DOI:
10.1039/C6QM00370B
(Research Article)
Mater. Chem. Front., 2017,
1, 1594-1599
Rapid hydrogel repair utilizing microgel architectures
Received
20th December 2016
, Accepted 29th March 2017
First published on 30th March 2017
Abstract
The usage of hydrogels has greatly expanded to a wide range of fields due to their eco-friendly properties. In this study, we demonstrated the rapid repair of broken hydrogels utilizing microgel architectures, which were prepared by the adhesion of oppositely charged microgels. The mechanical strength of the microgel architectures was able to be controlled by changing the charge density and/or mixing ratio of larger and smaller microgels. The rapid repair of broken microgel architectures and deletion-formed hydrogels was performed by gap filling using microgel paste. The reported microgel architectures would be useful for the do-it-yourself repair of soft materials including hydrogels and soft-tissues with green manufacturing processes.
Introduction
Hydrogels are fascinating materials because they swell in water, similarly to living soft-tissues. Therefore biodegradable and/or stimuli-responsive hydrogels have been studied for use in cell cultures,1 as drug delivery carriers,2 and as soft actuators.3–6 The fabrication of three-dimensional (3D) hydrogel architectures7 has recently received substantial attention because they allow the precise control of both the structure and material properties in a customizable fashion. The usage of hydrogels has greatly expanded to a wide range of fields due to their eco-friendly properties.
As a result, the repair of broken materials8–10 contributes to the science and technology of materials and a sustainable society because of the potential to decrease waste without damaging the environment. Adhesion is a key technology for the simple repair of broken materials, but it is generally difficult to adhere hydrogels.11–13 Although the self-healing property of cracked hydrogels has been reported,14–17 it is unsuitable for the repair of hydrogels with a large deficit because the precursors to hydrogels are often in solution and a template is needed for hydrogel preparation. Therefore, the reuse, recycle, and repair of hydrogels have not been examined in detail compared to those of thermoplastic polymers.
On the other hand, we recently reported the electrophoretic adhesion of hydrogels for building functional soft-materials.18–20 The rapid fabrication and reconstruction of hydrogels using oppositely charged microgels as building blocks was successfully achieved.21 Microgel architectures were rapidly fabricated by the electrophoretic adhesion of mouldable hydrogel precursor pastes without the need for a cross-linking reaction or the dripping of a pre-gel aqueous solution. Moreover, the green manufacturing process without chemical reactions or purification processes provides a way to repair materials in situ.
Herein, we demonstrate the rapid repair of broken hydrogels utilizing microgel architectures (Fig. 1). The regulation of the mechanical strength of the microgel architecture, which was prepared by the electrophoretic adhesion of oppositely charged microgels to each other, was investigated from the viewpoint of the charge and size ratio of the microgels. We demonstrated complete regeneration of the mechanical strength of the broken microgel architecture itself as well as other hydrogels. These materials would be useful as novel tools for the do-it-yourself repair of soft materials including hydrogels and soft-tissues with green manufacturing processes.
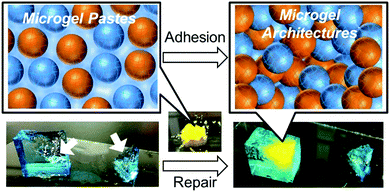 |
| | Fig. 1 Schematic representation of hydrogel repair by microgel architectures. Microgel architectures were prepared by the adhesion of oppositely charged microgel pastes. The white arrows in the picture indicate the broken parts of the hydrogels. | |
Experimental
Materials
Poly(vinyl alcohol) (PVA), glutaraldehyde, Span 80, paraffin oil, hexane, 2-propanol, and 1 mol L−1 HCl aqueous solution were purchased from Wako Pure Chemical Industries (Osaka, Japan) and used as purchased without further purification. Poly(styrene sulfonate) sodium salts (PSS) and poly(diallyldimethylammonium chloride) (PDDA) were purchased from Aldrich (USA) and used as purchased without further purification.
