
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of dendrobatid alkaloid (+)-167B and (+)-209D and the investigation of diastereoselectivity using DFT calculations†
Wen-Hua Chiou* and
Hao-Yu Chen
Department of Chemistry, National Chung Hsing University, Taichung, Taiwan 402, ROC. E-mail: wchiou@dragon.nchu.edu.tw; Fax: +886-4-22862547; Tel: +886-4-22840411-420
First published on 3rd January 2017
Abstract
The synthesis of dendrobatid alkaloid (+)-167B and (+)-209D has been developed on the basis of the effective preparation of chiral tropinone 7-azabicyclo[3.2.1]octan-6-ol. DFT calculations have been applied to explain the observed diastereoselectivity.
Recently, indolizidine alkaloids commonly found in amphibian skin have received a great deal of attention due to their medicinal interest and diverse physiological properties.1 These indolizidine alkaloids isolated from the dendrobatid family were found to be noncompetitive blockers of the neuromuscular transmission receptor and nicotinic acetylcholine receptors, which allowed these compounds to be promising drug candidates for epilepsy, schizophrenia, Parkinson disease and Alzheimer disease.2,3 Since Daly's pioneering work in the 1970s, numerous valuable indolizidines with interesting structures have been isolated from poison-dart frogs,4,5 and 5-mono, 3,5-di-substituted or 3,5-di-substituted indolizidines occupy a large portion of the discovered structures.6,7 (Fig. 1). Accordingly, novel strategies for the asymmetrical synthesis of these azabicycles continue to receive considerable attention from the synthetic community.8 As part of our interests in the synthesis of the dendrobatid alkaloids,9 we wish to demonstrate a general and efficient protocol to prepare these alkaloids. Here we report syntheses of (+)-167B and (+)-209D as an application of our efficient preparation of enantiomerical tropanol, and a rationale of the observed diastereoselectivity using the DFT calculations.
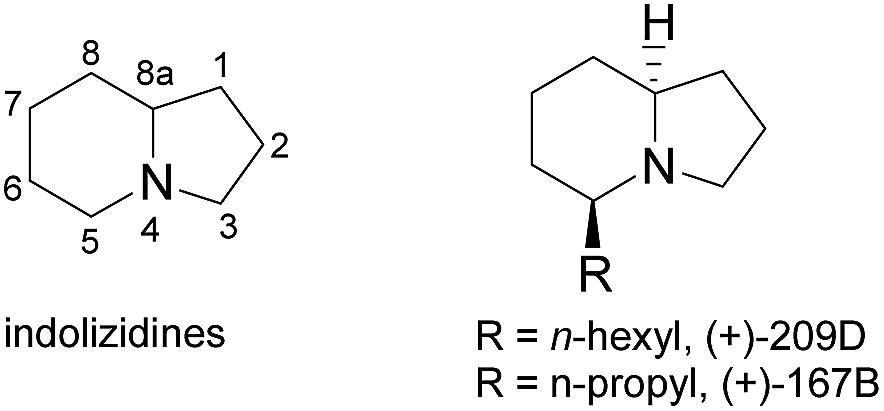 | ||
| Fig. 1 Substituted structure and numbering of indolizidines. | ||
Our approach to indolizidine 209D and 167B relies on efficient preparation of enantiopure 7-benzyloxycarbonylazabicyclo-[3.2.1]octan-6-one (1), readily available in both dextrorotatory and levorotatory forms by diastereomeric recrystallization of 7-azabicyclo[3.2.1]octan-6-ol with tartaric acid, and then protection of the free amine and subsequent oxidation of the hydroxyl group on basis of our previous progress (∼20% overall yield after 3 steps and resolution).10 The process proved to be an effective and reliable procedure for multi-gram scale production of enantiopure 7-azabicyclo[3.2.1]octan-6-ol with excellent chiral integrity. Bicyclic ketone 1 was subjected to Baeyer-Villiger oxidative ring expansion with m-CPBA in dichloromethane to give bicyclic (−)-lactone 2, which was a quite unstable substance in either acidic or oxidative conditions. Treatment of crude lactone 2 with a mixture of BF3·OEt2 and trimethylallylsilane resulted in cleavage of the oxabicyclic ring and formation of a transient N-acyl iminium ion which was captured by trimethylallylsilane, affording a 2,6-disubstituted piperidine 3 as a single diastereomer in 84% yield over two steps. The relative stereo configuration would be determined in the latter stage by comparison with known structures because the nOe signals of two key methines in piperidine 3 were so ambiguous (vide infra). Lactone 2 was a useful intermediate for syntheses of piperidine or quinolizidine compounds. For example, treatment with triethylsilane in the conditions would produce homopipecolic acid 4, which can be converted to (−)-lupinine, a quinolizidine alkaloids, according to the Davies's procedure.11 Here we would like to point out that enantiomerically pure 2,6-disubstituted piperidine 3 would be a versatile building block for syntheses of indolizidine or quinolizidine compounds, due to the two differential side chains for further construction of the second ring moiety (Scheme 1).
