DOI:
10.1039/C6RA25252D
(Paper)
RSC Adv., 2017,
7, 1877-1886
Structure and photocatalytic activity of a low band gap donor–acceptor–donor (D–A–D) type conjugated polymer: poly(EDOT–pyridazine–EDOT)
Received
14th October 2016
, Accepted 30th November 2016
First published on 12th January 2017
Abstract
A donor–acceptor–donor (D–A–D) type monomer (3,6-bis(2-(3,4-ethylenedioxy thiophene))pyridazine, EPE) with pyridazine as an intermediate unit (acceptor) and 3,4-ethylenedioxythiophene (EDOT) as a sealing unit (donor) was prepared. The monomer was then polymerized by adjusting various ratios of FeCl3/monomer to prepare a donor–acceptor–donor (D–A–D) type conjugated polymer: poly(EPE). The results from a structural analysis indicated that the conjugation length, doping level, thermal stability, crystallinity, as well as the morphologies of the poly(EPE)s can be affected by the oxidant![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :monomer ratio. The resulting products were characterized by 1H NMR, FT-IR, UV-vis, TGA, XRD, EDX and SEM. Studies were carried out on the photocatalytic performance of the poly(EPE)s for the photocatalytic degradation of different dyes (methylene blue (MB), methyl violet (MU), methyl orange (MO), rhodamine B (RhB) and phenol) (5 mg L−1) under xenon-lamp irradiation, and the same type of light source and different catalysts (poly(EPE)s and PEDOT) on the degradation efficiency of MU were investigated, which showed that poly(EPE) was an effective photocatalyst, and the presence of Fe ions in the polymer matrix played a significant role in enhancing the photocatalytic activity of poly(EPE). Among the poly(EPE)s, the doped poly(EPE)2 ([FeCl3]/[EPE] ratio of 8:1) achieved the highest degradation efficiency (95.53%) for MU dye under the simulation of a visible light source for 300 min.
:monomer ratio. The resulting products were characterized by 1H NMR, FT-IR, UV-vis, TGA, XRD, EDX and SEM. Studies were carried out on the photocatalytic performance of the poly(EPE)s for the photocatalytic degradation of different dyes (methylene blue (MB), methyl violet (MU), methyl orange (MO), rhodamine B (RhB) and phenol) (5 mg L−1) under xenon-lamp irradiation, and the same type of light source and different catalysts (poly(EPE)s and PEDOT) on the degradation efficiency of MU were investigated, which showed that poly(EPE) was an effective photocatalyst, and the presence of Fe ions in the polymer matrix played a significant role in enhancing the photocatalytic activity of poly(EPE). Among the poly(EPE)s, the doped poly(EPE)2 ([FeCl3]/[EPE] ratio of 8:1) achieved the highest degradation efficiency (95.53%) for MU dye under the simulation of a visible light source for 300 min.
1. Introduction
Over the last two decades, organic π-conjugated materials, particularly π-conjugated polymers, have become an important research topic in the field of functional polymers, and have been employed in several fields such as in sensors,1,2 light-emitting diodes,3 optical displays,4 electrochromic windows,5 photodetectors,6 transistors,7 molecular electronics8 and energy applications.9–11 Among the various type of π-conjugated polymers, π-conjugated systems with a low band gap have attracted special attention due to their electrical, optical and photoelectric characteristics, and they have found important applications in the fields of photovoltaic and optoelectronic devices.12 Usually, the synthesis of a low band gap structure of π-conjugated polymers requires the introduction of electron-withdrawing and electron-releasing units to the polymer chain,11 where the electron-releasing units serve as electron donors, whereas the electron-withdrawing units serve as electron acceptors.13–15 When the polymer forms a “donor–acceptor–donor” type structure (D–A–D type), the electron donors and electron acceptors form massive π-bonds, which can enhance the intramolecular charge transfer and form a zwitterionic resonance structure. Furthermore, in π-conjugated polymers, the donor units are connected with the acceptor units and this enhances the property of the double bonds. The enhancement of the double bonds broadens the material's valence bands and conduction bands; this can effectively reduce the energy gap, and thus this unique structure displays excellent optoelectronic properties.16,17
The pyridazine unit is one of the strongest electron-withdrawing units because it contains two electron-withdrawing imine nitrogens (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) in the structure.18 Furthermore, the lack of electrical characteristics of the pyridazine unit can reduce the minimum energy of the orbital energy level to effectively decrease the bandwidth of the polymer when the unit is combined with an electron-releasing unit.19 Among the electron-releasing units, 3,4-ethylenedioxythiophene (EDOT) is a major example of a thiophene derivative and can add interesting optoelectrical properties to the resulting polymer.20,21 Furthermore, EDOT has been universally used as a constitutional unit for several functional π-conjugated systems. Compared to the other thiophene derivatives, the higher electron-donating power of EDOT results in a stronger donor–acceptor interaction and a smaller band gap.22 Yamamoto et al. reported a π-conjugated monomer with pyridazine as the intermediate unit (electron acceptor) and thiophene as the sealing unit (electron donor), and this triplet compound was polymerized for achieving a low band gap D–A–D type conjugated polymer. They found that this type of D–A–D type conjugated polymer had the ideal charge transfer rate.19 Moon et al. reported a donor–acceptor–donor type liquid crystal with a pyridazine core (EDOT–pyridazine–EDOT) with two peripheral long alkyl chains, and the designated D–A–D type core structure could induce a distinct liquid crystalline smectic phase due to the strong intermolecular interaction between the donor and acceptor units.23
N–) in the structure.18 Furthermore, the lack of electrical characteristics of the pyridazine unit can reduce the minimum energy of the orbital energy level to effectively decrease the bandwidth of the polymer when the unit is combined with an electron-releasing unit.19 Among the electron-releasing units, 3,4-ethylenedioxythiophene (EDOT) is a major example of a thiophene derivative and can add interesting optoelectrical properties to the resulting polymer.20,21 Furthermore, EDOT has been universally used as a constitutional unit for several functional π-conjugated systems. Compared to the other thiophene derivatives, the higher electron-donating power of EDOT results in a stronger donor–acceptor interaction and a smaller band gap.22 Yamamoto et al. reported a π-conjugated monomer with pyridazine as the intermediate unit (electron acceptor) and thiophene as the sealing unit (electron donor), and this triplet compound was polymerized for achieving a low band gap D–A–D type conjugated polymer. They found that this type of D–A–D type conjugated polymer had the ideal charge transfer rate.19 Moon et al. reported a donor–acceptor–donor type liquid crystal with a pyridazine core (EDOT–pyridazine–EDOT) with two peripheral long alkyl chains, and the designated D–A–D type core structure could induce a distinct liquid crystalline smectic phase due to the strong intermolecular interaction between the donor and acceptor units.23
In addition, π-conjugated polymers with a favourable semiconductor band gap have achieved high photocatalytic activities.24 Patil et al. studied the degradation of azo dye OG by P3HT, MEH-PPV, and Degussa P25 under UV light and found that the highest photocatalytic rate of OG was approximately 80% after 60 min.24 Remita et al. found that the PEDOT nanostructures possessed better photocatalytic activity than P25-TiO2 for the degradation of MO under UV light, where the degradation efficiency reached 100% after 15 min.25 However, in real applications, these polymers may only achieve low photocatalytic activities because there is just 5% of UV light in visible light sources. Hou et al. reported a degradation efficiency of RhB by a conjugated polyene catalyst under visible light of only 20% after 160 min.26 In principle, shortening the forbidden band width of the catalyst to visible light absorption spectrum spreading is key to improving the photocatalytic activity of the catalyst.27 Therefore, a π-conjugated polymer with pyridazine as the intermediate unit (electron acceptor) and EDOT as the sealing unit (electron donor) with a low band gap and ideal charge transfer rate should be an ideal catalyst.
Moreover, the presence of a small amount of Fe ions in the polymer matrix is another important factor for enhancing the photocatalytic activity of the polymer.28 When the polymer absorbs visible light, it produces an electron (e−) that transfers to the conduction band of the Fe ions; this will lead to an enhancement in charge separation and the formation of hydroxyl and hyperoxide radicals (O2˙−, ˙OH). Accordingly, a high amount of radicals (O2˙−, ˙OH) leads to the polymer having a higher degradation efficiency for dyes under visible light. Furthermore, there are only a few reports in literature about the application of D–A–D type polymers in the field of dye degradation.
Herein, we prepared the donor–acceptor–donor (D–A–D) type monomer (3,6-bis(2-(3,4-ethylenedioxythiophene))pyridazine, EPE), which was then polymerized by adjusting various ratios of FeCl3/monomer to prepare a donor–acceptor–donor (D–A–D) type conjugated polymer: poly(EPE). Furthermore, the degradation of different types of dyes by different catalysts was explored under the simulation of visible light irradiation. In addition, the possible photocatalytic degradation mechanism of MU dye by poly(EPE) is presented herein.
2. Experimental
2.1 Materials
3,6-Dibromopyridazine (98%), 3,4-ethylenedioxythiophene (99%), n-butyl lithium, N-butyltin chloride (99%) and anhydrous ferric chloride were purchased from Aldrich. Pd(PPh3)4 was synthesized according to a method in the literature.29 All the other chemicals and solvents, including methylene blue, methyl violet, methyl orange, rhodamine B, phenol, methanol, and chloroform were used as received without further purification.
2.2 Instrumentation
The 1H NMR spectra of the products were obtained on Varian Inova 400 MHz with DMSO-d6 as the solvent. GPC analysis was carried on the Waters apparatus GPCV-1525 equipped with an RI detector using DMF as the eluent at 35 °C, and calibration was accomplished with polystyrene standards (Polysciences, JP). The FT-IR spectra of the samples were recorded on a BRUKER EQUINOX-55 Fourier transform infrared spectrophotometer using KBr pellets (Bruker, Billerica, MA, USA) (frequency range from 4000 to 400 cm−1). The UV-vis absorption spectra of the samples were measured with a UV-visible spectrophotometer (UV4802, Unico, USA). The thermal properties of the polymers were recorded via thermogravimetric analysis. The polymers were heated from 25 °C to 1000 °C at a heating rate of 10 °C min−1 under an argon atmosphere on a Hitachi STA7300 thermal analysis instrument. Phase characterization of the samples was done by X-ray powder diffraction (XRD, Bruker AXS D8 diffractometer) with monochromatic CuKα as the irradiation source (λ = 0.15418 nm), at a scan range (2θ) from 10° to 80°. SEM and EDX were conducted on a Hitachi SO8010 field emission scanning electron microscope.
