
A linear hydroxymethyl tetramate undergoes an acetylation–elimination process for exocyclic methylene formation in the biosynthetic pathway of pyrroindomycins†
Qingfei
Zheng‡
 a,
Zhuhua
Wu‡
a,
Peng
Sun‡
ab,
Dandan
Chen
ac,
Zhenhua
Tian
*a and
Wen
Liu
*ac
a,
Zhuhua
Wu‡
a,
Peng
Sun‡
ab,
Dandan
Chen
ac,
Zhenhua
Tian
*a and
Wen
Liu
*ac
aState Key Laboratory of Bioorganic and Natural Products Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: tiantou81@hotmail.com; wliu@mail.sioc.ac.cn; Fax: +86-21-64166128; Tel: +86-21-54925111
bResearch Center for Marine Drugs, School of Pharmacy, Second Military Medical University, 325 Guohe Road, Shanghai 200433, China
cHuzhou Center of Bio-Synthetic Innovation, 1366 Hongfeng Road, Huzhou 313000, China
First published on 6th December 2016
Abstract
We herein report the isolation and characterization of a key linear intermediate in the biosynthetic pathway of pyrroindomycins, the potent spirotetramate natural products produced by Streptomyces rugosporus. This polyene intermediate bears a γ-hydroxymethyl group that is exocyclic to the tetramate moiety, indicating that a serine residue serves as the three-carbon unit for tetramate formation and chain-elongation termination. The further conversion involves an acetylation–elimination of the exocyclic γ-hydroxymethyl group to generate a γ-methylene group, which is indispensable for intramolecular [4 + 2] cross-bridging to construct the characteristic pentacyclic core. The findings presented in this study provide new insights into the biosynthesis of pyrroindomycins, and thus suggest a common paradigm for both spirotetramates and spirotetronates in processing the exocyclic γ-hydroxymethyl group of the five-membered heterocycle.
The spirotetramate and spirotetronate natural products, which possess a similar pentacyclic core but differ in the heterocyclic atom of the five-membered ring, have long been appreciated as the target molecules of chemical synthesis.1 Benefiting from the structural complexity, both spirotetramates and spirotetronates display diverse biological activities and significant pharmaceutical potential. Although synthetic chemistry has shown great power in constructing the pentacyclic core of spirotetronates and spirotetramates,2,3 an understanding of the biosynthetic pathway of these natural products would facilitate the development of strategies for both combinatorial biosynthesis and biomimetic synthesis, thereby inspiring the production of new-type anti-infective drug leads via a synthetic biology-based approach.
Pyrroindomycins (PYRs) represent the first spirotetramates discovered in nature, which exhibit remarkable activities against various bacterial pathogens such as methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant Enterococcus faecium (VRE).4,5 Over the past few years, we and others have identified the gene clusters for the biosynthesis of PYRs6 and a number of spirotetronates including chlorothricin, tetrocarcin A, kijanimicin, lobophorins and versipelostatin A (Fig. 1),7–11 leading to the hypothesis that these structurally related natural products share a similar biosynthetic logic in the formation of a characteristic pentacyclic core. There are two cyclases that respectively catalyze two tandem intramolecular Diels–Alder [4 + 2] cycloadditions of a linear polyene intermediate, which contains a minimum of six double bonds for constituting two pairs of the 1,3-diene and alkene groups: five in the main carbon skeleton and the sixth exocyclic to the tetramate or tetronate moiety, as exemplified by 1 in PYR biosynthesis (Fig. 1).12 Specifically, the production of 1 involves a modular type I polyketide synthase (PKS) system that programs the assembly of its main carbon skeleton, followed by the activity of a non-ribosomal peptide synthase (NRPS) that mediates the incorporation of an amino acid residue for tetramate formation. However, which amino acid is incorporated needs to be determined, and the process of how the exocyclic γ-methylene group of 1 is formed remains elusive. To answer these questions, we herein re-analyzed the product profile of the mutant S. rugosporus strain WL2004,6 in which both the genes pyrD3 and pyrD4 have previously been deleted in frame, and subsequently dissected the following enzymatic transformations before the cross-bridging of 1.