Preparation of microgels
Glutaraldehyde (1 mol% in vinyl alcohol) and 1 mol L−1 HCl aqueous solution were added to 10 mL of 10 wt% PVA aqueous solution, and to this viscous solution was added 35 mL paraffin oil, followed by vigorous stirring. Span 80 was used as a stabilizer. The PVA microbeads were formed during this reverse suspension, and the cross-linking reaction was carried out at ambient temperature for 24 h with stirring. For the preparation of the cationic and anionic microbeads, 1 wt% PDDA or PSS was added into the reaction mixture, respectively. The PVA microbeads were lyophilized after washing with hexane, 2-propanol, and water, consecutively. The lyophilized microgels were separated between diameters of 100 to 150 μm, and 250 to 300 μm by a sieving mesh, and were used to prepare the microgel architectures.
Preparation of microgel architectures
Microgel architectures were prepared by the adhesion of cationic (C) and anionic (A) microgels. 20 mg of microgel powder was swollen in 1 mL of water, and then excess water was absorbed using Kimwipes to form microgel pastes. These pastes were moulded into strip shapes. After moulding, an AC voltage (10 V mm−1) was applied to a paste composed of cationic and anionic microgels. The microgels adhered to each other and formed an elastic gel-like material due to AC electric field application on the x, y, and z axes.
Characterization of microgel pastes and architectures
The viscoelastic properties of the microgel pastes were determined using a rheometer (Thermo Scientific HAAKE RheoStress 6000). The mechanical strengths of the microgel architectures were measured using a tensile tester (Shimadzu EZ-S10) in an ambient atmosphere. To visualize the microgel pastes and architectures, the cationic gels were stained with methyl orange, and the anionic gels were stained with methylene blue. The mechanical properties were measured without microgel staining.
Results and discussion
We prepared the microgel architectures by the electrophoretic adhesion of cationic and anionic microgels three-dimensionally. Cationic/anionic microgels with diameters of 150/300 μm were used for the preparation of microgel architectures. The chemical structures and diameter distributions of the microgels are shown in Fig. 2. At first, we measured the shear frequency dependence of the viscoelastic properties of the microgel pastes using a rheometer. The G′ values (storage modulus) of the microgel pastes were larger than the corresponding G′′ values (loss modulus) at 0.1 Hz, whereas the inverse phenomenon (G′′ < G′) was observed over ca. 0.3 Hz, suggesting a sol state. Therefore, microgel pastes are stable without dripping but are able to be scooped up for painting desired shapes (Fig. 3a). The microgel pastes which were stained blue, green, and yellow separately before electrophoresis were moulded into shapes by step-wise painting (Fig. 3b and c), and the microgel architectures were quickly fabricated within 10 s using only an electric field impression on the microgel paste.21 These results indicated that 3-D freeform fabrications were carried out without templates by the painting and adhesion of pastes. The prepared microgel architectures were stable without leakage of microgels in water after removal of the electric field, and were elastic and durable enough to handle with forceps as shown in Fig. 3d.