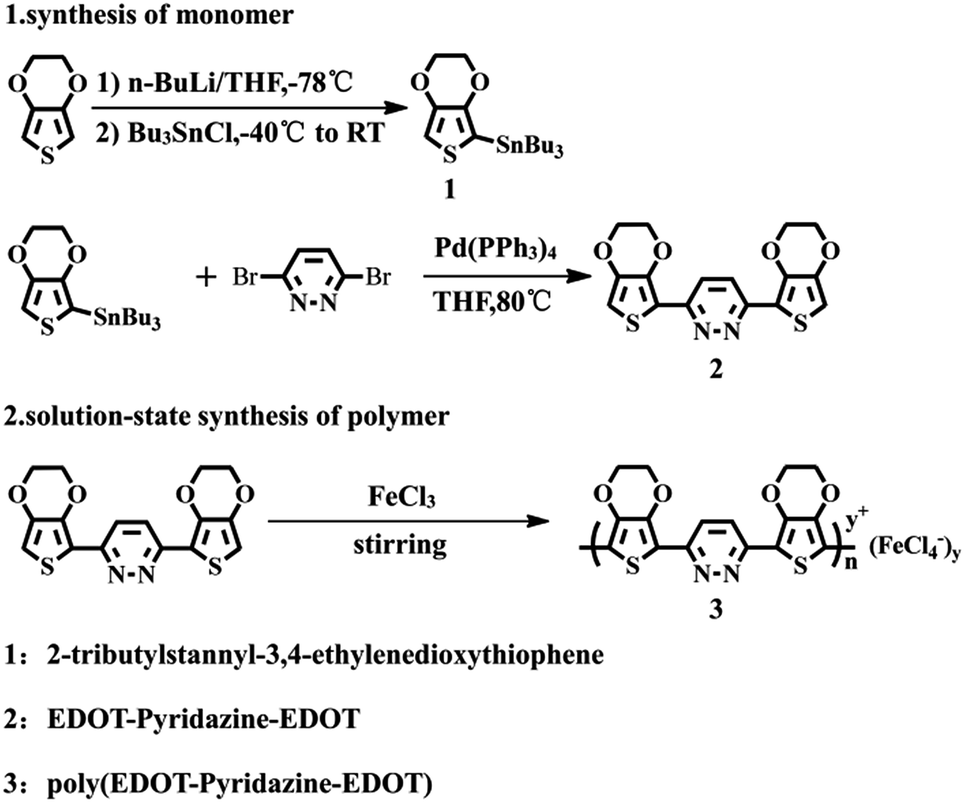
2.3 Synthesis of EPE
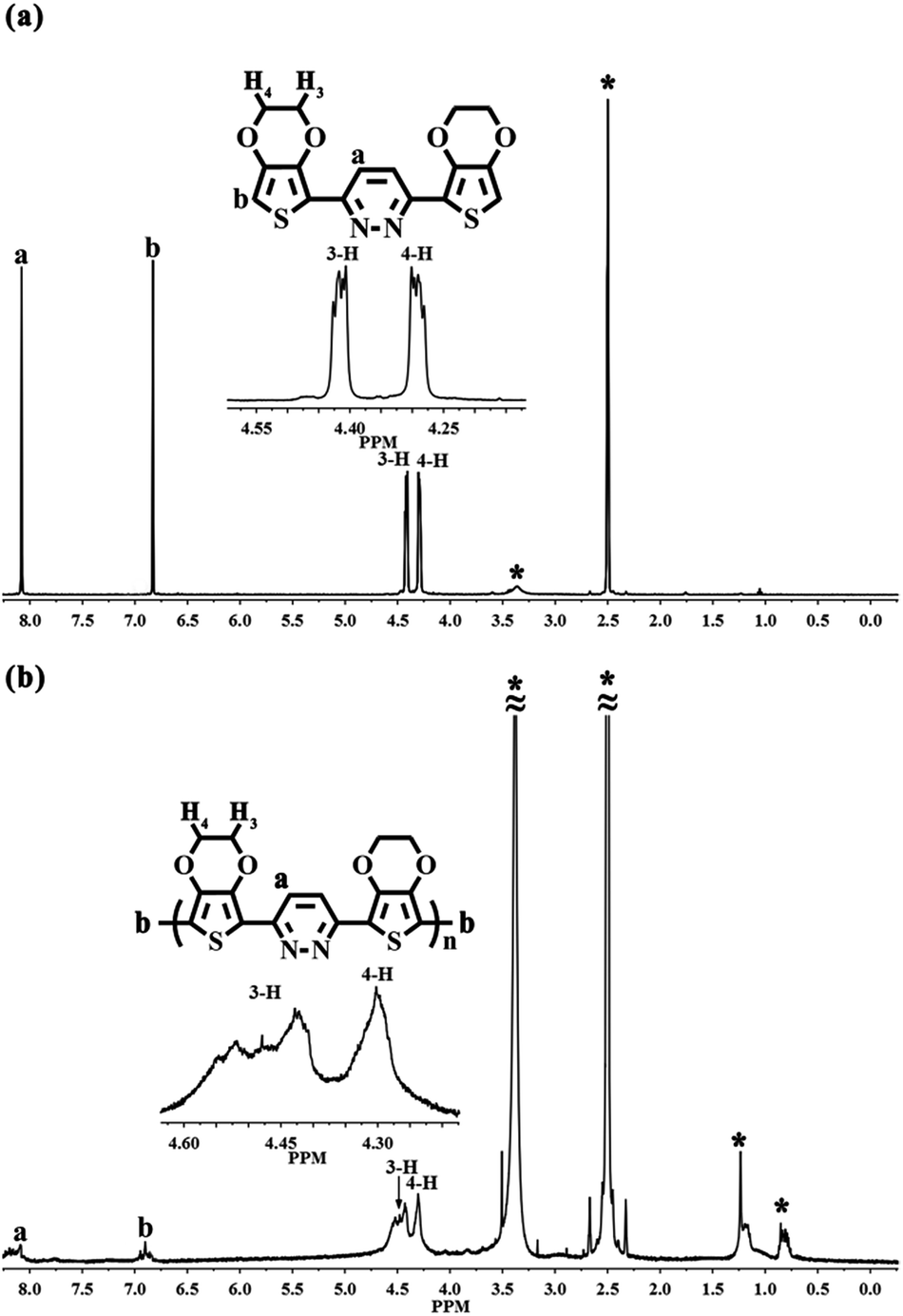
EPE was synthesized according to the procedure in ref. 23 and as shown in Scheme 1. 1H NMR (DMSO-d6, ppm): δ: 8.079 (m, 1H), 6.83 (m, 1H), 4.407 (m, 4H), 4.301 (m, 4H). The 1H NMR spectrogram is depicted in Fig. 2.
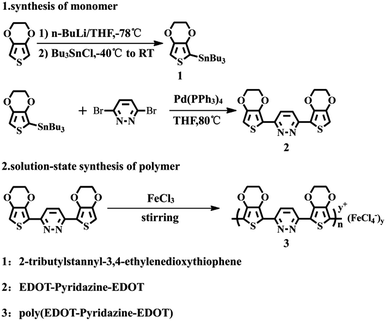 |
| | Scheme 1 Synthetic routes for the synthesis the monomer and polymer. | |
2.4 Liquid-phase oxidation polymerization of EPE
The simple solution oxidation polymerization process was carried out as follows: 10 mL chloroform and 0.179 g (1.1098 mmol) anhydrous ferric chloride were added to the polymerization bottle; 0.1 g (0.2774 mmol) EPE was dissolved in 10 mL chloroform solution and then slowly added into the abovementioned blend solution. After stirring the solution at room temperature for 12 h, a small amount of cold methanol was added to the final product. Finally, the product was removed from the mixture by repeatedly washing with methanol and distilled water until the filtrate was colorless. The powder was dried with a freeze drier at −55 °C for 24 h. The obtained polymer was noted as poly(EPE)1. In the same way, syntheses were carried out by adjusting the molar ratio of the oxidant/monomer (represented by [FeCl3]/[EPE]) to 8:1 and 12:1, respectively, and the resulting polymers were named as poly(EPE)2 and poly(EPE)3, respectively.
2.5 Measurements of the photocatalytic activities
The dyes methylene blue (MB), methyl violet (MU), methyl orange (MO), rhodamine B (RhB) and phenol were used as the model organic pollutants for the photocatalysis experiments. In addition, the efficacy of the as-prepared catalysts was estimated by the photodegradation of dyes under simulated visible light irradiation using a 300 W xenon lamp. Herein, 4 mg of samples was dispersed in 100 mL quartz beakers containing 40 mL of dye. Before irradiation, the dye solution was magnetically stirred for 60 min in the dark to ensure the establishment of an adsorption/desorption equilibrium, where the concentration of dye was designated as C0. Then, the mixed solutions were irradiated under a 300 W xenon lamp. At given intervals of illumination, the mixed solutions were sampled and centrifuged using an 800D high speed centrifuge at 4000 rpm for 1 min to remove the samples, which were then analyzed using a Unico 4802 recording spectrophotometer. In order to further understand the photocatalytic activities of the catalysts, we also studied the different catalysts for the degradation efficiency of MU dye. Different samples of (poly(EPE)1, poly(EPE)2, poly(EPE)3 and PEDOT) were tested the same way. In addition, in order to study the effect of Fe ions on the photocatalytic performance of the polymer, different doped states of the polymer (doped state and dedoped state) were performed in the same way as described above. Decolorization efficiencies of the dyes were estimated using the following equation:| | |
Degradation (%) = (C0 − Ct/C0) × 100%
| (1) |
where C0 is the initial concentration of the MU dye and Ct is the concentration at time t.