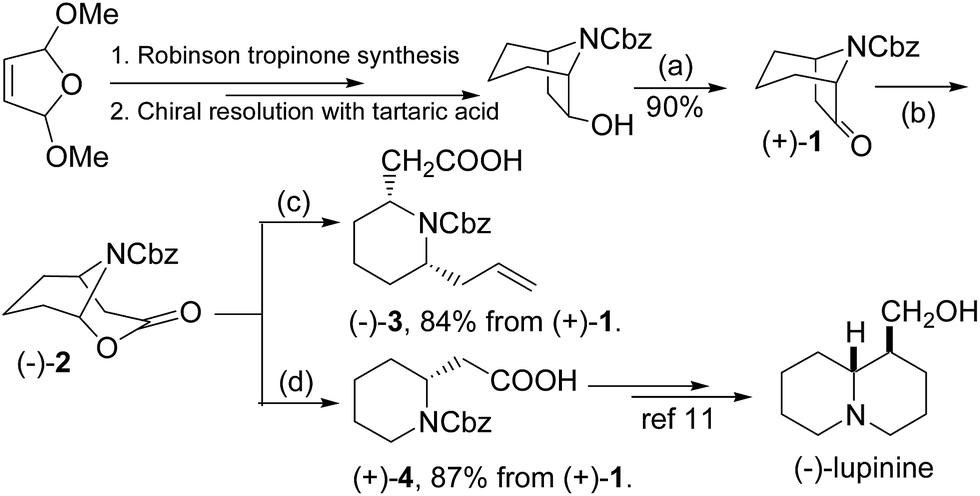 | ||
Scheme 1 Preparation of cis-2,6-disubstituted piperidine and piperidinyl acetic acid from chiral 6-tropanol derivative. (a) TEMPO, NaOCl, KBr, NaHCO3, acetone, 0 °C. (b) mCPBA, Na2HPO4, CH2Cl2, rt. (c) BF3–Et2O, Me3SiCH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2, CH2Cl2, −78 °C. (d) BF3–Et2O, Et3SiH, CH2Cl2, −78 °C. CH2, CH2Cl2, −78 °C. (d) BF3–Et2O, Et3SiH, CH2Cl2, −78 °C. | ||
To complete the synthesis of the target molecules, treatment of acid 3 with SOCl2 in cold methanol resulted in the formation of methyl ester 5 in 92% yield. The allyl portion of 18 was converted to a linear alcohol group using the modified Kabalka's hydroboration–oxidation procedure,12 i.e. sodium perborate, to furnish alcohol 6 in 80% overall yield. The temperature control was critical in this reaction. If the reaction temperature was higher, i.e. 0 °C, it would resulted in yield loss to 40%, probably due to the reduction of the ester moiety.13 Alcohol 6 was reacted with MsCl in the presence of Et3N in dichloromethane to produce mesylate 7, which was treated under ambient hydrogen pressure to remove the benzyl group, and initiated cyclization to yield indolizidine 7 in 88% yield. Having achieved the synthesis of the crucial indolizidine intermediate 7, we continued to use the product to complete the syntheses of 209D and 167B. Reduction of ester 8 group with LiAlH4 yielded 89% yield of primary alcohol 9, which was further converted to the corresponding tosylate 10 in 92% yield. The resulting tosylate 10 was alkylated with dibutylcyanocuprate, a high order cyanocuprate prepared by treatment of cupper cyanide with two-fold butylithium, to furnish butylated product 11a in 64% yield. The 13C signals of 11a were identical to the