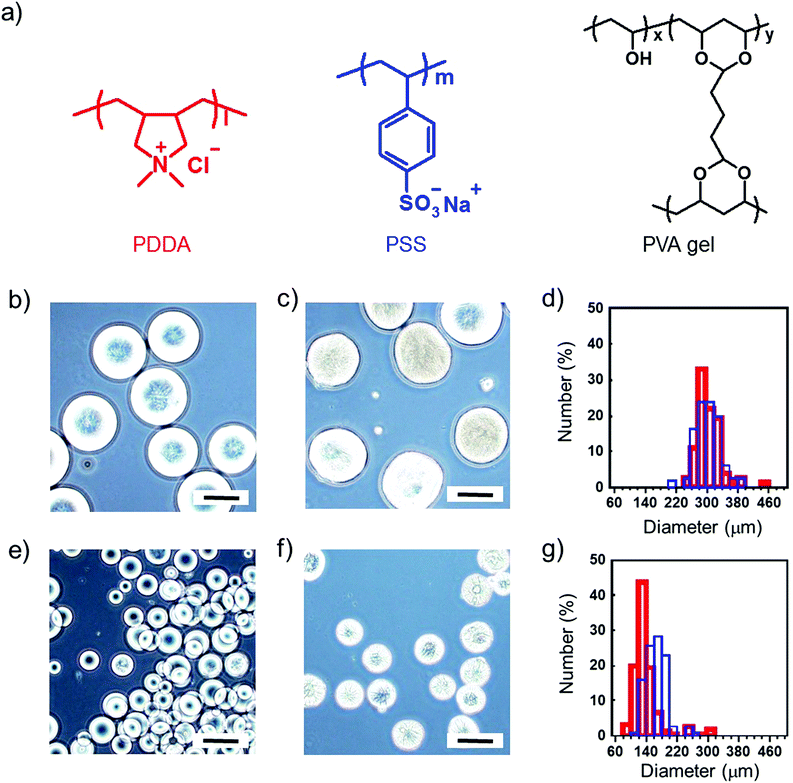 |
| | Fig. 2 (a) Chemical structures and (b–g) characterization of cationic (PDDA) and anionic (PSS) PVA microgels. The lyophilized microgels were separated between diameters of (b–d) 100 to 150 μm, and (e–g) 250 to 300 μm by a sieving mesh, and swelled in water. (d and g) Size distribution of the microgels. The red and blue bars represent cationic (b and e) and anionic (c and f) microgels, respectively. Data were collected from 50 objects. All scale bars are 200 μm. | |
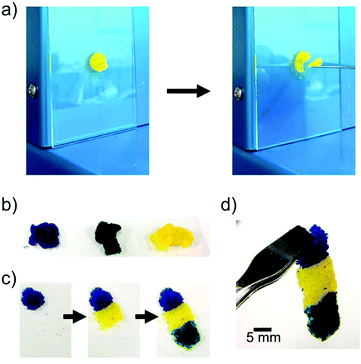 |
| | Fig. 3 Preparation of microgel architectures by the electrophoretic adhesion of microgels. (a) Microgel pastes are stable without dripping but are able to be scooped up. (b) Pastes which consisted of oppositely charged microgels (stained blue, green, and yellow). (c) The moulding process by painting. (d) The microgel architecture was elastic and durable enough to handle with forceps by electrophoretic adhesion. | |