3. Results and discussion
3.1 FT-IR and 1H NMR spectra
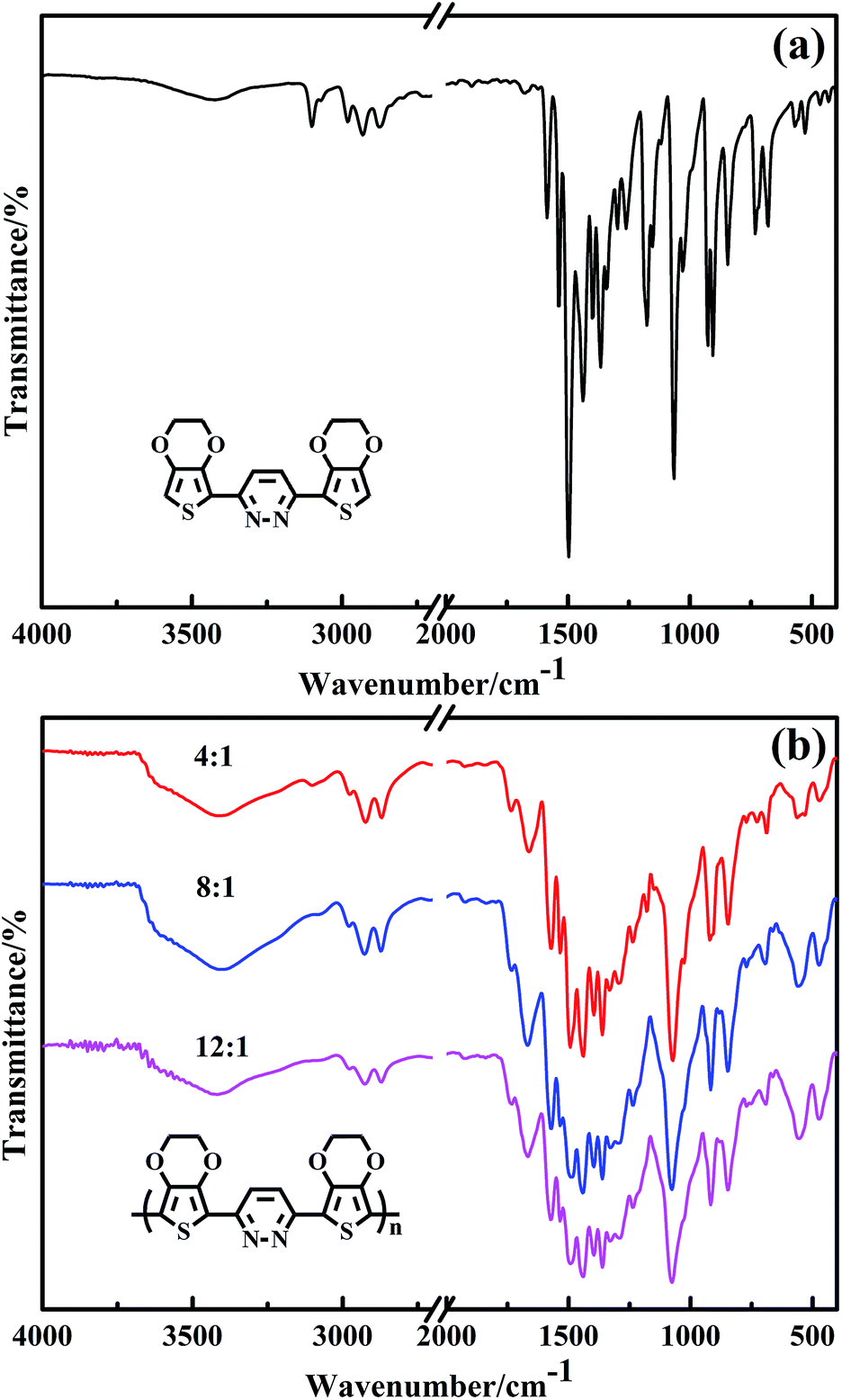
Fig. 1 shows the FT-IR spectra of EPE and poly(EPE)s powders obtained with various oxidant/monomer molar ratios. As can be seen in Fig. 1, all the poly(EPE)s show similar spectra. The characteristic bands of the pyridazine ring at about 1670, 1573 and 1496 cm−1 can be observed, while at about 1442, 1396, 1361, 1238, 1076, 918, 848, 771 and 690 cm−1 are the characteristic bands of 3,4-ethylenedioxy-thiophene (EDOT).19 The marked reduction in the relative intensities of the vibration absorption bands at 3021–3129 cm−1, corresponding to the C–H in the pyridazine ring in the poly(EPE)s, suggests that the poly(EPE)s are partly in the doped state.30 The bands at approximately 1496, 1442, 1396 and 1361 cm−1 are attributed to the asymmetric stretching vibration of CC and the stretching vibration of C–C in the ring, respectively. The 1238 and 1076 cm−1 bands are assigned to the presence of ν(C–O–C) in the ethylenedioxy group. There are also C–S–C characteristic stretching vibrations bands at 918, 848 and 690 cm−1 in the thiophene ring.31–33 In addition, characteristic bands at approximately 1439 cm−1 (ring stretching vibration) and 848 cm−1 (C–H out-of-plane vibration) are due to α,α′-coupled thiophene rings. A ν(CN) peak appears at about 1573 cm−1, which is a characteristic of the pyridazine unit.34 According to the previous report, the intensity ratios (Isym/Iasym) of the bands at 1438–1442 cm−1 compared to the bands at 1489–1494 cm−1 are indicative of the conjugated length.35 To compare with others, the intensity ratio (Isym/Iasym) is the highest in poly(EPE)2 ([FeCl3]/[EPE] ratio of 8:1), suggesting that the longest conjugated length is in poly(EPE)2 compared to that of the others.
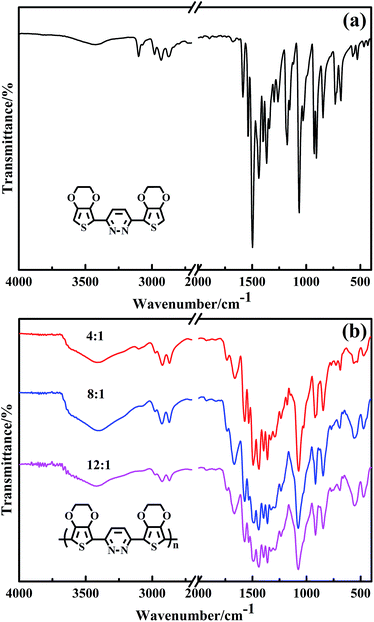 |
| | Fig. 1 FT-IR spectra of (a) EPE and (b) poly(EPE)s in different [FeCl3]/[EPE] ratios: 4:1; 8:1; 12:1. | |
Fig. 2 presents the comparison of the 1H NMR spectra of EPE and poly(EPE)1 ([FeCl3]/[EPE] ratio of 4:1) in DMSO-d6. The 1H NMR spectrum of EPE shows the typical resonance lines for this monomer, and their assignments are shown in Fig. 2(a). As depicted in Fig. 2(b), poly(EPE)1 displays the characteristics of two aromatic rings signal at ∼δ 8.1 and 6.8, which are attributed to protons from the pyridazine and thiophene rings, respectively. Furthermore, the aliphatic peaks at ∼δ 4.3 and 4.4 are ascribed to the O–CH2 protons of the O–CH2–CH2–O band.36 Except for the characteristic signals for the polymer, the peaks in the range of δ 1.5–0.7 are assigned to the alkyl groups of some impurities in the polymer matrix. In order to understand the molecular weight of the polymers, GPC was performed on the samples. The presence of hydrogen atoms at the α-position of the polymer indicates the low molecular weight of the soluble fractions of the polymer, which was further evidenced by the gel permeation chromatography measured results. As shown in Table 1, the weight-average molecular weights (Mw) of the poly(EPE)s were in the range from 2902 to 3955. However, all the polymers possessed a rather low molecular weight with low solubility as the molecular structures of the polymers do not have alkyl chains, while the backbone of the polymers possess conjugate structures, which together lead to a low solubility and high rigidity.37 Furthermore, most of the soluble parts come from the oligomer of the polymers, and the lower molecular weights of the poly(EPE)s are probably ascribable to the increased stability of the radical cations formed from EDOT, which slows the polymerization rate and leads to low molecular weight polymers.33
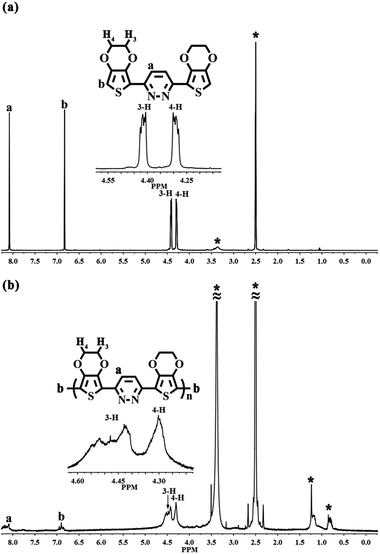 |
| | Fig. 2 1H NMR spectra of (a) EPE and (b) poly(EPE)1 ([FeCl3]/[EPE] ratio of 4:1) in DMSO-d6. Starred peaks come from H2O, alkyl groups and DMSO-d6. | |
Table 1 Molecular weight and polydispersity of the polymers
| Polymers |
[FeCl3]/[EPE] |
Mn |
Mw |
PDI |
| Poly(EPE)1 |
4:1 |
2063 |
2902 |
1.40 |
| Poly(EPE)2 |
8:1 |
2576 |
3955 |
1.53 |
