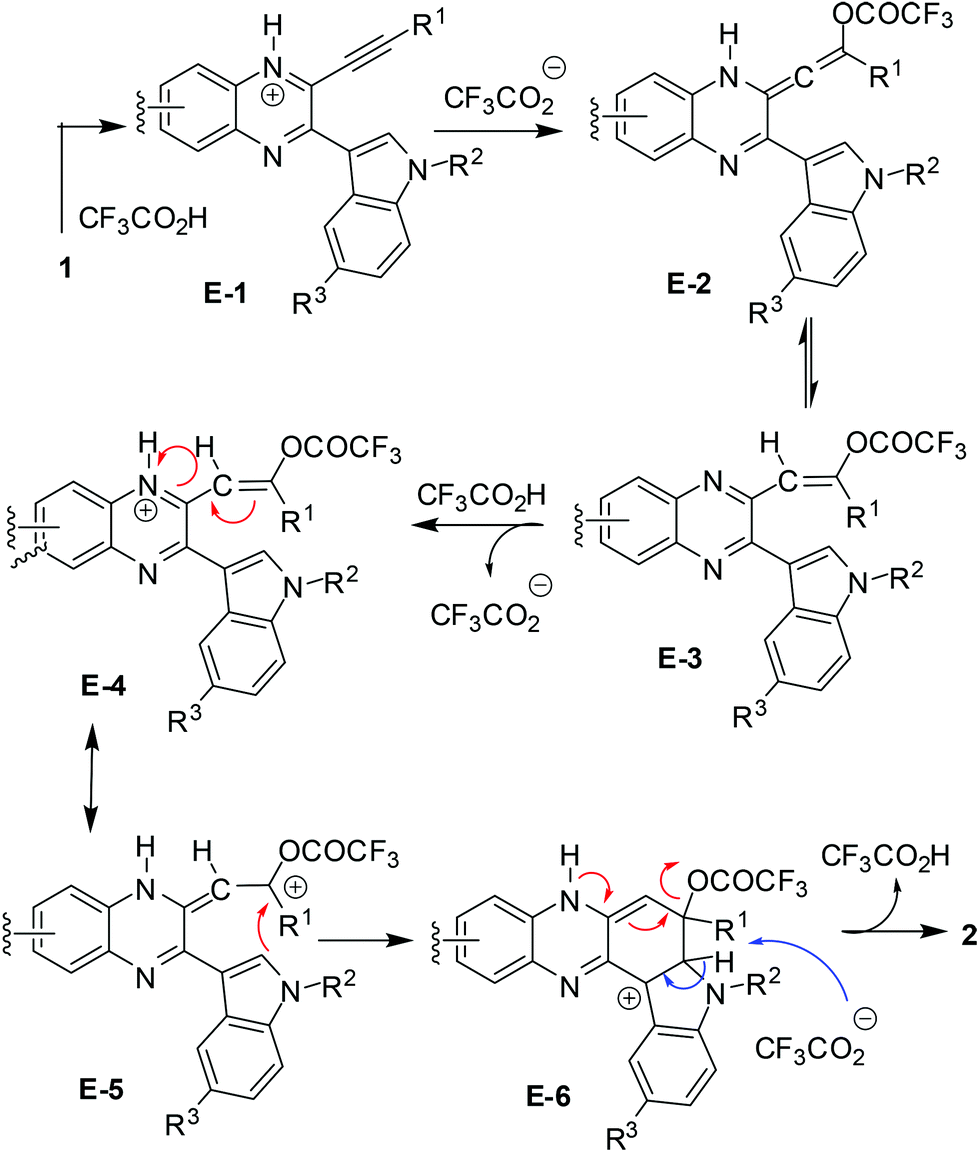 | ||
| Scheme 2 The proposed reaction mechanism. | ||
The results shown in Table 2 also indicated that the present reaction required strong acidic conditions (entries 8 and 11, Table 2) and appeared to be an acid mediated process rather than an acid catalyzed one. Additionally, the progress of the reaction seemed to be greatly aided by the counter anion released from TFA. Thus the protonated species E-1 generated was transformed (by the trifluoroacetate anion) into an allenic species E-2, the tautomeric form of which i.e.E-3 then underwent further protonation to give E-4 leading to its resonating carbocationic species E-5. This facilitated the intramolecular attack by C-2 of the proximate indole ring thereby resulting in cyclization to afford the intermediate E-6. A double aromatization involving the indole part of E-6 aided by the trifluoroacetate anion and subsequent removal of another molecule of TFA afforded the desired product 2. A similar pathway can be proposed for the AlCl3 mediated method (see the ESI†). While TFA seemed to be the source of the C-6 proton in product 2, a separate experiment was performed for further confirmation. Thus compound 1m was treated with a solution of 4:1 TFA–D2O (prepared 2 h before to ensure the in situ generation of CF3CO2D) as shown in Scheme 3 and 2m was obtained in 65% yield with significant incorporation of deuterium at the C-6 position (H/D ratio = 50/50).