reported values of cis-(5R,8aS)-5-hexyl-indolizidine,14 while the specific rotation of the compound ([α]25D +92.6° (c: 1.0, CH2Cl2), literature value: +86.6° (c: 1.3, CH2Cl2)) also compared favourably.15 Similarly, tosylate 10 was also reacted with dimethylcyanocuprate to produce the product 11b in 61% yield, in which the 13C signals and the specific rotation of 11b were consistent with the reported values of cis-(5R,8aS)-5-propyl indolizidine16 ([α]25D +115.0° (c: 1.3, CH2Cl2), literature value for its enantiomer: −111.3° (c: 1.3, CH2Cl2)) (Scheme 2).15
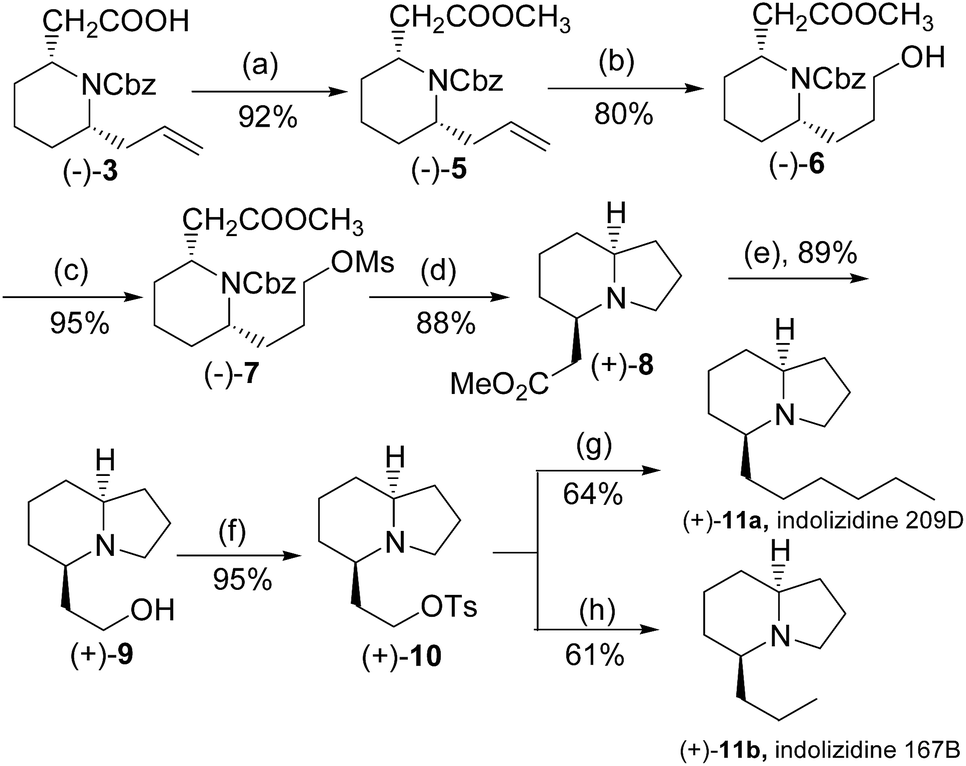 | ||
| Scheme 2 Synthesis of indolizidine 167B and 209D. (a) SOCl2, MeOH, 0 °C. (b) (i) BH3–THF, THF, −50 °C, (ii) NaBO3, H2O, rt. (c) MsCl, Et3N, CH2Cl2, rt. (d) Pd/C, H2, Et3N, MeOH, rt. (e) LiAlH4, THF, rt. (f) TsCl, Et3N, CH2Cl2, rt. (g) CuCN, MeLi, Et2O, 0 °C. (g) CuCN, n-BuLi, Et2O, −78 °C. | ||
To explain the resulting stereochemistry outcome, we propose the addition proceeded in a two-step manner.17 BF3-mediated coordination on the lactone moiety immediately results in the ring cleavage, and therefore the formation of the transient N-acyliminium intermediate. Subsequent silane addition on the intermediate furnishes the cis-2,6-disubstituted piperidine stereoselectively. First of all, we consider the prototype reaction in which trimethylallylsilane reacts on a substituent-free N-methoxycarbonyl Δ1-piperideinium. Trimethylallylsilane may take either axial or equatorial approach to react on the piperideinium bearing either s-trans or s-cis configuration, which is achieved by rotation of the carbamate bond.18 Thus, we perform the DFT calculations at the level of B3LYP/6-31++G (d, p) to obtain all four possible TS geometries during the silane addition (Fig. 2).