| Poly(EPE)3 |
12:1 |
2495 |
2902 |
1.16 |
3.2 Ultraviolet-visible absorption spectra
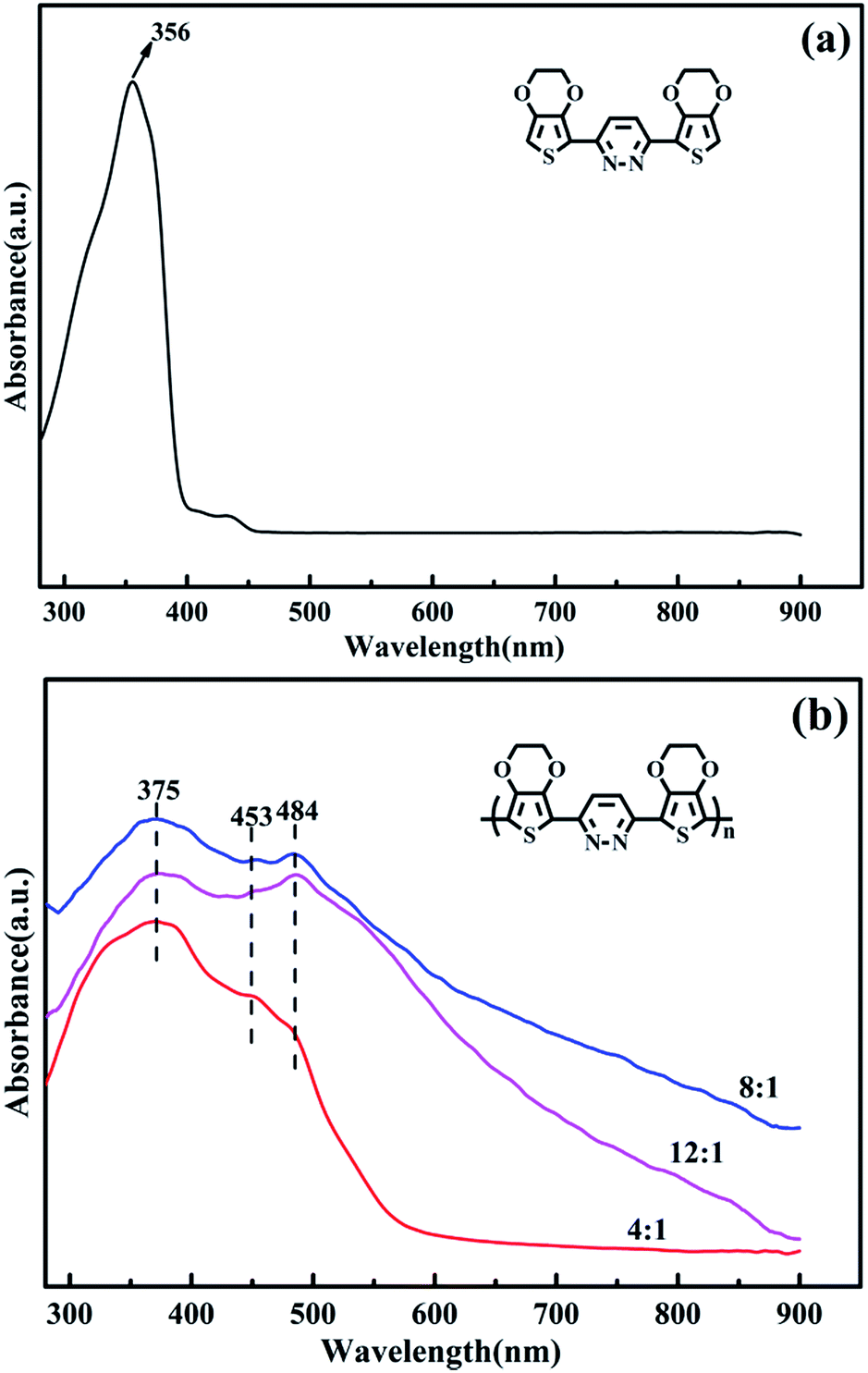
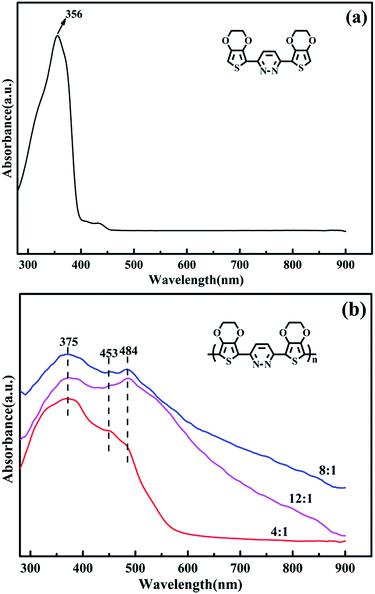
The UV-vis absorption spectra of the EPE and poly(EPE)s recorded in N-methyl-2-pyrrolidinone (NMP) solvent are displayed in Fig. 3. As depicted in Fig. 3, EPE shows a peak at 356 nm, assigned to the π–π* transition of the EDOT and pyridazine units,19 whereas poly(EPE)s have three absorption bands at ∼375, 453 and 484 nm from the conjugated segments having different conjugation lengths,38 which correspond to π–π* transitions in the polymer backbone.39 Compared with poly(EPE)1([FeCl3]/[EPE] ratio of 4:1), the relative intensity of the absorption bands at 453 and 484 nm are higher in the case of poly(EPE)2 and poly(EPE)3, implying that a higher content of oxidant in the reaction medium leads to a higher conjugation length in the polymer chains.
 |
| | Fig. 3 UV-vis spectra of (a) EPE and (b) poly(EPE)s in different [FeCl3]/[EPE] ratios: 4:1; 8:1; 12:1. | |
Furthermore, the intensity ratio of the absorption band at 484 nm to the band at 375 nm is the highest in the case of poly(EPE)3 ([FeCl3]/[EPE] ratio of 12:1) among the poly(EPE)s. This means that poly(EPE)3 has the highest conjugation length among the poly(EPE)s. However, compared with that of poly(EPE)2, an upward trend is observed in the spectrum of poly(EPE)3 from 600 to 900 nm, which is a feature typically seen in the partially doped state of polymer chains.40 Therefore, it can be concluded that poly(EPE)2 has a higher doping level than poly(EPE)3. Furthermore, the optical band gaps (Eoptg) for poly(EPE)1, poly(EPE)2 and poly(EPE)3 were determined to be 2.25 eV, 2.06 eV and 2.04 eV, respectively, from their solution absorption edges (λonset) (Table 2).
Table 2 Summary of the optical data, including maximum absorption peak, absorption edge wavelength (λonset) and optical band gap (Eoptg) of poly(EPE)1, poly(EPE)2 and poly(EPE)3
| UV-vis absorption spectra |
| |
Solution |
Eoptg (eV) |
| λmax (nm) |
λonset (nm) |
| Poly(EPE)1 |
375 |
550 |
2.25 |
| Poly(EPE)2 |
375 |
602 |
2.06 |
| Poly(EPE)3 |
375 |
609 |
2.04 |
3.3 Thermal stability
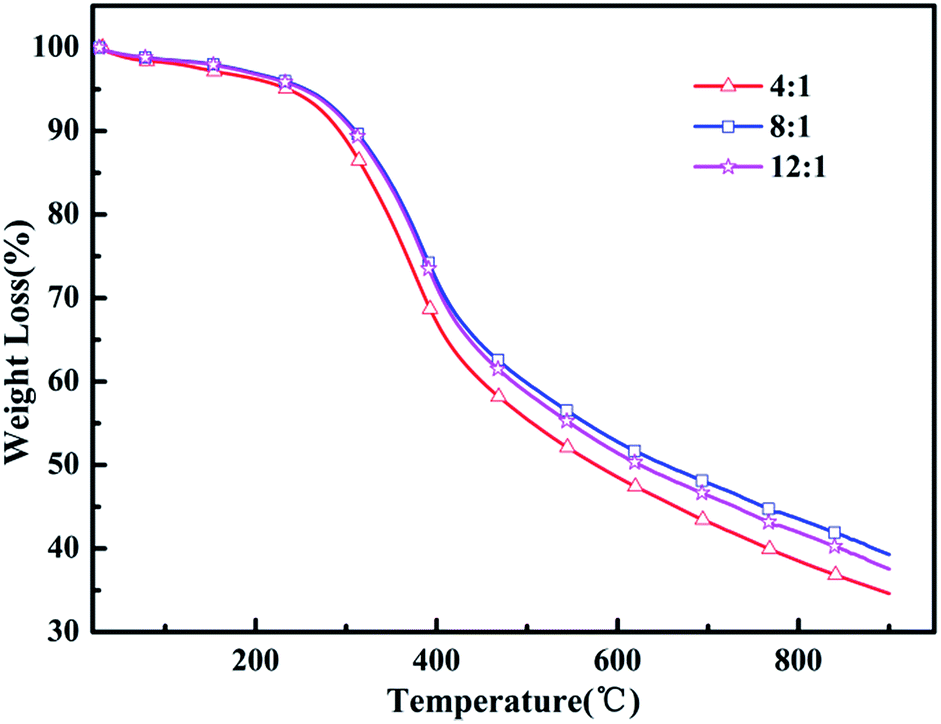
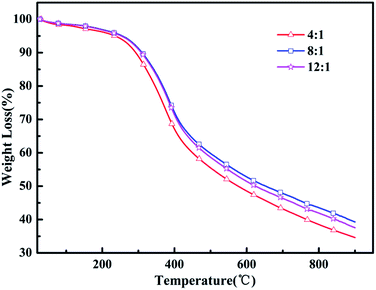
Heat resistance is an important feature to measure the quality of the polymer to adapt to the environment. Thermogravimetric analysis (TGA) was utilized to evaluate the thermal properties of the resulting polymers. Fig. 4 shows the TGA curves analyses of the poly(EPE)s and reveals that all the polymers lost 5% of their weight at about 236–253 °C under an argon atmosphere. In addition, poly(EPE)s gave a high residue at 900 °C, of 34–39 wt%, indicating that the poly(EPE)s have good thermal stability. The poly(EPE)s exhibited three important weight loss steps. The initial weight loss of less than 5% is attributed to the evaporation of solvents, moisture, and oligomers of EPE. The second step, from 236–253 °C to 420–425 °C, may be due to the loss of the pendant ethylenedioxyl chain and decomposition of the polymer framework. The last step, occurring from 420–425 °C to 900 °C is attributed to the loss from thermal destruction of the polymer framework. A comparison study indicated that the initial 5% weight loss temperature of poly(EPE)2 was higher than that for others, and the residue at 900 °C for poly(EPE)2 was also higher than that for the others. Based on the above discussion, the higher initial 5% weight loss temperature of poly(EPE)2 suggests that the amount of oligomers for EPE in the poly(EPE)2 matrix is lower than that of the others. This phenomenon can be a result of the longer conjugated length of this matrix than that of the others. In addition, the high content of oxidant (initiator) favours chain propagation, which can result in longer polymer chains, consequently increasing the conjugation length.41 Therefore, according to the results of the FT-IR and UV-vis studies, the lower residue at 900 °C for poly(EPE)1 than for the others may be due to the lower conjugation length in poly(EPE)1.