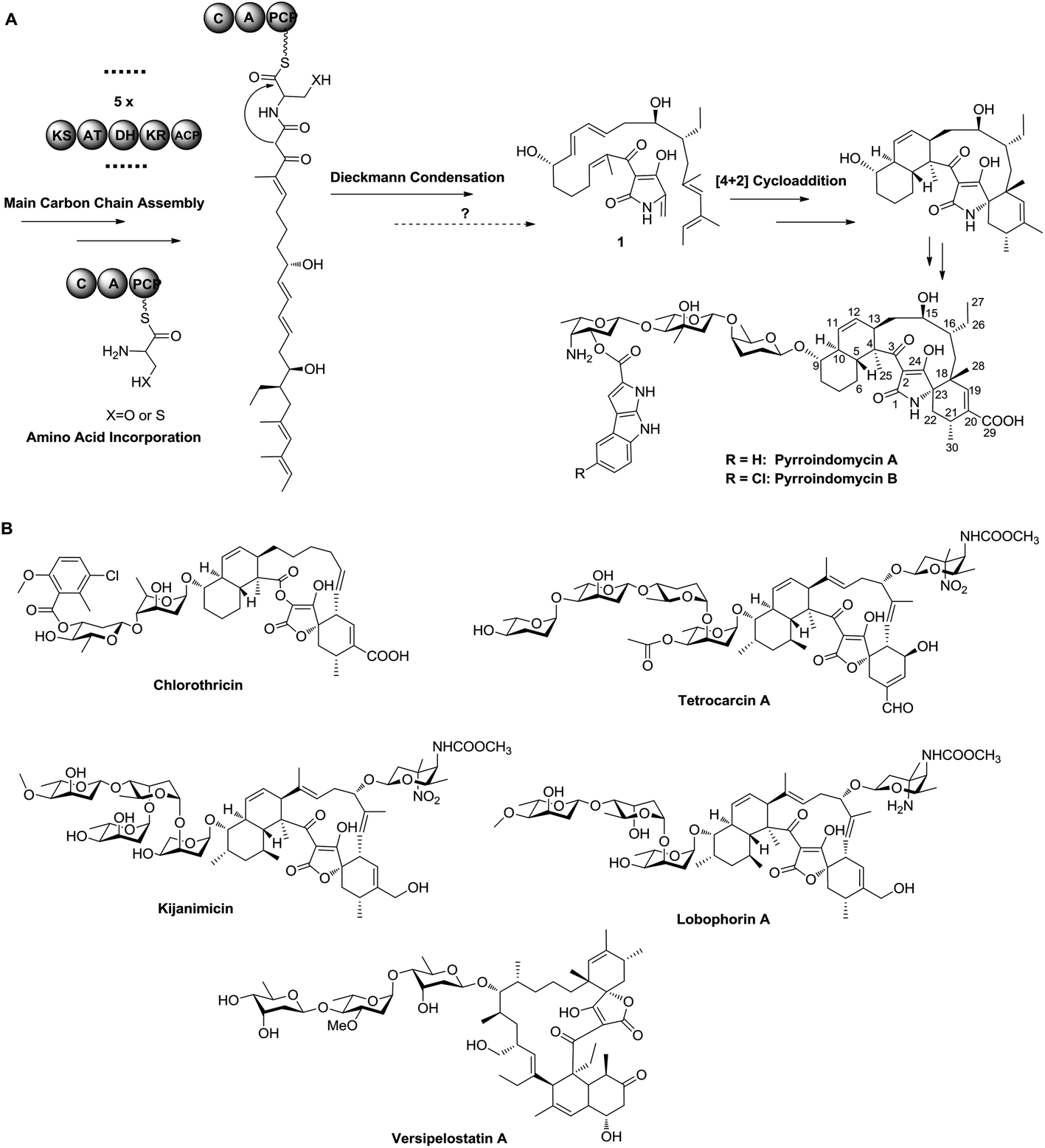 | ||
| Fig. 1 Biosynthetic pathway and chemical structures. (A) Assembly of the aglycone of pyrroindomycins that involves a hybrid PKS/NRPS system. At least five PKS modules share the organization KS-AT-DH-KR-ACP for the generation of five double bonds, and the NRPS contains a single C-A-PCP module for three-carbon unit incorporation. KS, ketosynthase; AT, acyltransferase; DH, dehydrogenase; KR, ketoreductase; ACP, acyl carrier protein; C, condensation domain; A, adenylation domain; PCP, peptide carrier protein. (B) Structures of representative spirotetronates. | ||
The gene pyrD3 encodes a protein that is homologous to various acyltransferase E2 subunits of 2-oxoacid dehydrogenase complexes, whereas pyrD4 encodes a protein that belongs to the α/β-hydrolase fold enzyme superfamily. Initial in vivo investigations into PYR biosynthesis showed that the mutant S. rugosporus strains in which either pyrD3 or pyrD4 was inactivated still produced PYRs, albeit in lower yields.6 In contrast, the simultaneous inactivation of pyrD3 and pyrD4 completely abolished PYR production; however, no related product was detected in the resulting double mutant strain WL2004.6 Using a method that has been newly developed for the fermentation and characterization of 1,12 we re-analyzed the metabolite profile of WL2004 in this study and obtained a new product (2), which is more polar than 1 and appeared in HPLC with a shorter elution time (Fig. 2).
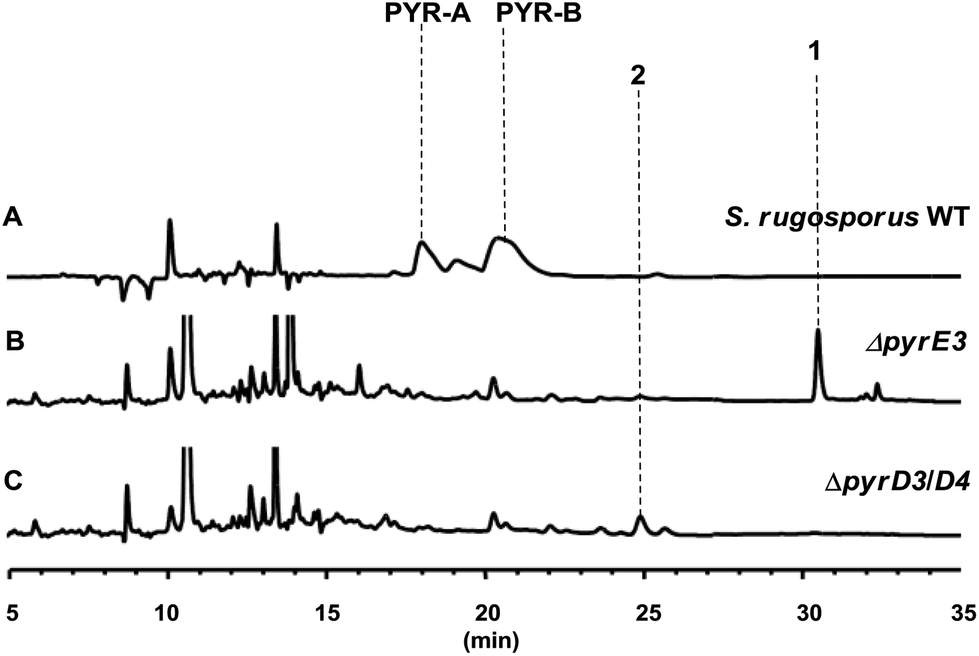 | ||
| Fig. 2 HPLC profiles of the metabolites of S. rugosporus wild-type and mutant strains: (A) S. rugosporus wild-type; (B) ΔpyrE3 mutant; (C) ΔpyrD3/D4 mutant. | ||