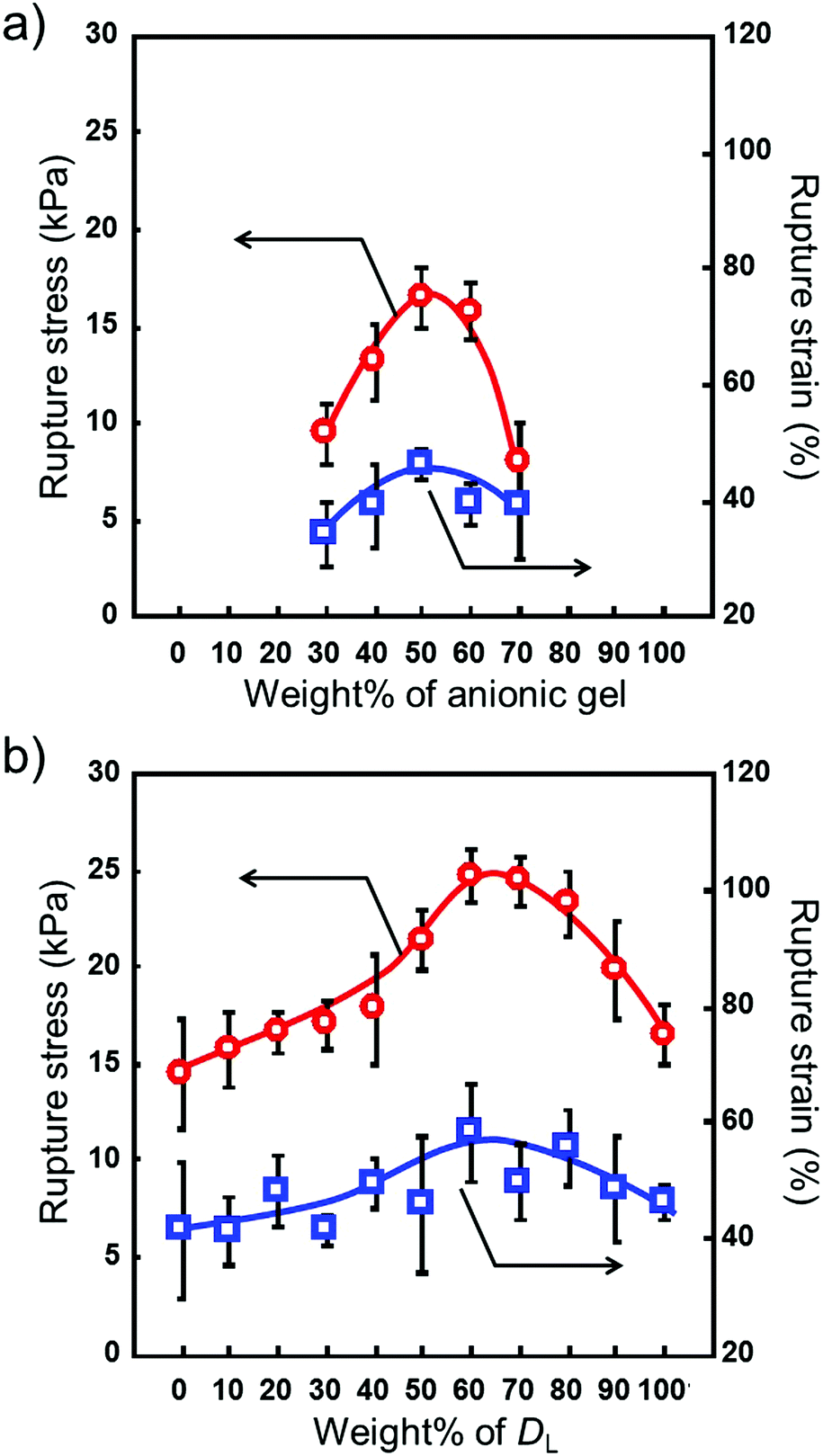
We prepared microgel architectures using a microgel with a diameter of 300 μm by changing the charge ratio. The charge ratio defined the amount of cationic (C) and anionic (A) microgels (C/A ratio). Hydrogel constructs did not form with a C/(C + A) ratio of up to 0.2. As shown in Fig. 4a, the rupture stress of microgel architectures increased with increasing C/(C + A) values (from 0.3 to 0.5), and the maximum value of rupture stress was obtained at 0.5. When the C/(C + A) values were over 0.5, the rupture stress decreased with increasing C/(C + A) values, and microgel architectures did not form over 0.8. Similar results were obtained when a microgel with a 150 μm diameter was used for the preparation of microgel architectures. These results indicate that microgel architectures were formed by the adhesion of cationic and anionic microgels, and more than a critical number of oppositely charged microgels is necessary for the formation of 3D networks.