 |
| | Fig. 4 TGA of poly(EPE)s in different [FeCl3]/[EPE] ratios: 4:1; 8:1; 12:1. | |
3.4 XRD structural analysis
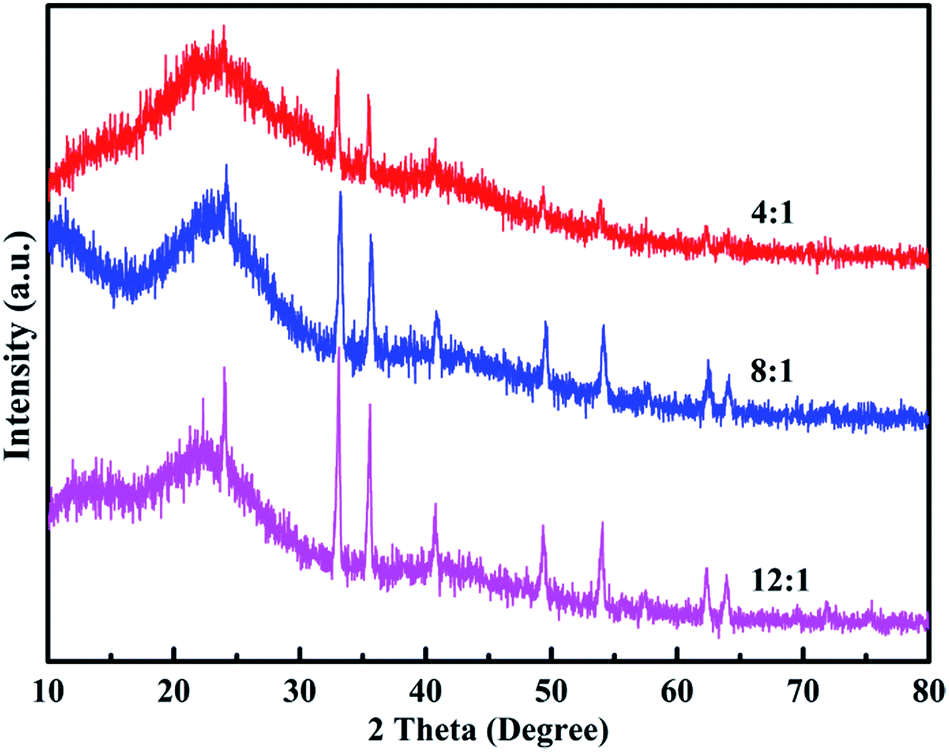
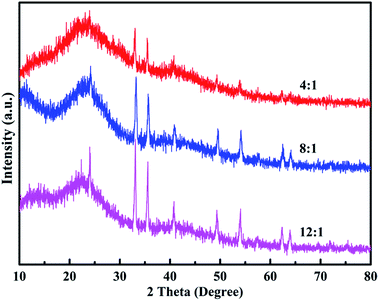
Fig. 5 shows the XRD patterns of the poly(EPE)s. As shown in Fig. 5, all the polymers display a rather broad diffraction peak with a low intensity at approximately 2θ = 24° due to the intermolecular internal π → π* stacking or the (020) reflection. Compared with that of poly(EPE)1, the other two poly(EPE)s display diffraction peaks between ∼2θ = 10° and 13°, which can be assigned to the (200) reflection. All these diffraction peaks are similar to the characteristic diffraction peak of PEDOT; this indicates the existence of a crystalline phase.42 Therefore, it can be concluded that poly(EPE)1 is more amorphous than the others. Moreover, the sharp diffraction peaks at 2θ = 33°, 35°, 49° and 54° with low intensity are present in all the polymers, which could be attributed to the existence of the doping agent FeCl4−. According to the related literature reports, the synthesized polymers are in a partially doped state using FeCl3 as an oxidizing agent for the oxidative polymerization of thiophene, and the FeCl4− ion is most likely the counter anion from the FeCl3 doping into the polymer structure.42–44
 |
| | Fig. 5 XRD of poly(EPE)s in different [FeCl3]/[EPE] ratios: 4:1; 8:1; 12:1. | |
3.5 Energy-dispersive X-ray spectroscopy
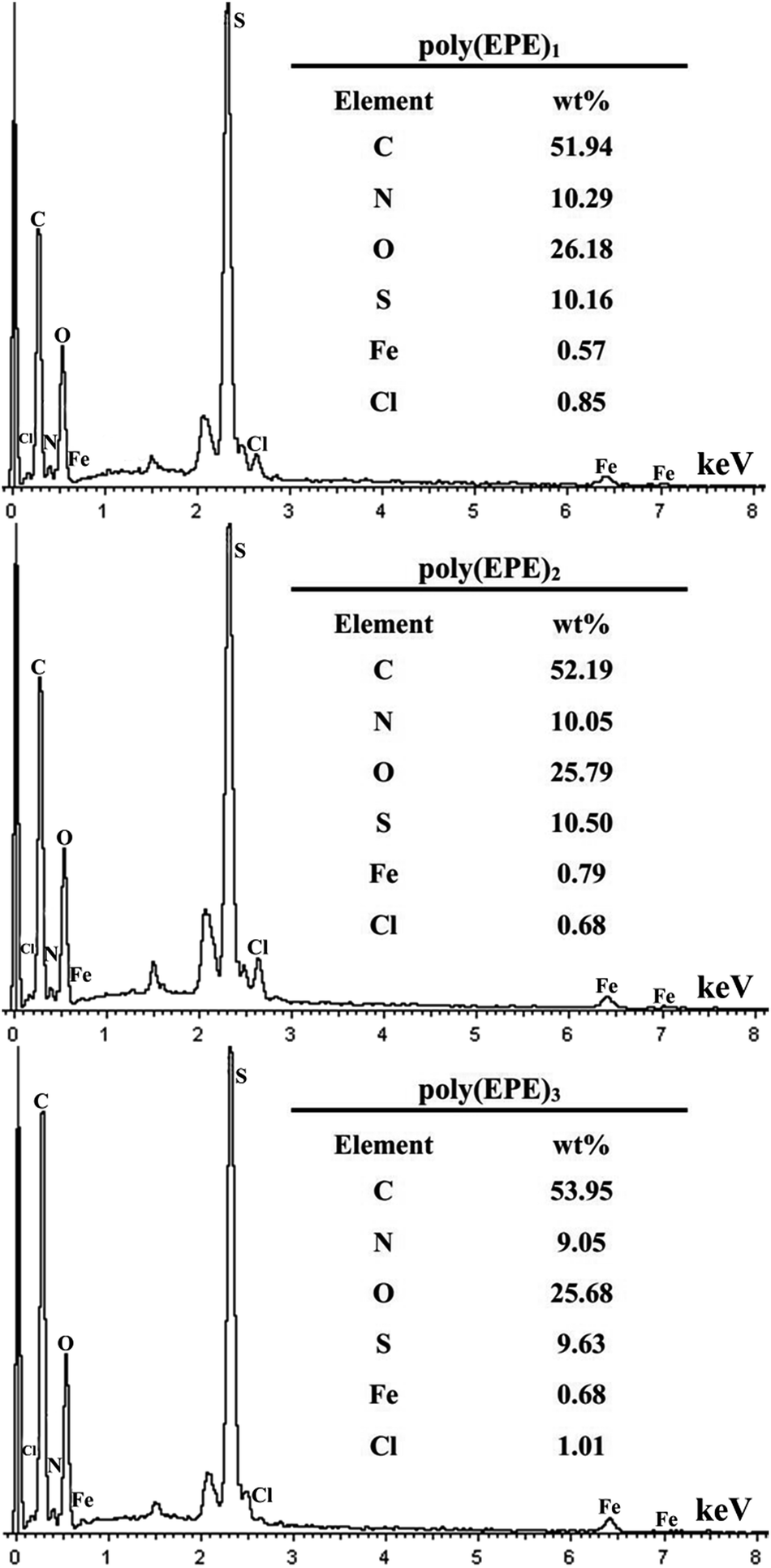
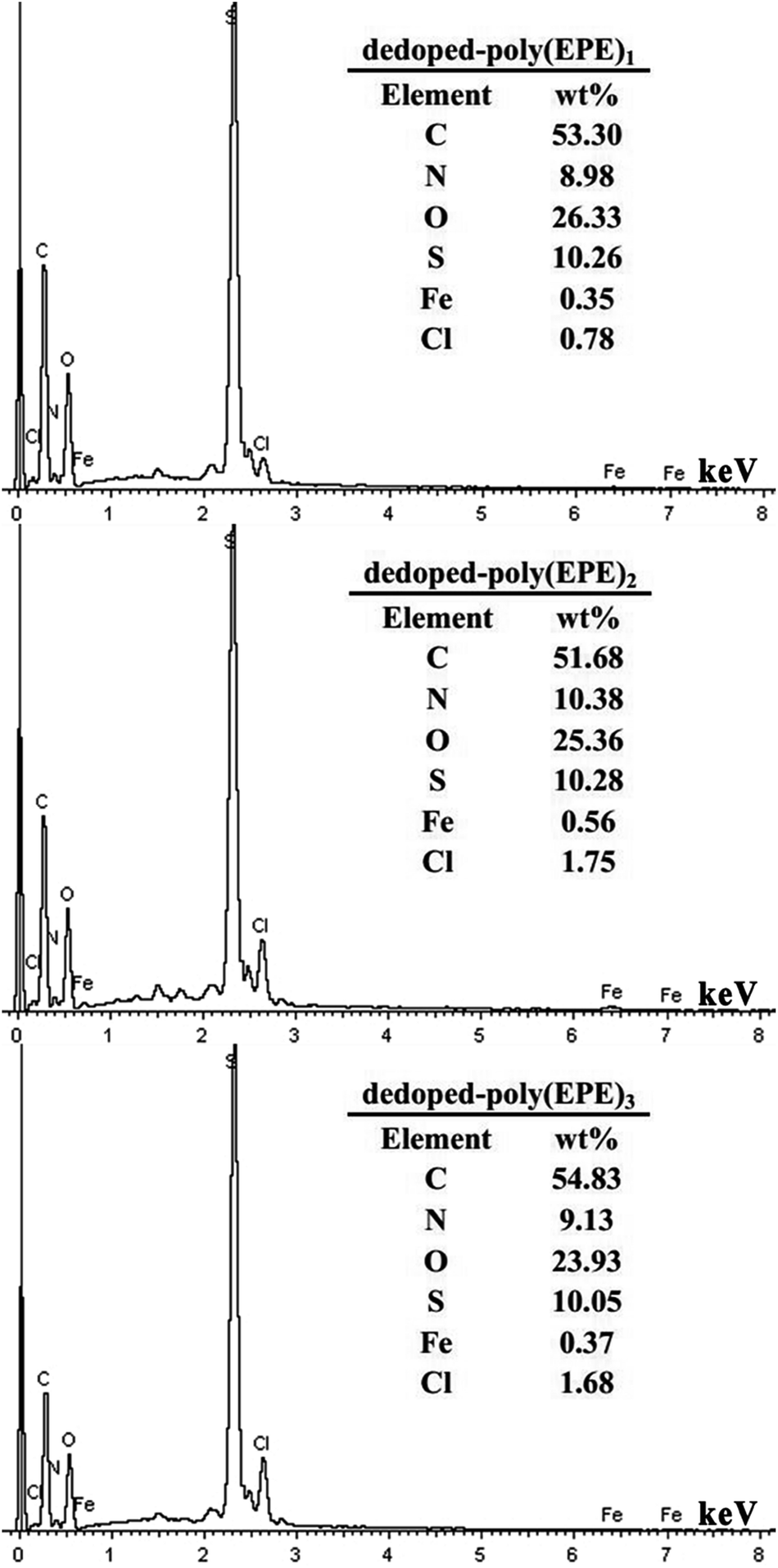
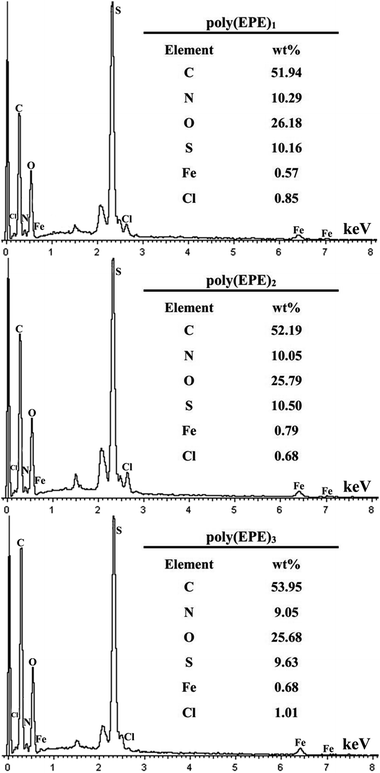
Fig. 6 shows the EDX spectra of the polymers. The peaks corresponding to C, N, O, S, Cl and Fe are clearly identified. Furthermore, the peaks for Cl and Fe ions, which result from the doping agent of FeCl4−, and the content of Fe ions are 0.57, 0.79 and 0.68 wt%, respectively. The percentage of Fe ions in the polymers is in the order of poly(EPE)2 > poly(EPE)3 > poly(EPE)1. In addition, based on the results of the UV-vis study, compared with that of poly(EPE)2, an upward trend is observed in the spectrum of the other two from 600 to 900 nm, which is a feature typically seen in the partially doped state of the polymer chains.40 Therefore, it can be concluded that poly(EPE)2 has the highest doping level. Comparisons showed that the EDX results are consistent with the UV-vis results.
 |
| | Fig. 6 EDX of poly(EPE)s in different [FeCl3]/[EPE] ratios: 4:1; 8:1; 12:1. | |
3.6 Morphology
The morphologies of the polymers were investigated by scanning electron microscopy (SEM). Fig. 7 displays different morphologies present in the various oxidant/monomer molar ratios. As shown in Fig. 7, the morphology of poly(EPE)1 shows an irregular coral shape, having tentacles extending in a random fashion, accompanied with the accumulation of a large number of particles. Moreover, with the increase in concentration of the oxidant (initiator), poly(EPE) has a more regular coral-like morphology, with low accumulated particles. Furthermore, the tentacles have the tendency to adhere to each other with the increase in the oxidant (initiator), particularly in the case of poly(EPE)3. The possible reasons for such morphologies appearing in the poly(EPE)s may be correlated with the structural properties of EPE and the proportion of FeCl3/EPE. EDOT units in the triplet unit can immobilize the conjugated chain segment by intramolecular covalent bond interaction and by the strong electron-transport effect of EDOT in EPE, and this synergism of EDOT will cause the polymers structure to have a more regular and rigid structure.45 Moreover, as the concentration of the oxidant (initiator) increases, the internal diffusion rate of the monomer decreases, and the induction time of polymerization could be shortened. Furthermore, based on the previous report, when the polymerization time elongates, the EDOT units containing the monomer, which newly form nano-fibriform oligomers, serve as a soft template to form the nano-fibriform structures.19 Therefore, according to the abovementioned inference, the poly(EPE) from [FeCl3]/[EPE] = 4:1 forms the irregular coral-like shape mostly as a result of crooked nanofibers growing together. As the content of the oxidant (initiator) increases, the concentration of the monomer and the newly formed nano-fibriform oligomers is relatively reduced. As for the poly(EPE)2, the branches of neighboring corals will link together to cause a shape of hybrid tentacles with particles, whereby the clusters of coral-like morphology of poly(EPE)3 will be conducive to this formation.
 |
| | Fig. 7 SEM images of (a) poly(EPE) (4:1), (b) poly(EPE) (8:1) and (c) poly(EPE) (12:1). | |
3.7 Photocatalytic activity
Organic dyestuffs containing methyl blue (MB), methyl violet (MU), methyl orange (MO), rhodamine B (RhB) and phenol dyes have been widely applied in textile industries, but they raise concerns as they are toxic to aquatic life and create serious pollution to the environment. In this study, MB, MU, MO, RhB and phenol were used as model organic pollutants to demonstrate the effectiveness of poly(EPE)2 under a 300 W xenon lamp. In addition, comparative experiments were conducted on MU solution with the presence of different catalysts and poly(EPE)2 in different doping states.
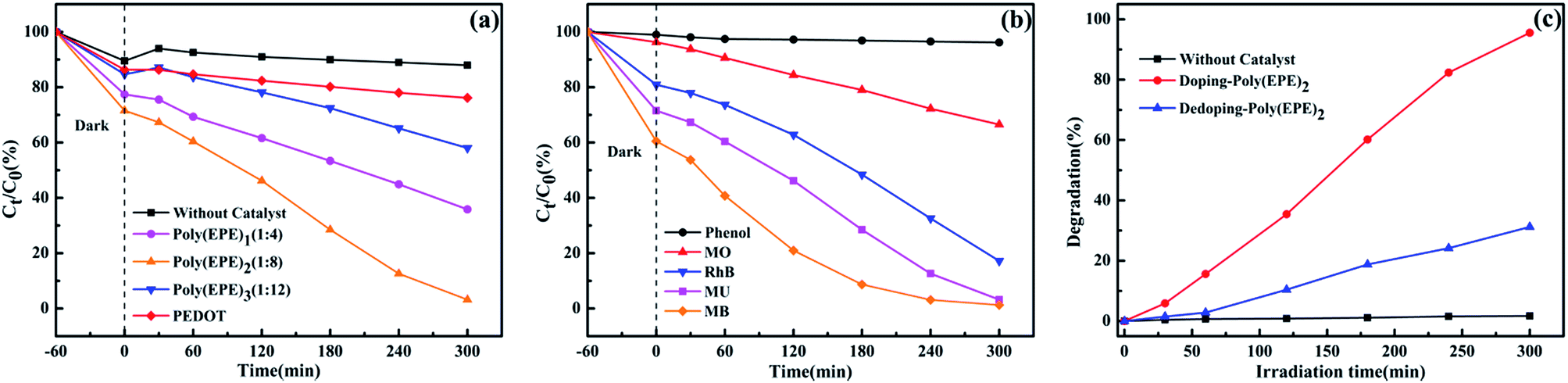
Fig. 8(a) displays the time-dependent absorption spectra of MU dye degraded by different catalysts and without a catalyst. The concentration of the dye shows a different degree of reduction in the presence of PEDOT, poly(EPE)1, poly(EPE)2 and poly(EPE)3 catalysts for 300 min under a xenon lamp, with the photodegradation rate being 11.54%, 53.7%, 95.53% and 31.45%, respectively. It is clear that the catalytic activity of poly(EPE)s is much higher than that of a PEDOT and MU aqueous solution containing 4 mg poly(EPE)2 as it exhibits the highest photodegradation efficiency among the four groups of catalysts. Moreover, the results show that the [FeCl3]/[EPE] ratio of 8:1 is the optimum proportion to use for the photocatalyst. Furthermore, the photocatalytic activity of different dyes is shown in Fig. 8(b), and it can be seen that in the presence of poly(EPE)2, the photodegradation rate for MB, MU, MO, RhB and phenol is 97.88%, 95.53%, 30.93%, 78.73% and 3.63% in 300 min, respectively, which indicates that the photocatalyst is selective to degrading different dyes. Furthermore, the photocatalytic performance of poly(EPE) was compared to that of the other catalysts for the photodegradation of MO, RhB and MB and so on, and the results are shown in Table 3.46–54 The results indicate that, in comparison to the others, the D–A–D type polymer is an efficient photocatalyst. In addition, Fig. 8(c) illustrates the photocatalytic degradation of MU dye with simulated visible light, in the presence of Fe-doped poly(EPE)2 and Fe-dedoped poly(EPE)2. As shown in Fig. 8(c), the decomposition efficiency of the MU dye in the presence of Fe-doped poly(EPE)2 is 95.53% in 300 min, and for Fe-dedoped poly(EPE)2, under the same simulated visible light irradiation, only about 30.17% of the MU dye is decomposed within the same time. This result shows that the Fe ions as oxidants play a very major role in improving the degradation efficiency of the MU dye.