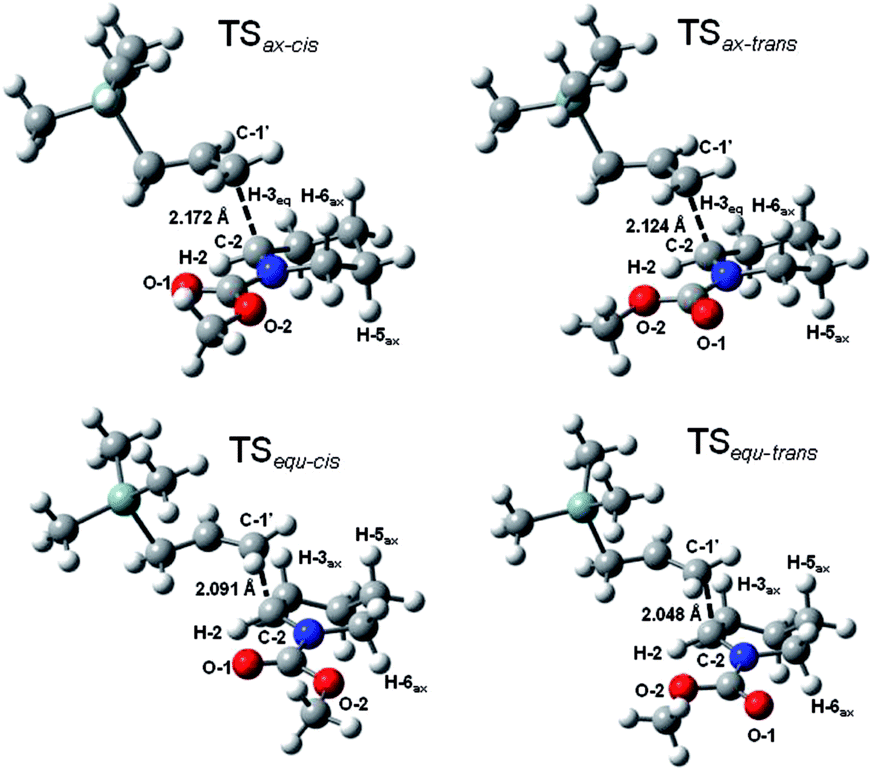 | ||
| Fig. 2 Four TS geometries describing axial and equatorial silane additions on N-methoxycarbonylpiperidenium with s-cis and s-trans configuration. | ||
The results in Table 1 have disclosed the stabilities of these four transition states appear to be concerned with the piperideinium conformations and electrostatic attractions between two major dipoles, the iminium and the carbonyl groups. The s-cis-configuration guarantees best coulombic attraction by disposing the most positive iminium carbon atom (C-2) as close in space to the most negative carbonyl oxygen atom (O-1) as possible.19 In addition, with a pseudo-chair conformation, TSax-cis and TSax-trans structures involved with the axial approach are more stable than TSequ-cis and TSequ-trans which bears with a twist boat conformation. In fact, the conformation distortion can be best characterized and evaluated by the dihedral angle ϕ1, the angle of the iminium hydrogen and the equatorial hydrogen at the C-3 position (Table 1). Severe deviation from the typical gauche angle (∼60°) is noticed (Δϕ1 ∼ 45°) and strong eclipsing interaction is also expected. A large angle deviation implies a local eclipse conformation, and naturally results in a relative unstable twist boat conformation, while a small deviation ensures a stable pseudo-chair conformation. Thus, we conclude that the axial addition on the s-cis-carbamate substrate dominates among these four possible reaction routes since the TSax-cis is the most stable among the four possible transition state geometries. It is worthy to point out that the electrostatic interactions and the ring conformations seem to contribute roughly equally in the stabilities of these transition states, and a longer reaction distance in the transition state geometries will be allowed in case of that either beneficial factor is hold.