To elucidate the structure of 2, we fermented WL2004 on a large scale and obtained 2 by intensive purification processes. The molecular formula of 2 was deduced to be C30H45NO6 by analyses of the HR-ESI-MS data (m/z calcd for C30H45NO6Na: 538.3145 [M + Na]+, found: 538.3137) and NMR spectra. The 1H and 13C NMR spectra of 2 (Table S1†) resembled greatly the reported spectra of compound 1, indicating that they share high structural similarities. Detailed analyses of the 2D NMR data revealed that compound 2 possesses a linear 30-carbon skeleton containing the Δ10,11, Δ12,13-diene, the Δ18,19, Δ20,21-diene, and a tetramate moiety (Fig. 3). Instead of the terminal γ-methylene tetramate moiety in compound 1, a hydroxymethyl group (δC, 60.5, CH2, C-22; δH 3.61, H2-22) exocyclic to the tetramate moiety was observed. This structural feature was further supported by the DEPT spectrum, COSY correlation between H-23 (δH 3.75) and H2-22, and HMBC correlation from H2-22 to C-23 (δC, 63.5). Consequently, compound 2 is a linear polyene intermediate of PYRs that harbors a γ-hydroxymethyl tetramate moiety. The absolute configurations of chiral centers in 2 are biogenetically supposed to be the same as those in 1. These results indicate that a serine residue, other than a cysteine residue that was previously proposed according to in vitro assays,6 is incorporated into the formation of the five-membered tetramate heterocycle during PYR biosynthesis and facilitates the release of the nascent linear intermediate from the PKS–NRPS hybrid assembly line. Similar inconsistency between in vivo and in vitro experiments was also reported in the studies regarding the biosynthesis of isoquinoline alkaloid.13
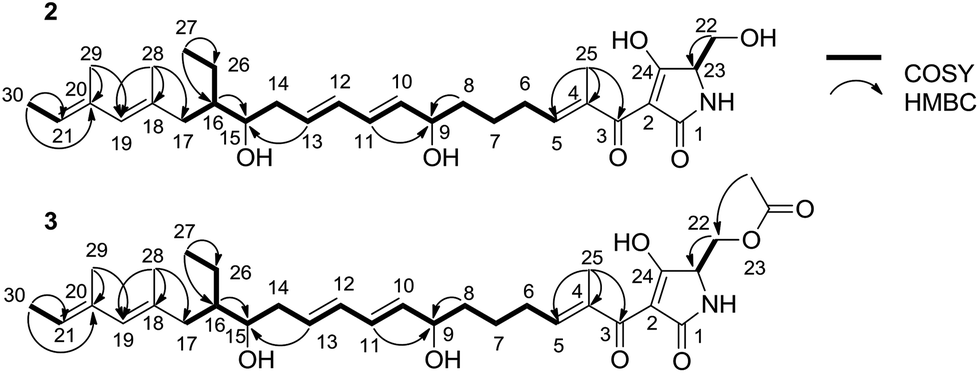 | ||