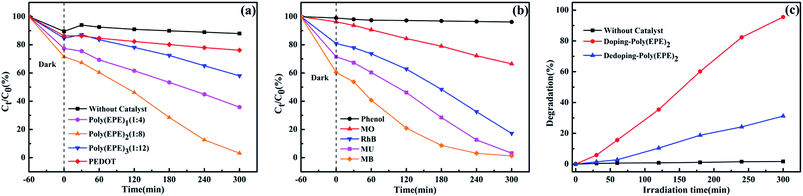 |
| | Fig. 8 (a) Photocatalytic degradation of MU dye with the change in Ct/C0 in the presence of different catalysts. (b) Photocatalytic degradation of MB, MU, MO, RhB and phenol dyes with the change in Ct/C0 of poly(EPE)2. (c) Time-dependent absorption spectra of MU dye degraded by different doped states of poly(EPE)2. | |
Table 3 Comparison of poly(EPE)2 photocatalyst with other photocatalysts recently used for the degradation of dyes
| Sample |
Dye |
D.Ta |
Db% |
Irradiation source |
(mg)sample/(mg(mol) L−1)Dye × (mL)Dye |
Ref. |
| Degradation time. Degradation percentage. |
| TiO2–Fe3O4/RGO |
MB |
180 min |
100 |
500 W xenon lamp |
10 mg/5.35 × 10−5 (mol L−1) × 40 mL |
46 |
| 0.05 FeT |
4-NP |
300 min |
92 |
150 W Philips CFL bulb |
0.5 mg/10 (mg L−1) × 1 mL |
47 |
| 5% Pt–TiO2/G |
AO7 |
360 min |
91.06 |
Visible light |
10 mg/35 (mg L−1) × 60 mL |
48 |
| PANI/ZnO |
MB |
360 min |
94 |
Natural sunlight |
0.4 mg/1 × 10−5 (mol L−1) × 1 mL |
49 |
| MG |
360 min |
98 |
Natural sunlight |
0.4 mg/1 × 10−5 (mol L−1) × 1 mL |
| FNZP/PANI |
MB |
300 min |
99.47 |
Natural sunlight |
0.25 mg/1.5 × 10−5 (mol L−1) × 1 mL |
50 |
| PNA/ZnO |
MB |
140 min |
22 |
300 W xenon arc lamp |
200 mg/10 (mg L−1) × 100 mL |
51 |
| CdS–TiO2–Au |
MO |
300 min |
98 |
400 W metal halogen lamp |
150 mg/10 (mg L−1) × 100 mL |
52 |
| PMPD |
AR249 |
600 min |
2.2 |
300 W tungsten halogen lamp |
50 mg/50 (mg L−1) × 50 mL |
53 |
| PFT |
MO |
120 min |
40 |
Visible light |
100 mg/20 (mg L−1) × 100 mL |
54 |
| Poly(EPE)2 |
MB |
300 min |
97.88 |
300 W xenon lamp |
4 mg/5 (mg L−1) × 40 mL |
This work |
| Poly(EPE)2 |
MU |
300 min |
95.53 |
300 W xenon lamp |
4 mg/5 (mg L−1) × 40 mL |
| Poly(EPE)2 |
RhB |
300 min |
78.73 |
300 W xenon lamp |
4 mg/5 (mg L−1) × 40 mL |
| Poly(EPE)2 |
MO |
300 min |
30.93 |
300 W xenon lamp |
4 mg/5 (mg L−1) × 40 mL |
3.8 Postulated mechanism
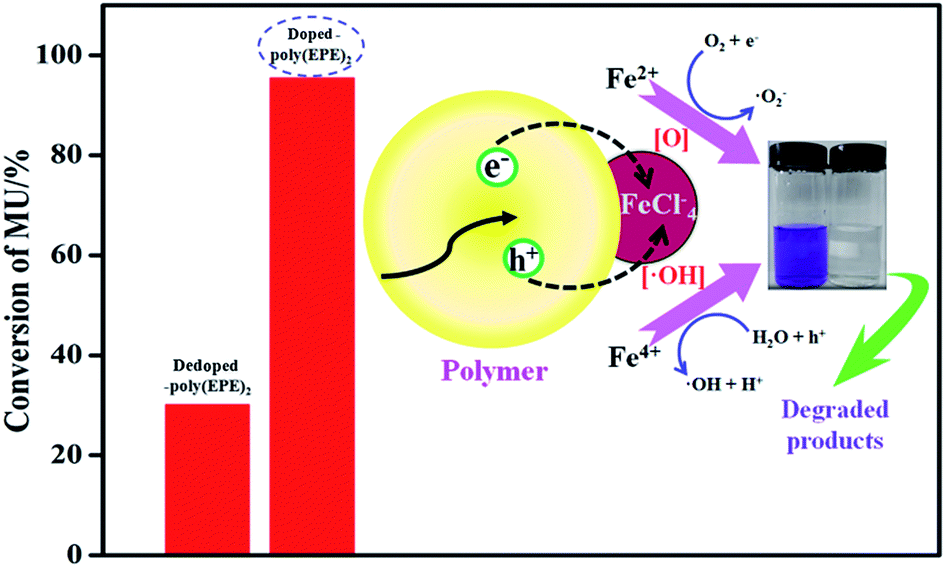
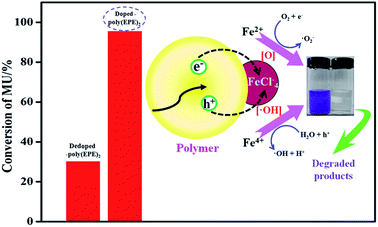
Based on the results from the EDX spectra of the doped polymers (Fig. 6) and dedoped polymers (Fig. 9), the percentage of Fe ions for the doped poly(EPE)2 and dedoped poly(EPE)2 are in the order of: doped poly(EPE)2 > dedoped poly(EPE)2. This phenomenon further indicates that the degradation efficiency of MU dye by dedoped polymers is lower than that of the doped polymers. As it has already been reported, a small number of Fe ions can narrow the band gap of the polymer and postpone the electron–hole pair recombination over time, which would be beneficial to an improvement in the photocatalytic activity of the polymer.55,56 Therefore, it can be concluded that a higher percentage of Fe ions in the polymer can lead to a more efficient photodegradation of MU dye, and that the Fe ions in the polymer matrix play a major role in improving the degradation efficiency of the MU dye.
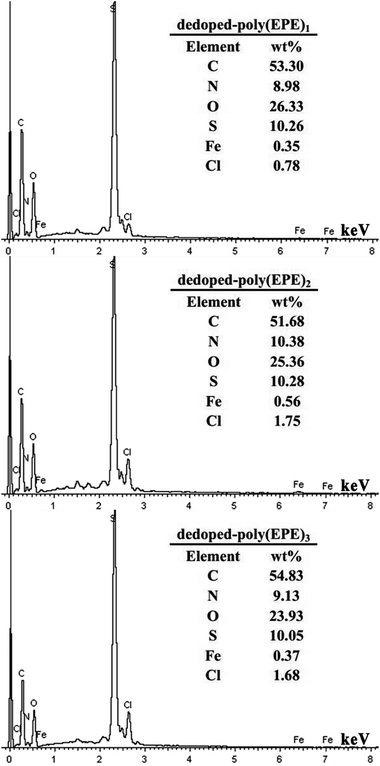 |
| | Fig. 9 EDX of dedoped poly(EPE)s in different [FeCl3]/[EPE] ratios: 4:1; 8:1; 12:1. | |
A schematic of MU dye decomposition is depicted in Fig. 10. If the electrons and holes cannot be timely captured, they will restructure together in a very short period of time, which will reduce the photocatalytic activity of polymers. However, for doping Fe ions in the polymers, the presence of a small amount of Fe ions in the structure means Fe ions can come not only into an electron capture position but also a hole capture position, which can cause a decrease in the electron–hole pair recombination. Then, when the polymers are illuminated with simulated visible light, both Fe ions and polymers absorb the photons at their interface, and then charge separation occurs at the interface. Since the conduction band of Fe ions and the LUMO level of the polymer are well matched for the charge transfer, the electrons are promoted from the π → π* absorption band of the outside polymers upon visible light illumination and are easily injected into the conduction band of Fe ions. In addition, Fe ions doping in polymers can result in three types of valence states: Fe2+, Fe3+ and Fe4+, where Fe3+ is the most stable state among them.28 Irradiation leads to the generation of electron–hole pairs [eqn (2)], and then Fe ions can trap them to form Fe2+ and Fe4+ ions [eqn (3) and (4)]. Finally, the Fe2+ and Fe4+ ions generate hydroxyl radicals (˙OH) and hyperoxide radicals (O2˙−), and the Fe3+ is formed again [eqn (5) and (6)]. These reactions may delay the recombination of electrons and holes,57 and then Fe ions lead to a continuation of the degradation reaction by hydroxyl and hyperoxide radicals.28
| | |
Polymers/Fe3+ + ħν ⇒ ħ+ + e−
| (2) |
| | |
Fe3+ + ħ+ ⇒ Fe4+ + hole capture
| (3) |
| | |
Fe3+ + e− ⇒ Fe2+ + electrons capture
| (4) |
| | |
Fe4+ + OH− ⇒ Fe3+ + ˙OH
| (5) |
| | |
Fe2+ + O2 ⇒ Fe3+ + O2˙−
| (6) |
 |
| | Fig. 10 Schematic mechanism of MU dye degradation. | |
4. Conclusion
In this study, a D–A–D type monomer, namely EPE combining ethylenedioxythiophene donor units with a pyridazine acceptor unit was prepared. In addition, a D–A–D type conjugated polymer, namely poly(EPE), was prepared by adjusting the ratios of FeCl3/monomer. The results of a structural analysis indicated that an increase in the content of the oxidant (initiator) in the reaction medium could lead to an increase in the formation of oligomers. However, it also favours the chain propagation, which could increase the conjugation length. As a result, the poly(EPE)s from FeCl3/monomer ratios of 8:1 and 12:1 displayed a higher conjugation length, thermal stability, doping level and crystallinity than the poly(EPE) from the FeCl3/monomer ratio of 4:1. The results also showed that the morphology of the polymers could be affected by the ratios of FeCl3 to the monomer. The poly(EPE)2 showed a good selectivity for the degradation of different dyes under a xenon lamp. In addition, studies on the photocatalytic degradation of MU dye displayed that poly(EPE)2 was an effective photocatalyst. However, in the presence of PEDOT, the degradation rate was only 13.04%. Furthermore, the presence of Fe ions in the polymer matrix was the main factor for enhancing the photocatalytic activity of poly(EPE). The presence of Fe ions in the catalyst structure increased the number of active sites on the grid. Among the poly(EPE)s with the highest doping level and crystallinity as well as effective conjugation length, the polymer from the FeCl3/monomer ratio of 8:1 displayed a higher photocatalytic activity in the light source illumination. In summary, this study indicated the feasible and potential use of a new D–A–D type of polymer as a new class of photocatalyst in the photocatalytic degradation of organic dye waste waters, and it enriches the knowledge of the photocatalytic behavior of such polymers.