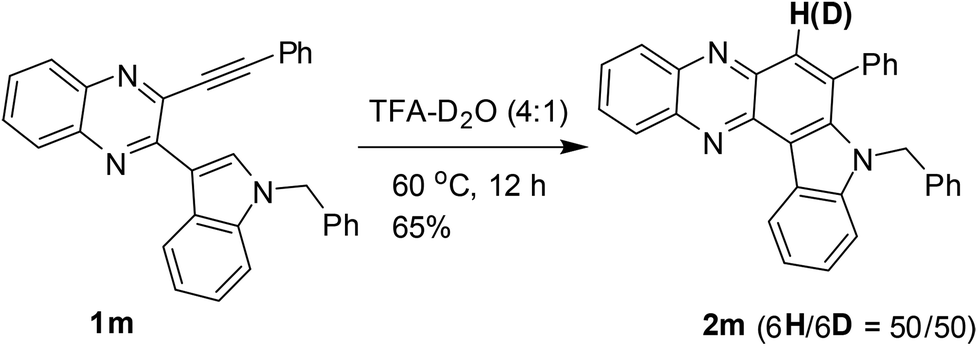 | ||
| Scheme 3 Conversion of 1m to 2m in 4:1 TFA–D2O. | ||
All the synthesized indolophenazine derivatives were tested for their cytotoxic properties against TZM-BL (human cervical carcinoma cells) and A549 (human lung carcinoma cells) cancer cell lines at 10 μM using the sulphorhodamine B (SRB) assay with gemcitabine as a reference compound. Among all the compounds tested, 2d, 2g and 2l showed good activity against both the cell lines. These three compounds showed 90–98% inhibition against TZM-BL (Fig. 4) and 79–85% inhibition against A549 cell lines (Fig. 5) indicating the potential of this class of compounds as new cytotoxic agents.
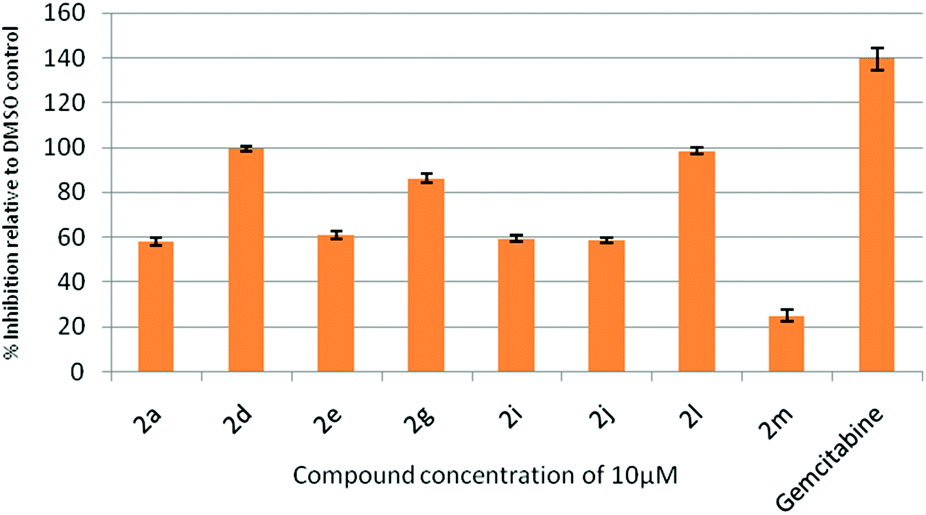 | ||
| Fig. 4 Effect of compound 2 on the growth of the TZM-BL cell line at 10 μM. | ||
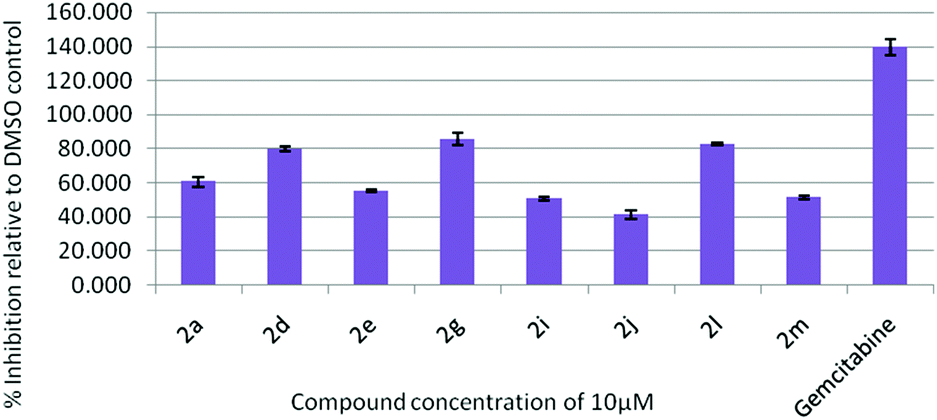 | ||
| Fig. 5 Effect of compound 2 on the growth of the A549 cell line at 10 μM. | ||
In conclusion, TFA alone when employed as a solvent was found to be remarkably effective for the intramolecular cyclization of 3-alkynyl substituted 2-(indol-3-yl)quinoxalines leading to 8H-indolo[3,2-a]phenazine derivatives. The reaction does not require the use of any additional metal catalyst, promoter or reagent and followed IMHA of alkyne as a key step. The use of excess TFA was found to be crucial for the success of