| Fig. 3 Key COSY and HMBC correlations of compounds 2 and 3. | ||
Both PyrD3 and PyrD4 have previously been shown to possess activity that accelerates the intramolecular Dieckmann condensation to form a tetramate moiety after amino acid incorporation.6 In fact, in silico sequence alignments revealed that PyrD3 and PyrD4 share high sequence similarities to the enzyme pair Agg4/Agg5 in RK-682 biosynthesis and QmnD3/QmnD4 in quartromicin biosynthesis, both of which are involved in an acetylation-elimination process for the conversion of the γ-hydroxymethyl group of tetronate into the γ-methylene group.14,15 Here, the structural characterization of the intermediate 2 led to an intriguing query of whether a similar functionalization strategy is employed to process its γ-hydroxymethyl group of tetramate. To validate this hypothesis, we expressed and purified PyrD3 and PyrD4 from Escherichia coli as previously described,6 and then performed the in vitro assays with 2 as the substrate. In the presence of PyrD3 and PyrD4, acetyl-CoA and Mg2+, 2 was rapidly converted into a product with an identical retention time and UV profile to those of compound 1 on HPLC (Fig. 4C). The HR-ESI-MS result showed that this product exhibited a molecular formula of C30H43NO6 (m/z calcd for C30H43NO5: 480.3105 [M + H − H2O]+, found: 480.3089) identical to that of compound 1 produced from the fermentation extract of the ΔpyrE3 mutant.12
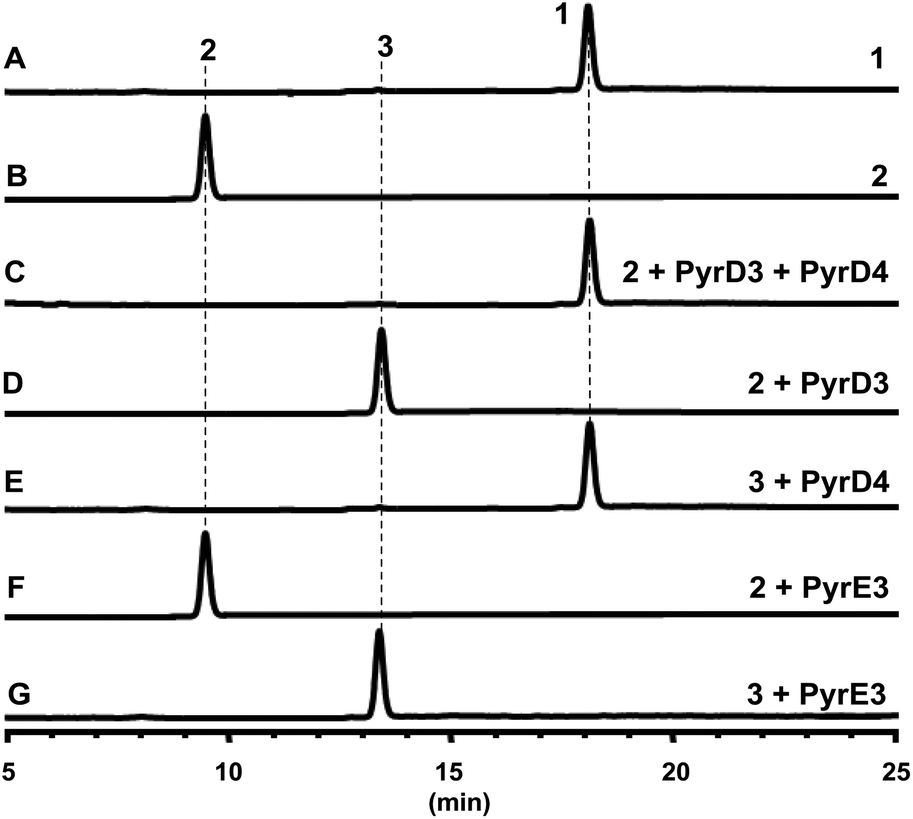 | ||
| Fig. 4 HPLC analyses of in vitro assays: (A) standard 1; (B) standard 2; (C) incubation of 2 with PyrD3, PyrD4, acetyl-CoA and Mg2+; (D) incubation of 2 with PyrD3, acetyl-CoA and Mg2+; (E) incubation of 3 with PyrD4; (F) incubation of 2 with PyrE3; (G) incubation of 3 with PyrE3. | ||