Acknowledgements
The authors are grateful to the National Natural Science Foundation of China (No. 21464014).
Notes and references
- K. J. Albert, N. S. Lewis, C. L. Schauer, G. A. Sotzing, S. E. Stitzel, T. P. Vaid and D. R. Walt, Chem. Rev., 2000, 100, 2595 CrossRef CAS PubMed.
- D. T. McQuade, A. E. Pullen and T. M. Swager, Chem. Rev., 2000, 100, 2537 CrossRef CAS PubMed.
- E. J. List, R. Guentner, P. Scanducci de Freitas and U. Scherf, Adv. Mater., 2002, 14, 374 CrossRef CAS.
- K. Bange and T. Gambke, Adv. Mater., 1990, 2, 10 CrossRef CAS.
- C. D. Dimitrakopoulos and P. R. Malenfant, Adv. Mater., 2002, 14, 99 CrossRef CAS.
- Y. Yao, Y. Liang, V. Shrotriya, S. Xiao, L. Yu and Y. Yang, Adv. Mater., 2007, 19, 3979 CrossRef CAS.
- Z. Chen, Y. Zheng, H. Yan and A. Facchetti, J. Am. Chem. Soc., 2008, 131, 8 CrossRef PubMed.
- R. L. McCreery and A. J. Bergren, Adv. Mater., 2009, 21, 4303 CrossRef CAS PubMed.
- Z. Yin and Q. Zheng, Adv. Energy. Mater., 2012, 2, 179 CrossRef CAS.
- L. Schmidt-Mende, A. Fechtenkötter, K. Müllen, E. Moons, R. H. Friend and J. MacKenzie, Science, 2001, 293, 1119 CrossRef CAS PubMed.
- H. Van Mullekom, J. Vekemans, E. Havinga and E. Meijer, Mater. Sci. Eng., R, 2001, 32, 1 CrossRef.
- H. Sirringhaus, N. Tessler and R. H. Friend, Science, 1998, 280, 1741 CrossRef CAS PubMed.
- T. Yamamoto, Z.-H. Zhou, T. Kanbara, M. Shimura, K. Kizu, T. Maruyama, Y. Nakamura, T. Fukuda, B.-L. Lee and N. Ooba, J. Am. Chem. Soc., 1996, 118, 10389 CrossRef CAS.
- C. Kitamura, S. Tanaka and Y. Yamashita, Chem. Mater., 1996, 8, 570 CrossRef CAS.
- T. Yamamoto and B.-L. Lee, Macromolecules, 2002, 35, 2993 CrossRef CAS.
- T. Michinobu, K. Okoshi, H. Osako, H. Kumazawa and K. Shigehara, Polymer, 2008, 49, 192 CrossRef CAS.
- M. Nikolou, A. L. Dyer, T. T. Steckler, E. P. Donoghue, Z. Wu, N. C. Heston, A. G. Rinzler, D. B. Tanner and J. R. Reynolds, Chem. Mater., 2009, 21, 5539 CrossRef CAS.
- I. Nenner and G. Schulz, J. Chem. Phys., 1975, 62, 1747 CrossRef CAS.
- T. Yasuda, Y. Sakai, S. Aramaki and T. Yamamoto, Chem. Mater., 2005, 17, 6060 CrossRef CAS.
- L. Groenendaal, G. Zotti, P. H. Aubert, S. M. Waybright and J. R. Reynolds, Adv. Mater., 2003, 15, 855 CrossRef CAS.
- L. Groenendaal, F. Jonas, D. Freitag, H. Pielartzik and J. R. Reynolds, Adv. Mater., 2000, 12, 481 CrossRef CAS.
- J. Roncali, P. Blanchard and P. Frère, J. Mater. Chem., 2005, 15, 1589 RSC.
- Y. S. Park, D. Kim, H. Lee and B. Moon, Org. Lett., 2006, 8, 4699 CrossRef CAS PubMed.
- B. Muktha, G. Madras, T. Guru Row, U. Scherf and S. Patil, J. Phys. Chem. B, 2007, 111, 7994 CrossRef CAS PubMed.
- S. Ghosh, N. A. Kouame, S. Remita, L. Ramos, F. Goubard, P.-H. Aubert, A. Dazzi, A. Deniset-Besseau and H. Remita, Sci. Rep., 2015, 5, 18002 CrossRef CAS PubMed.
- H. Li, W. Hou, X. Tao and N. Du, Appl. Catal., B, 2015, 172–173, 27 CrossRef CAS.
- M. Pelaez, N. T. Nolan, S. C. Pillai, M. K. Seery, P. Falaras, A. G. Kontos, P. S. M. Dunlop, J. W. J. Hamilton, J. A. Byrne and K. O'Shea, Appl. Catal., B, 2012, 125, 331 CrossRef CAS.
- S. Naghibi, S. Vahed, O. Torabi, A. Jamshidi and M. H. Golabgir, Appl. Surf. Sci., 2015, 327, 371 CrossRef CAS.
- H. Meng, D. F. Perepichka, M. Bendikov, F. Wudl, G. Z. Pan, W. Yu, W. Dong and S. Brown, J. Am. Chem. Soc., 2003, 125, 15151 CrossRef CAS PubMed.
- S. Breda, I. Reva, L. Lapinski, M. Nowak and R. Fausto, J. Mol. Struct., 2006, 786, 193 CrossRef CAS.
- C. Deetuam, C. Samthong, S. Thongyai, P. Praserthdam and A. Somwangthanaroj, Compos. Sci. Technol., 2014, 93, 1 CrossRef CAS.
- Y. Xiao, J. Wu, G. Yue, J. Lin, M. Huang, Z. Lan and L. Fan, Electrochim. Acta, 2012, 85, 432 CrossRef CAS.
- D. Zhang, J. Qin and G. Xue, Synth. Met., 1999, 100, 285 CrossRef CAS.
- R. Hoogenboom, D. Wouters and U. S. Schubert, Macromolecules, 2003, 36, 4743 CrossRef CAS.
- A. Buzarovska and L. Arsov, Polym. Bull., 2003, 50, 161 CrossRef CAS.
- T. Yamamoto, K. Shiraishi, M. Abla and I. Yamaguchi, Polymer, 2002, 43, 711 CrossRef CAS.
- G. Zhang, J. Zhang, G. Ding, J. Guo, H. Lu, L. Qiu and W. Ma, Polymer, 2016, 93, 213 CrossRef CAS.
- A. Ali, R. Jamal, A. Rahman, Y. Osman and T. Abdiryim, Prog. Nat. Sci., 2013, 23, 524 CrossRef.
- T. Nie, K. Zhang, J. Xu, L. Lu and L. Bai, J. Electroanal. Chem., 2014, 717, 1 CrossRef.
- T. Yamamoto, T. Shimizu and E. Kurokawa, React. Funct. Polym., 2000, 43, 79 CrossRef CAS.
- N. Gospodinova and L. Terlemezyan, Prog. Polym. Sci., 1998, 23, 1443 CrossRef CAS.
- T. Y. Kim, C. M. Park, J. E. Kim and K. S. Suh, Synth. Met., 2005, 149, 169 CrossRef CAS.
- C. Wang, M. E. Benz, E. LeGoff, J. L. Schindler, J. Allbritton-Thomas, C. R. Kannewurf and M. G. Kanatzidis, Chem. Mater., 1994, 6, 401 CrossRef CAS.
- J. Xu, H. Chan, S. Ng and T. Chung, Synth. Met., 2002, 132, 63 CrossRef CAS.
- T. Abdiryim, R. Jamal, C. Zhao, T. Awut and I. Nurulla, Synth. Met., 2010, 160, 325 CrossRef CAS.
- Z.-Q. Li, H.-L. Wang, L.-Y. Zi, J.-J. Zhang and Y.-S. Zhang, Ceram. Int., 2015, 41, 10634 CrossRef CAS.
- S. Sood, A. Umar, S. K. Mehta and S. K. Kansal, J. Colloid Interface Sci., 2015, 450, 213 CrossRef CAS PubMed.
- S. H. Hsieh, W. J. Chen and C. T. Wu, Appl. Surf. Sci., 2015, 340, 9 CrossRef CAS.
- V. Eskizeybek, F. Sarı, H. Gülce, A. Gülce and A. Avcı, Appl. Catal., B, 2012, 119–120, 197 CrossRef CAS.
- S. Kant, S. Kalia and A. Kumar, J. Alloys Compd., 2013, 578, 249 CrossRef CAS.
- S. Ameen, M. S. Akhtar, Y. S. Kim, O. B. Yang and H.-S. Shin, Colloid Polym. Sci., 2010, 288, 1633 CAS.
- T. Lv, L. Pan, X. Liu and Z. Sun, Electrochim. Acta, 2012, 83, 216 CrossRef CAS.
- Y.-G. Peng, J.-L. Ji, Y.-L. Zhang, H.-X. Wan and D.-J. Chen, Environ. Prog. Sustainable Energy, 2014, 33, 123 CrossRef CAS.
- R. Qiu, D. Zhang, Y. Mo, L. Song, E. Brewer, X. Huang and Y. Xiong, J. Hazard. Mater., 2008, 156, 80 CrossRef CAS PubMed.
- J. Chen, Y. Qian and X. Wei, J. Mater. Sci., 2010, 45, 6018 CrossRef CAS.
- Y. Lu, P. R. Chang, P. Zheng and X. Ma, Chem. Eng. J., 2014, 255, 49 CrossRef CAS.
- W. Zhao, W. Fu, H. Yang, C. Tian, M. Li, J. Ding, W. Zhang, X. Zhou, H. Zhao and Y. Li, Nano-Micro Lett., 2011, 3, 20 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a