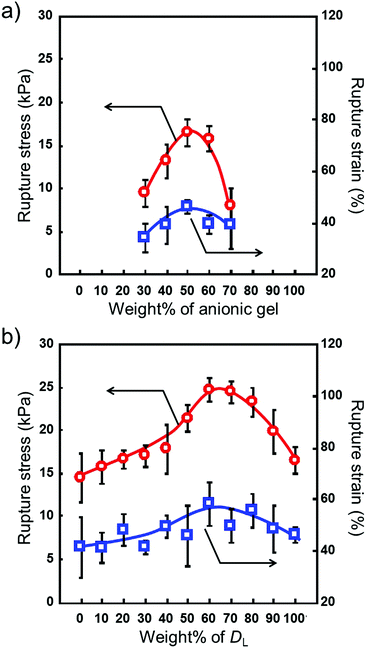 |
| | Fig. 4 Rupture stress and strain of microgel architectures prepared by (a) changing the amount of cationic/anionic microgels and (b) using a larger/smaller ratio. The circle and square symbols represent the rupture stress and strain, respectively. (a) Microgel architecture prepared using 300 μm microgels. (b) The C/A ratio is 1/1. Data are expressed as the mean ± SD (n = 3). | |
The microgel architectures were prepared singly and as a mixture of two diameters. The C/A ratios were fixed to 1.0. The average diameter of the microgels used here was 150 (smaller microgels: DS) and 300 (larger microgels: DL) μm. Although no significant difference was observed for the single diameter architectures, the rupture stress for a mixture of large and small microgels was higher than that for the microgel architectures prepared singly. As shown in Fig. 4b, the mechanical strength of the microgel architectures increased with an increasing amount of larger microgels. Upon reaching the maximum value around 60% of larger microgels, a decrease in mechanical strength was observed. This result indicates that the mechanical strength of microgel architectures is controlled by the mixing ratio of larger and smaller microgels. To study the change in the mechanical strength of microgel architectures, we utilized the random packing model proposed by Furnas.22,23 For an ideal binary mixture consisting of particles with infinitely different sizes, the maximum packing density (fmax) is given by eqn (1), and this condition occurs at a composition of xmax = fL/fmax.
| | | fmax = fL + (1 − fL)fS | (1) |
where
xmax and
fL are the percentage and packing density of larger particles, respectively, and the remaining interstitial volume (1 −
fL) is filled by the smaller particles at a density of
fS. At compositions with more or fewer large particles than
xmax, the packing densities follow
eqn (2) and (3).
| | | f = fS/(fSx + 1 − x) for x < xmax | (2) |
where
x is the percentage of larger particles.
Eqn (1)–(3) indicate that
f increased to
fmax, and then decreased below
fmax with an increasing value of
x. Therefore, the tendency of the rupture stress of microgel architectures with the binary mixture observed here is in good agreement with the packing density of Furnas's model. In this combination of larger (
DL) and smaller (
DS) microgels, the maximum value of rupture stress was 24.7 ± 1.4 kPa when
DL/
DS was 3/2, indicating a high degree of microgel packing. The rupture strain shows a tendency to increase with the increasing rupture stress of microgel architectures (
Fig. 4; square symbol). Although direct observation of microgel packing inside the microgel architecture is difficult because of the 3D structure, the mixing ratio of larger and smaller microgels does not contradict the literature.
24 Therefore, we can control the mechanical strength with a constant charge density by changing the mixing ratio of larger and smaller microgels, although the charge ratio as well as the mechanical strength changed when the
C/
A ratio was changed during the preparation of the microgel architectures.