We then dissected the activities of PyrD3 and PyrD4. Compound 2 was converted to a new product (3) when incubated with PyrD3, acetyl-CoA and Mg2+ (Fig. 4D). To elucidate its structure, we scaled up the enzymatic biotransformation and purified 3 by semi-preparative HPLC. The HR-ESI-MS result showed that compound 3 exhibited a molecular formula of C32H47NO7 (m/z calcd for C32H47NO7Na: 580.3250 [M + Na]+, found: 580.3239), 42 Da greater than that of 2. Comparison of the 1H and 13C NMR spectra of 3 with those of 2 indicated great structural similarities of these two compounds. The only difference observed was the presence of an additional acetyl group (δC 172.7, C and 20.7, CH3; δH 2.02, s). A downfield-shifted phenomenon of H2-22 (δH 3.61 in 2, 4.44 in 3) indicated that the acetyl group could be linked to C-22, which was also supported by the HMBC correlation between H3-OAc and C-22. Consequently, 3 is the acetylated derivative of compound 2, which is produced via a regioselectively reaction mediated by PyrD3. The absolute configurations of chiral centers in 3 are biogenetically supposed to be the same as those in 1 and 2. In the presence of PyrD4, compound 3 was transformed to compound 1 completely, without any co-factors (Fig. 4E). Therefore, the intrinsic activities of PyrD3 and PyrD4 were dissected, providing direct evidence that both spirotetramates and spirotetronates share a common acetylation–elimination strategy to generate exocyclic γ-methylene in linear precursor preparation, despite the fact that the eliminated hydroxyl groups arise from different three-carbon units (1,3-bisphosphoglycerate for spirotetronates and serine for spirotetramates).