| Rel E | Imag ν | ϕ1 | C2–C1′ | |
|---|---|---|---|---|
| a Energy in kcal mol−1, imagine frequency in cm−1, ϕ1 is the dihedral angle ∠H2–C2–C3–H3equ, distance in Å. | ||||
| TSax-cis | 0.0 | 208 | 55.0 | 2.172 |
| TSax-trans | + 2.3 | 232 | 54.5 | 2.174 |
| TSequ-cis | + 3.0 | 246 | 15.2 | 2.091 |
| TSequ-trans | + 5.8 | 259 | 15.8 | 2.048 |
Next, we consider two possible syn and anti approach in the axial addition manner on the Z-N-methoxycarbonyl Δ1-piperideinium bearing an acetic group at the C-6 position on the basis of the favored transition state TSax-Z, and obtain two TS structures TSsyn and TSanti which lead the cis- and trans-2,6-disubstituted adduct respectively (Fig. 3). The results show the TSsyn is favoured over TSanti by 8.1 kcal mol−1, indicating the silane addition prefers to proceed in “syn addition” to yield the cis-2,6-disubstituted adduct.
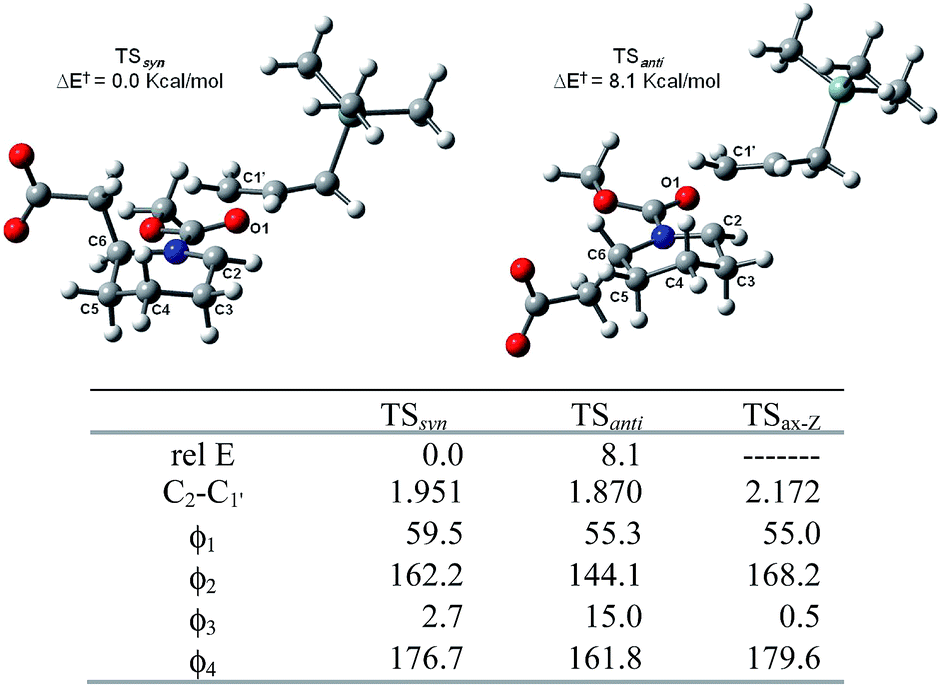 | ||
| Fig. 3 syn and anti approach TS geometries with axial silane addition on 6-substituted N-methoxycarbonyl Δ1-piperideinium. Energy in kcal mol−1, distance in Å, ϕ1: ∠H2–C2–C3–H3equ, ϕ2: ∠C5ax–C5–C6–C6ax, ϕ3: ∠C2–N–CO1, ϕ4: ∠C2–N–C–OMe. | ||
In the TSsyn structure, the piperideinyl ring with the axial substituent on the C-6 position keeps a less strained conformation because of small deviations from the parent structure (Δϕ2 = 6°). In addition, analysis of the TSsyn structure reveals good planar geometry in the carbamate group (ϕ3 = 2.7° and ϕ4 = 176.7°), suggesting the double bond character in the carbamate remains nearly intact during silane approaching. The distortion of the piperideinium ring is best illustrated by dihedral angle ϕ2, the angle of the two axial substituents at the C-5 and C-6 position, while dihedral angle ϕ3 and ϕ4 are used to describe the planarity of the carbamate. Since the syn approach to the cis-2,6-disubstituted piperidine adduct does not distort either the planarity or the ring conformation, a long distance between C1′–C2 in transition state has been allowed. Comparing to the TSsyn geometry, the TSanti geometry shows that the anti addition does not only alter the dihedral angle ϕ2 to 144 degree, but also change the dihedral angle ϕ4 to 162 degree, significantly attenuate the planarity of the carbamate group (ϕ3 = 15.0° and ϕ4 = 161.8°) and twist the conformation of the Δ1-piperideinium ring to suffer more strain compared to that in the non-substituent case (Δϕ2 = 24.1°). Both the piperideinyl moiety and the carbamate moiety need to be distorted to accommodate the equatorial substituent as the anti approach progressed. Thus, anti addition will result in the instability of the corresponding transition state, and the absence of the 2,6-trans product. The traditional explanation argues that a stable reactant conformation dominates stereo outcome: since the A1,3-strain exerted from a substituent on the nitrogen has imposed an pseudoaxial orientation on the C-6 substituent as a stable reactant conformation, only an nucleophilic addition is only allowed from the least hindered face, i.e. an axial addition, to afford the cis-2,6-disubstituted product.20
Our calculations provide different perspectives, in which the stable transition state is crucial to determine the possible reaction routes. Three major factors, distortion in the ring conformation, intactness of carbamate planarity and electrostatic attraction, contribute to the stabilities of the transition states and dominate the silane addition to proceed in the syn-approach on the C-6 substituted Δ1-piperideinium with s-cis-carbamate configuration in the axial manner.
In conclusion, we have presented syntheses of two dendrobated alkaloids, (+)-indolizidine 209D and (+)-indolizidine 167B, featuring applications of readily available enantiomerical 6-tropinonol to the 2,6-disubstituted piperidines and 5-substituted indolizidines via Baeyer-Villiger oxidation. The DFT results described above provides a transition state-based rationale of the observed cis-selectivity for the formation of only cis-2,6-disubstituted piperidine product. Further applications to interesting targets and extension of calculations are under active investigation.
Acknowledgements
The research was supported by the National Science Council, Taiwan (NSC104-2113-M-005-004), and National Center for High-Performance Computing for computation facilities.Notes and references
- (a) J. P. Michael, Nat. Prod. Rep., 2008, 25, 139 RSC; (b) J. W. Daly and T. F. Spande, Alkaloids: Chemical and Biological Perspectives: Amphibian Alkaloids: Chemistry, Pharmacology and Biology, Wiley Interscience, New York, 1986 Search PubMed.
- N. Toyooka, T. Hirosho, K. Soushi, Z. Dejun, K. Masashi, K. Ikuto, S. Toshiyasu and N. Hideo, Curr. Chem. Biol., 2007, 1, 87 Search PubMed.
- A. M. Lourenco, P. Maximo, L. M. Ferreira and M. M. A. Pereira, Stud. Nat. Prod. Chem., 2002, 27, 233 CrossRef CAS.
- J. W. Daly, H. M. Garraffo, T. F. Spande, M. W. Decker, J. P. Sullivan and M. Williams, Nat. Prod. Rep., 2000, 17, 131 RSC.
- J. W. Daly, T. F. Spande and H. M. Grraffo, J. Nat. Prod., 2005, 68, 1556 CrossRef CAS PubMed.
- R. A. Saporito, T. F. Spande, H. M. Garraffo and M. A. Donnelly, Heterocycles, 2009, 79, 277 CrossRef CAS.
- F. Abeis, C. Lindermann, E. Koch and C. Schneider, Org. Lett., 2012, 14, 5972 CrossRef PubMed.
- For selected excellent reviews of syntheses of enantionmerical indiolizidines, see (a) S. M. Bronner, G.-Y. J. Im and N. K. Garg, in Heterocycles in Natural Product Synthesis, ed. K. C. Majumdar and S. K. Charropadhyay, 2011, pp. 221–265 Search PubMed; (b) R. Lazzaroni and R. Settambolo, Chirality, 2011, 23, 730 CrossRef CAS PubMed; (c) A. Brandi, F. Cardona, S. Cicchi, F. M. Cordero and A. Goti, Chem.–Eur. J., 2009, 15, 7808 CrossRef CAS PubMed; (d) S. Agarwal, S. Cämmerer, S. Filali, W. Fröhner, J. Knöll, M. P. Krahl, K. R. Reddy and H.-J. Knölker, Curr. Org. Chem., 2005, 9, 1601 CrossRef CAS; (e) S. Hanessian, E. Therrien, J. S. Warrier and G. Charron, Heterocycles, 2006, 70, 461 CrossRef CAS; (f) M. S. M. Timmer, S. H. L. Werheist, G. M. Grotenbreg, M. Overhand and H. S. Overkleefe, Pure Appl. Chem., 2005, 77, 1173 CrossRef CAS.