this reaction. This simple method afforded a range of indolophenazines in good yields accomplishing a new synthesis of this class of N-heteroarenes. Three of these compounds showed cytotoxic properties when tested against two cancer cell lines. Overall, in addition to highlighting the development of an operationally simple and straightforward method for a new and easy access to indolophenazines, our study also suggests that the described framework could be a useful template for the design and discovery of potential anticancer agents.
KSK thanks University Grants Commission (UGC), New Delhi, for the award of Assistant Professorship under FRP and DST, India for the financial support (SR/FTP/CS-017/2012). B. B. thanks CSIR, New Delhi for a Junior Research Fellowship.
Notes and references
- For antifungal agents, see: (a) Y. Ge, D. Pei, Y. Zhao, W. Li, Sh. Wang and Y. Xu, Curr. Microbiol., 2007, 54, 277 CrossRef CAS PubMed; (b) H. Liu, Y. He, H. Jiang, H. Peng, X. Huang, X. Zhang, L. S. Thomashow and Y. Xu, Curr. Microbiol., 2007, 54, 302 CrossRef CAS PubMed.
- For antimicrobial agents, see: H. Hussain, S. Specht, S. R. Sarite, M. Saeftel, A. Hoerauf, B. Schulz and K. Krohn, J. Med. Chem., 2011, 54, 4913 CrossRef CAS PubMed.
- For antimycobacterial agents, see: S. G. Franzblaul and J. F. O'sullivan, Antimicrob. Agents Chemother., 1988, 32, 1583 CrossRef.
- For anticancer agents, see: (a) L. Marler, M. Conda-Sheridan, M. A. Cinelli, A. E. Morrell, M. Cushman, L. Chen, K. Huang, R. B. van Breemen and J. M. Pezzuto, Anticancer Res., 2010, 30, 4873 CAS; (b) S. Imai, K. Furihata, Y. Hayakawa, T. Noguchi and H. Seto, J. Antibiot., 1989, 42, 1196 CrossRef CAS PubMed; (c) S. Imai, T. Noguchi and H. Seto, J. Antibiot., 1993, 46, 1232 CrossRef CAS PubMed; (d) M. Matsumoto and H. Seto, J. Antibiot., 1991, 44, 1471 CrossRef CAS PubMed.
- K. A. Nolan, D. J. Timson, I. J. Stratforda and R. A. Bryce, Bioorg. Med. Chem. Lett., 2006, 16, 6246 CrossRef CAS PubMed.
- (a) J. Bian, X. Qian, B. Deng, X. Xu, X. Guo, Y. Wang, X. Li, H. Sun, Q. You and X. Zhang, RSC Adv., 2015, 5, 49471 RSC; (b) K. A. Scott, J. Barnes, R. C. Whitehead, I. J. Stratford and K. A. Nolan, Inhibitors of NQO1: identification of compounds more potent than dicoumarol without associated off-target effects, Biochem. Pharmacol., 2011, 81, 355 CrossRef CAS PubMed.
- T. A. Shoker, K. I. Ghattass, J. C. Fettinger, M. J. Kurth and M. J. Haddadin, Org. Lett., 2012, 14, 3704 CrossRef CAS PubMed.
- For a review, see: V. Cadierno, in Green Synthetic Approaches for Biologically Relevant Heterocycles, ed. G. Brahmachari, Elsevier, 2015, pp. 87–90 Search PubMed.
- For recent reports, see: (a) R. S. Menon, A. D. Findlay, A. C. Bissember and M. G. Banwell, J. Org. Chem., 2009, 74, 8901 CrossRef CAS PubMed; (b) L. Alonso-Marañón, M. M. Martínez, L. A. Sarandeses and J. P. Sestelo, Org. Biomol. Chem., 2015, 13, 379 RSC.
- (a) S. Kumar, R. K. Saunthwal, M. Mujahid, T. Aggarwal and A. K. Verma, J. Org. Chem., 2016, 81, 9912 CrossRef CAS PubMed; (b) A. V. Gulevskaya, Eur. J. Org. Chem., 2016, 4207 CrossRef CAS.
- For selected references, see: (a) K. S. Kumar, D. Rambabu, S. Sandra, R. Kapavarapu, G. R. Krishna, M. V. B. Rao, K. Chatti, C. M. Reddy, P. Misra and M. Pal, Bioorg. Med. Chem., 2012, 20, 1711 CrossRef CAS PubMed; (b) K. S. Kumar, D. Rambabu, B. Prasad, M. Mujahid, G. R. Krishna, M. V. B. Rao, C. M. Reddy, G. R. Vanaja, A. M. Kalle and M. Pal, Org. Biomol. Chem., 2012, 10, 4774 RSC; (c) A. Nakhi, S. Archana, G. P. K. Seerapu, K. S. Chennubhotla, K. L. Kumar, P. Kulkarni, D. Haldar and M. Pal, Chem. Commun., 2013, 49, 6268 RSC; (d) A. Nakhi, Md. S. Rahman, G. P. K. Seerapu, R. K. Banote, K. L. Kumar, P. Kulkarni, D. Haldar and M. Pal, Org. Biomol. Chem., 2013, 11, 4930 RSC; (e) K. S. Kumar, S. R. Meesa, B. Rajesham, B. Bhasker, M. A. Ashfaq, A. A. Khan, S. S. Rao and M. Pal, RSC Adv., 2016, 6, 48324 RSC.
- Crystallographic data (excluding structure factors) for 2h and 2l have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication numbers CCDC 1503459 and 1503460, respectively.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectral data for all new compounds, and copies of spectra. CCDC 1503459 and 1503460. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ob02340a |
| This journal is © The Royal Society of Chemistry 2017 |