The newly elucidated compounds 2 and 3 share with 1 the same internal pair of the 1,3-diene and alkene groups for dialkyldecalin formation, which proceeds through an intramolecular [4 + 2] cycloaddition mediated by PyrE3, the dialkyldecalin synthase. To validate whether the dialkyldecalin formation is dependent or independent of the functionality exocyclic to the tetramate moiety, which is necessary to generate the terminal pair of 1,3-diene and alkene groups for spiro-conjugate formation, we incubated 2 and 3 individually with PyrE3. As a result, no cycloaddition reaction occurred when either compound 2 or 3 was used as the substrate (Fig. 4F and G), indicating that the formation of terminal γ-methylene precedes the formation of dialkyldecalin (Fig. 5).
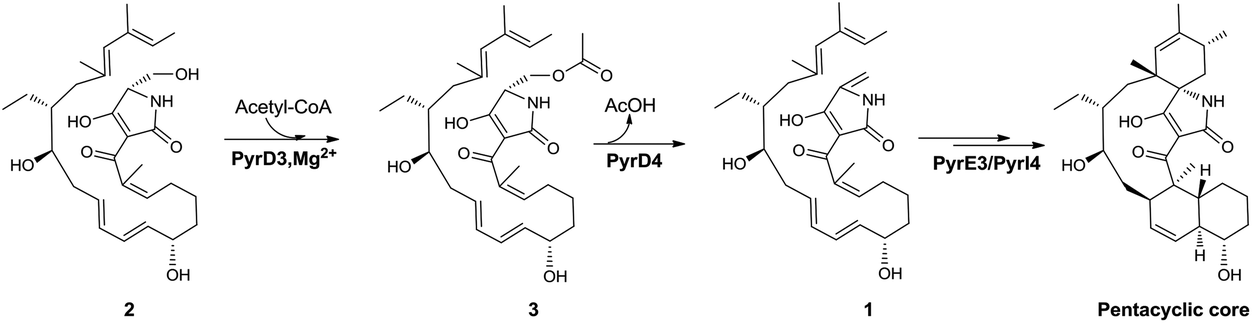 | ||
| Fig. 5 Post-PKS–NRPS assembly modifications of PYRs by enzymes PyrD3 and PyrD4. | ||
In summary, we isolated and characterized compound 2, a γ-hydroxymethyl tetramate-containing polyene intermediate in PYR biosynthesis that is most likely off-loaded immediately from the PKS–NRPS assembly line. By in vitro assays and structure elucidation of compound 3, we provided evidence that PyrD3 and PyrD4 are involved in the formation of the exocyclic double bond of tetramate via an acetylation–elimination process, thus validating their intrinsic functions in addition to the activity previously found for catalyzing Dieckmann cyclization. The in vitro biochemical studies described herein indicate that spirotetramates and spirotetronates share a similar mechanism for the formation of the exocyclic γ-methylene, although their five-membered heterocyclic atoms are derived from distinct three-carbon units.