- W.-H. Chiou and Y.-M. Chiang, RSC Adv., 2014, 4, 11444 RSC.
- W.-H. Chiou, G.-T. Chen, C.-L. Kao and Y.-K. Gao, Org. Biomol. Chem., 2012, 10, 2518 CAS.
- S. G. Davies, A. M. Fletcher, E. M. Foster, I. T. T. Houlsby, P. M. Roberts, T. M. Schofiled and J. E. Thomson, Chem. Commun., 2014, 50, 8309 RSC.
- G. W. Kabalka, T. M. Shoup and N. M. Goudgaon, J. Org. Chem., 1989, 54, 5930 CrossRef CAS.
- E.-K. Shin, H. J. Kim, Y. Kim, Y. Kim and Y. S. Park, Tetrahedron Lett., 2006, 47, 1865 CrossRef.
- (a) For the reported 13C-NMR peak values of cis-209D, see R. T. Yu, E. E. Lee, G. Malik and T. Rovis, Angew. Chem., Int. Ed., 2009, 48, 2379 CrossRef CAS PubMed; (b) For the reported 13C-NMR peak values of trans-209D, see C. Alegret and A. Riera, J. Org. Chem., 2008, 73, 8661 CrossRef CAS PubMed.
- P. G. Reddy and S. Baskaran, J. Org. Chem., 2004, 69, 3093 CrossRef CAS PubMed.
- (a) For the reported 13C-NMR peak values of cis-167B, see A. Kapat, E. Nyfeler, G. T. Giuffredi and P. Renaud, J. Am. Chem. Soc., 2009, 131, 17746 CrossRef CAS PubMed; (b) For the reported 13C-NMR peak values of trans-167B, see A. K. Saikia, K. Indukuri and J. Das, Org. Biomol. Chem., 2014, 12, 7026 RSC.
- (a) W. N. Speckamp and M. J. Moolenaar, Tetrahedron, 2000, 56, 3817 CrossRef CAS; (b) R. Remuson, Beilstein J. Org. Chem., 2007, 3, 1 CrossRef PubMed; (c) M. Node, A. Itoh, Y. Masaki and K. Fuji, Heterocycles, 1991, 32, 1705 CrossRef CAS.
- Calculations also discloses that the s-cis conformer is more stable than the s-trans one by 0.6 kcal mol−1 in free rotation of carbon-nitrogen bond of a substituent-free N-methoxycarbonyl Δ1-piperideinium. It can be also rationalized by the concept of “dipole cancellation”, a critical factor in nonpolar solvents. The rotation energy barriers from the s-cis form to the s-trans form are about 6.1 to 6.3 kcal mol−1 depending on which ways of two possible rotations of the carbonyl group, moving through either the equatorial substituent side at the C-6 or the axial one.
- (a) I. Flaming, Molecular Orbitals and Organic Chemical Reactions, John Wiley & Sons, 2009 Search PubMed; (b) E. V. Anslyn and D. A. Dougherty, in Modern Physical Organic Chemistry, University Science Books, 2006, p. 123 Search PubMed.
- (a) T. Luker, H. Hiemstra and W. N. Speckamp, J. Org. Chem., 1997, 62, 3592 CrossRef CAS; (b) M. P. Cassidy and A. Padwa, Org. Lett., 2004, 6, 4029 CrossRef CAS PubMed; (c) S.-S. P. Chou, T.-H. Yang, W.-S. Wu and T.-H. Chiu, Synthesis, 2011, 759 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, 1H, 13C NMR spectra, stereochemistry assignment and calculated geometry coordinates. See DOI: 10.1039/c6ra24960d |
| This journal is © The Royal Society of Chemistry 2017 |