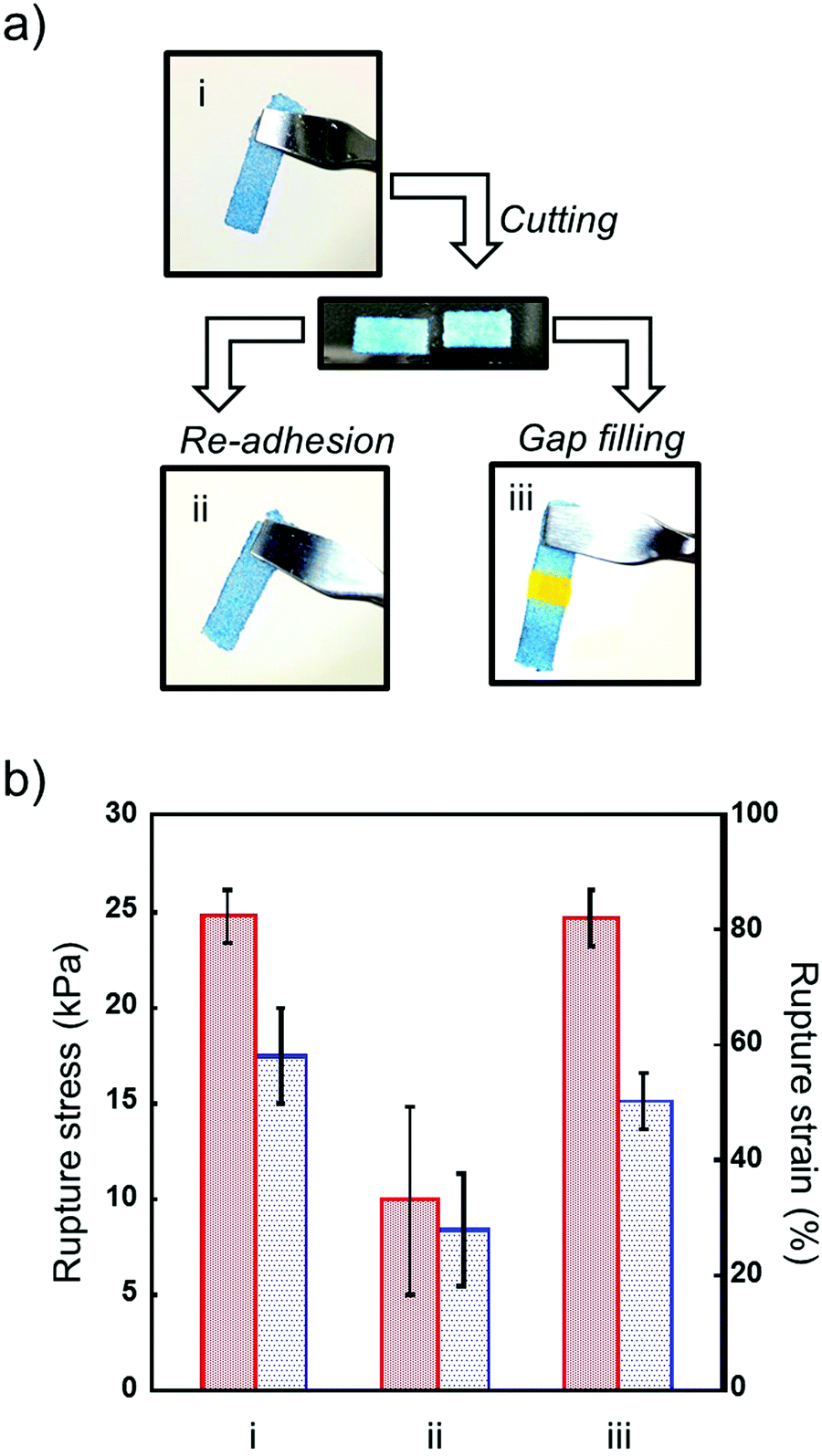
Next, we investigated the mechanical strength of repaired microgel architectures. Microgel architectures which were prepared by adhesion of microgels were cut into to two pieces and used for the repairing experiment (Fig. 5a). Unlike self-healing hydrogels, cut microgel architectures did not show self-healing properties. However, we can rapidly repair a broken microgel architecture by the re-application of voltage on both of the hydrogel edges. The mechanical strength of the re-adhered microgel architectures decreased to 40% of the initial value due to insufficient network formation. Previously, we reported that the microgel architectures turned back into a microgel paste by grinding to obliterate the adhesion interfaces, and microgel architectures could be reconstructed using reused microgel.21 However, the mechanical strength was significantly decreased by reusing microgel pastes. Therefore, we tried repairing cut microgel architectures by gap filling with microgel paste between the two broken pieces. Interestingly, the mechanical strength recovered to almost 100% of the initial value by the gap-filling and adhesion of microgels (Fig. 5b). These results indicate that the repair of broken hydrogels can be performed many times by the removal of broken parts and gap-filling using virgin microgel pastes.
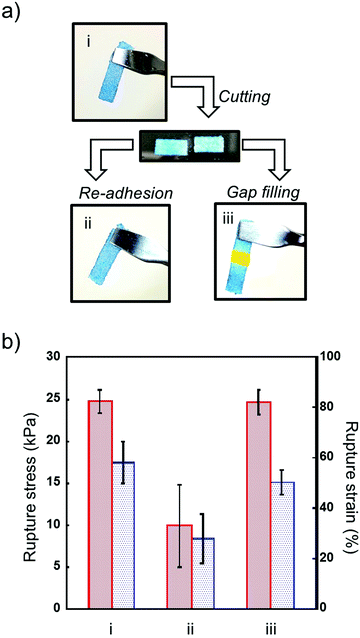 |
| | Fig. 5 (a) Representation of the repair of broken microgel architectures. (b) Rupture stress (left) and strain (right) of the as-prepared (i), re-adhered (ii), and repaired (iii) microgel architectures. The composition of microgel architectures is DL/DS = 3/2 and C/A = 1/1. Data are expressed as the mean ± SD (n = 3). | |
Although the self-healing property of cracked hydrogels has been reported, it is difficult to repair hydrogels with large amounts of deletion. However, in this study, pastes with good formability were able to repair broken hydrogels in a do-it-yourself manner (Fig. 6). We prepared a deletion-formed hydrogel construct, and a defective part of the hydrogel was filled with microgel paste and repaired by the adhesion of microgels (Fig. 6a). The mechanical strength of the hydrogel before and after repairing was investigated by tensile strength measurement (Fig. 6b). The rupture stress of the defective hydrogel decreased to 30% of the initial value, and we observed a propagation of the fracture from the crack edge. When the defective part of the hydrogel was repaired by the microgel paste, the mechanical strength of the hydrogel recovered to almost 100% of the initial value because the propagation of the fracture was suppressed by microgel packing. Unlike gel-formation from the aqueous pre-gel solution, the template for the preparation and purification of hydrogels such as the washing of unreactive monomers is not required, because the microgel architectures were formed by the adhesion of microgels. Moreover, excess swelling/deswelling was not observed after the preparation of the microgel architectures. Therefore, this method is useful for the do-it-yourself repair of broken hydrogels because of the template-free fabrication based on the painting of microgel pastes as shown in Fig. 1.
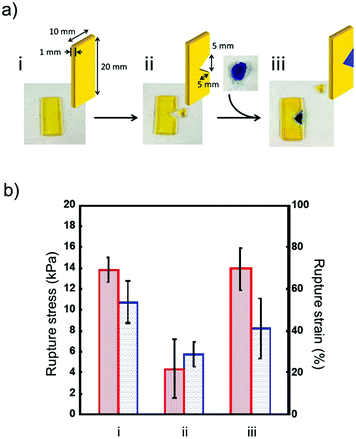 |
| | Fig. 6 (a) Representation of the repair of broken hydrogels. (b) Rupture stress (left) and strain (right) of the as-prepared (i), broken (ii), and repaired (iii) hydrogels. Data are expressed as the mean ± SD (n = 3). | |
Conclusions
We demonstrated a do-it-yourself method for the repair of broken hydrogels using microgel architectures, which were prepared by the adhesion of oppositely charged microgels. The mechanical strength of the microgel architectures was easily controlled by changing the charge density and mixing ratio of larger and smaller microgels. The rapid repair of broken microgel architectures was performed by gap filling using microgel paste between two broken pieces. Deletion-formed hydrogels were also repaired by the filling of microgel architectures into defective parts of the hydrogels. This reported novel green manufacturing process for hydrogel repair contributes to a sustainable society.
Acknowledgements
This study was supported by the Futaba Electronics Memorial Foundation, the Sumitomo Foundation, and JSPS KAKENHI Grant Numbers JP16H06507 in Scientific Research on Innovative Area “Nano-Material Optical-Manipulation”. We thank Profs. Takashi Tsujimoto and Hiroshi Uyama at Osaka University for helpful rheology measurement. We also thank Profs. Tetsuro Shinada and Yutaka Amao at Osaka City University for helpful discussion.
Notes and references
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869 CrossRef CAS PubMed.
- T. Vermonden, R. Censi and W. E. Hennink, Chem. Rev., 2012, 112, 2853 CrossRef CAS PubMed.
- Y. Osada, H. Okuzaki and H. Hori, Nature, 1992, 355, 242 CrossRef CAS.
- Z. Hu, X. Zhang and Y. Li, Science, 1995, 269, 525 CAS.
- S. Maeda, Y. Hara, T. Sakai, R. Yoshida and S. Hashimoto, Adv. Mater., 2007, 19, 3480 CrossRef CAS.
- T. Asoh, M. Matsusaki, T. Kaneko and M. Akashi, Adv. Mater., 2008, 20, 2080 CrossRef CAS.
- T. Jungst, W. Smolan, K. Schacht, T. Scheibel and J. Groll, Chem. Rev., 2016, 116, 1496 CrossRef CAS PubMed.
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. R. Sriram, E. N. Brown and S. Viswanathan, Nature, 2001, 409, 794 CrossRef CAS PubMed.
- K. S. Toohey, N. R. Sottos, J. A. Lewis, J. S. Moore and S. R. White, Nat. Mater., 2007, 6, 581 CrossRef CAS PubMed.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698 CrossRef CAS PubMed.
- H. Tamagawa, F. Nagato, S. Umemoto, N. Okui, S. Popovic and M. Taya, Bull. Chem. Soc. Jpn., 2002, 75, 383 CrossRef CAS.
- A. Harada, R. Kobayashi, Y. Takashima, A. Hashidzume and H. Yamaguchi, Nat. Chem., 2011, 3, 34 CrossRef CAS PubMed.
- S. Rosel, A. Prevoteau, P. Elzière, D. Hourdet, A. Marcellan and L. Leibler, Nature, 2014, 505, 382 CrossRef PubMed.
- Q. Wang, J. L. Mynar, M. Yoshida, E. Lee, M. Lee, K. Okuro, K. Kinbara and T. Aida, Nature, 2010, 463, 339 CrossRef CAS PubMed.
- N. Holten-Andersen, M. J. Harrington, H. Birkedal, B. P. Lee, P. B. Messersmith, K. Y. C. Lee and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 265 CrossRef PubMed.
- K. Haraguchi, K. Uyama and H. Tanimoto, Macromol. Rapid Commun., 2011, 32, 1253 CrossRef CAS PubMed.
- T. L. Sun, T. Kurokawa, S. Kuroda, A. B. Ihsan, T. Akasaki, K. Sato, M. A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932 CrossRef CAS PubMed.
- T. Asoh and A. Kikuchi, Chem. Commun., 2010, 46, 7793 RSC.
- T. Asoh, K. Takaishi and A. Kikuchi, J. Mater. Chem. B, 2015, 3, 6740 RSC.
- M. Kato, Y. Tsuboi, A. Kikuchi and T. Asoh, J. Phys. Chem. B, 2016, 120, 5042 CrossRef CAS PubMed.
- T. Asoh and A. Kikuchi, Chem. Commun., 2012, 48, 10019 RSC.
- C. C. Furnas, Ind. Eng. Chem., 1931, 23, 1052 CrossRef CAS.
- R. M. German, Metall. Trans. A, 1992, 23, 1455 CrossRef.
- K. Woo, D. Kim, J. S. Kim, S. Lim and J. Moon, Langmuir, 2009, 25, 429 CrossRef CAS PubMed.
Footnote |
| † Present address: Department of Applied Chemistry, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: asoh@chem.eng.osaka-u.ac.jp |
|
| This journal is © the Partner Organisations 2017 |
Click here to see how this site uses Cookies. View our privacy policy here.