Acknowledgements
The research work was financially supported by NSFC (81573342, 31430005, and 21520102004), STCSM (14JC1407700 and 15JC1400400), and MST (2012AA02A706) of China.References
- L. Vieweg, S. Reichau, R. Schobert, P. F. Leadlay and R. D. Sussmuth, Nat. Prod. Rep., 2014, 31, 1554–1584 RSC.
- M. H. Lacoske and E. A. Theodorakis, J. Nat. Prod., 2015, 78, 562–575 CrossRef CAS PubMed.
- N. A. Butt and C. J. Moody, Org. Lett., 2011, 13, 2224–2227 CrossRef CAS PubMed.
- M. P. Singh, P. J. Petersen, N. V. Jacobus, M. J. Mroczenski-Wildey, W. M. Maiese, M. Greenstein and D. A. Steinberg, J. Antibiot., 1994, 47, 1258–1265 CrossRef CAS PubMed.
- W. D. Ding, D. R. Williams, P. Northcote, M. M. Siegel, R. Tsao, J. Ashcroft, G. O. Morton, M. Alluri, D. Abbanat, W. M. Maiese and G. A. Ellestad, J. Antibiot., 1994, 47, 1250–1257 CrossRef CAS PubMed.
- Q. Wu, Z. Wu, X. Qu and W. Liu, J. Am. Chem. Soc., 2012, 134, 17342–17345 CrossRef CAS PubMed.
- X. Jia, Z. Tian, L. Shao, X. Qu, Q. Zhao, J. Tang, G. Tang and W. Liu, Chem. Biol., 2006, 13, 575–585 CrossRef CAS PubMed.
- J. Fang, Y. Zhang, L. Huang, X. Jia, Q. Zhang, X. Zhang, G. Tang and W. Liu, J. Bacteriol., 2008, 190, 6014–6025 CrossRef CAS PubMed.
- H. Zhang, J. A. White-Phillip, C. E. Melançon, H.-J. Kwon, W.-L. Yu and H.-W. Liu, J. Am. Chem. Soc., 2007, 129, 14670–14683 CrossRef CAS PubMed.
- S. Li, J. Xiao, Y. Zhu, G. Zhang, C. Yang, H. Zhang, L. Ma and C. Zhang, Org. Lett., 2013, 15, 1374–1377 CrossRef CAS PubMed.
- T. Hashimoto, J. Hashimoto, K. Teruya, T. Hirano, K. Shin-ya, H. Ikeda, H.-W. Liu, M. Nishiyama and T. Kuzuyama, J. Am. Chem. Soc., 2015, 137, 572–575 CrossRef CAS PubMed.
- (a) Z. Tian, P. Sun, Y. Yan, Z. Wu, Q. Zheng, S. Zhou, H. Zhang, F. Yu, X. Jia, D. Chen, A. Mándi, T. Kurtán and W. Liu, Nat. Chem. Biol., 2015, 11, 259–265 CAS; (b) Q. Zheng, Y. Guo, L. Yang, Z. Zhao, Z. Wu, H. Zhang, J. Liu, X. Cheng, J. Wu, H. Yang, H. Jiang, L. Pan and W. Liu, Cell Chem. Biol., 2016, 23, 352–360 CrossRef CAS PubMed; (c) Q. Zheng, Z. Tian and W. Liu, Curr. Opin. Chem. Biol., 2016, 31, 95–102 CrossRef CAS PubMed.
- J. A. Baccile, J. E. Spraker, H. H. Le, E. Brandenburger, C. Gomez, J. W. Bok, J. Macheleidt, A. A. Brakhage, D. Hoffmeister, N. P. Keller and F. C. Schroeder, Nat. Chem. Biol., 2016, 12, 419–424 CrossRef CAS PubMed.
- C. Kanchanabanca, W. Tao, H. Hong, Y. Liu, F. Hahn, M. Samborskyy, Z. Deng, Y. Sun and P. F. Leadlay, Angew. Chem., Int. Ed., 2013, 52, 5785–5788 CrossRef CAS PubMed.
- L. Wu, H. He, H. Pan, L. Han, R. Wang and G. Tang, Org. Lett., 2014, 16, 1578–1581 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ob02567f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |