
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanisms and stereoselectivities of the DABCO-catalyzed Rauhut–Currier reaction of α,β-unsaturated ketones and aryl acrylates: a computational investigation†
Yan Li*,
Ruixue Tian and
Shiwen Du
School of Petroleum and Chemical Engineering, Dalian University of Technology, Panjin Campus, Panjin, 124221, China. E-mail: yanli_101@dlut.edu.cn; Tel: +86 15142738951
First published on 13th January 2017
Abstract
It has been experimentally reported that a great acceleration of the Rauhut–Currier reaction of aryl acrylates with α,β-unsaturated ketones is achieved in the presence of phenol (PhOH) [W. Liu, G. Zhao, Org. Biomol. Chem., 12 (2014) 832–835]. A theoretical investigation on the mechanisms as well as stereoselectivities of the 1,4-diazabicyclo[2.2.2]octane (DABCO)-catalyzed RC reaction of phenyl acrylate R1 with (E)-2-benzoyl-3-phenyl-acrylonitrile R2 in the absence and in the presence of PhOH has been performed using density functional theory (DFT). Multiple possible reaction pathways (associated with RRS, RSR, SRS and SSR configurations) have been explored. The whole reaction process can be characterized by four steps: (1) the conjugate addition of DABCO to R1, (2) the nucleophilic attack at R2, (3) the 1,3-H-shift process (direct H-shift in the absence of PhOH and stepwise H-shift in the presence of PhOH) and (4) the catalyst release affording the final product. The calculated results show a relatively high energy barrier (42.2–46.0 kcal mol−1, corresponds to the direct 1,3-H-shift step) for the reaction in the absence of PhOH. For the reaction in the presence of PhOH, the pathway leading to the (SSR)-product is the most favorable pathway. The first step of the hydrogen-shift process is found to be the rate-determining step, whereas the second step is the stereoselectivity determining step. The role of PhOH on the title reaction is discussed.
1. Introduction
It is well known that the Rauhut–Currier (RC) reaction (also known as the vinylogous Morita–Baylis–Hillman (MBH) reaction), addition of the α-position of one activated alkene to the β-position of a second alkene in the presence of a nucleophilic catalyst, is one of the effective methods in creating carbon–carbon bonds.1–6 Due to its high atom- and step-economy, the RC reaction has become an increasingly active area, since it was first discovered by Rauhut and Currier in 1963.1 Up to now, a large number of experimental studies have been reported on the RC dimerization of acrylonitrile,7–9 vinyl ketones,10 acrylates,11 and nitroalkenes.12 The cross coupling versions of the RC reaction have also been reported.13–20 Furthermore, the enantioselective RC reactions have been studied and significant progress has been achieved.21–27 Moreover, the RC reaction has been developed for the synthesis of many natural products and pharmaceuticals.28–33However, compared to the rich knowledge of experimental studies, much less theoretical studies have been reported on the RC reaction. In 2011, Gu, Xiao and co-workers explored the enantioselective crossed RC reaction between nitroolefins and α,β-unsaturated esters by using a combined experimental and theoretical approach, and their calculation results support the experimental findings (reaction mechanism and enantioselectivity).34 Subsequently, Houk and co-workers investigated the enantioselective cysteine-catalyzed RC reaction using the density functional theory method. Their computational study demonstrated that the water and counterions (K+/Na+) play an important role in determining the stereochemistry of the reaction.35
Recently, Zhao and co-workers investigated 1,4-diazabicyclo[2.2.2]octane (DABCO)-catalyzed cross RC reaction of aryl acrylates with α,β-unsaturated ketone, which serve as a key step for the synthesis of pyran-2-ones (Scheme 1).36 Pyran-2-ones as structural motifs are often found in many natural products, bioactive molecules, and organic intermediates.37–41 Preliminary experimental results led Zhao et al. to propose the possible catalytic cycle. As depicted in Scheme 2, the originally proposed mechanism consists of four steps: the nucleophilic addition of DABCO to aryl acrylate to form a zwitterionic intermediate A, addition of the zwitterion A to α,β-unsaturated ketone to form B, 1,3-hydrogen shift affording C and catalyst regeneration. On the experimental observations, Zhao and coworkers demonstrated that phenol (PhOH) could be used as a Brønsted acid additive to accelerate the reaction. Zhao and co-workers also suggested that addition of the zwitterion A to α,β-unsaturated ketone is the rate-determining step (RDS). Although the mechanism for the DABCO-catalyzed RC reaction between aryl acrylates and α,β-unsaturated ketone has been proposed, the details are still unclear. Particularly unclear issues concern the following questions: how does each of the catalytic steps take place? What is the role of PhOH? How the PhOH promote the reaction? To the best of our knowledge, no theoretical investigation on the mechanism of the current RC reaction has been reported up to now.
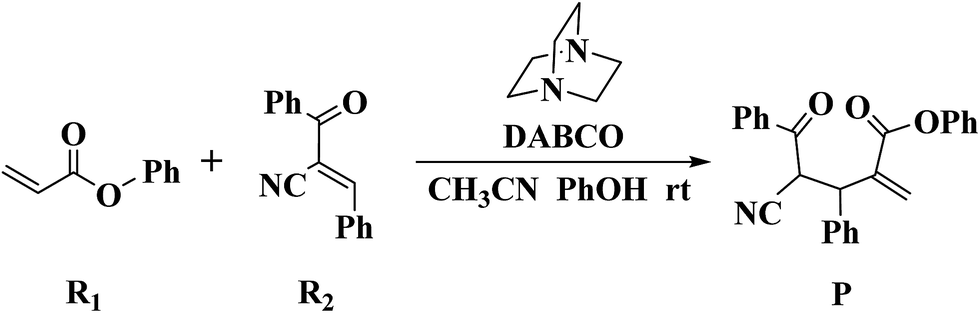 | ||
| Scheme 1 DABCO-catalyzed Rauhut–Currier (RC) reaction. | ||
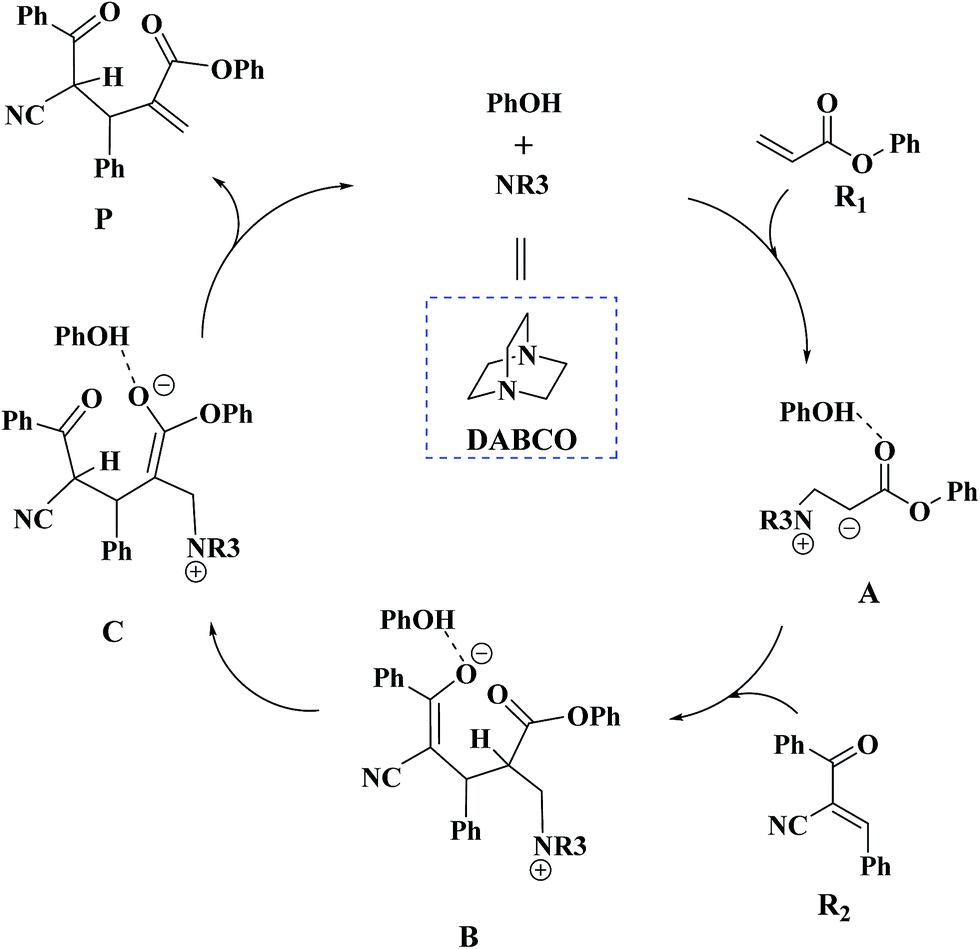 | ||
| Scheme 2 The proposed mechanism for the DABCO-catalyzed RC reaction in experiment. | ||
In this work, we investigated the mechanism of the DABCO-catalyzed RC reaction between phenyl acrylate and (E)-2-benzoyl-3-phenyl-acrylonitrile with the aid of density functional theory (DFT) calculations and try answer the questions raised above. For this purpose, the reaction pathways both in the absence and presence of PhOH have been studied.
The remainder of this paper is organized as follows: we first describe the Computational methods in Section 2. Then we present the Results and discussion in Section 3, followed by the Comparison with experiments in Section 4. Finally, we finish with some concluding remarks in Section 5.
2. Computational method
The geometry optimizations of the critical points (reactants, products, intermediates and transition states) were carried out using the M06-2X42 method in conjunction with 6-31G(d) basis set.43 It has been shown that M06-2X provide a better description of kinetics and thermodynamics.44–49 Frequency calculations were performed at the same level of theory, to check whether the obtained species was a minimum (with all real frequencies) or a transition state (with only one imaginary frequency), as well as to obtain zero point energy (ZPE) and thermodynamic corrections at 298.15 K and 1 atm. Intrinsic reaction coordinate (IRC) calculations were performed to ensure that the transition state is connected with its designated isomers.50 Single point energy calculations were performed at the M06-2X/6-311++G(d,p) level of theory. The solvation effects of acetonitrile (CH3CN) were considered during geometry optimization, frequency calculation and single point energy calculation by using the Cramer–Truhlar continuum solvation model SMD.51 The Gibbs free energies were used to discuss the reaction mechanisms. The natural bond orbital (NBO) analysis was applied to calculate the charge distributions.52–54 The Gaussian 09 program package was used for all DFT calculations described herein.553. Results and discussion
As noted in the introduction, Zhao et al.'s experimental studies indicated that phenol (PhOH) can promote the RC reaction.36 To examine the role of PhOH, the DABCO-catalyzed RC reaction of phenyl acrylate R1 with (E)-2-benzoyl-3-phenyl-acrylonitrile R2 in both the absence and the presence of PhOH have been investigated with the aid of DFT calculations.3.1 Reaction mechanism in the absence of PhOH
On the basis of previous experimental studies36 and our calculation results, the reaction mechanism of the RC reaction between R1 and R2 is depicted in Scheme 3. The corresponding potential energy profile is shown in Fig. 1. In the following part of this section, we will detail the reaction mechanism step by step.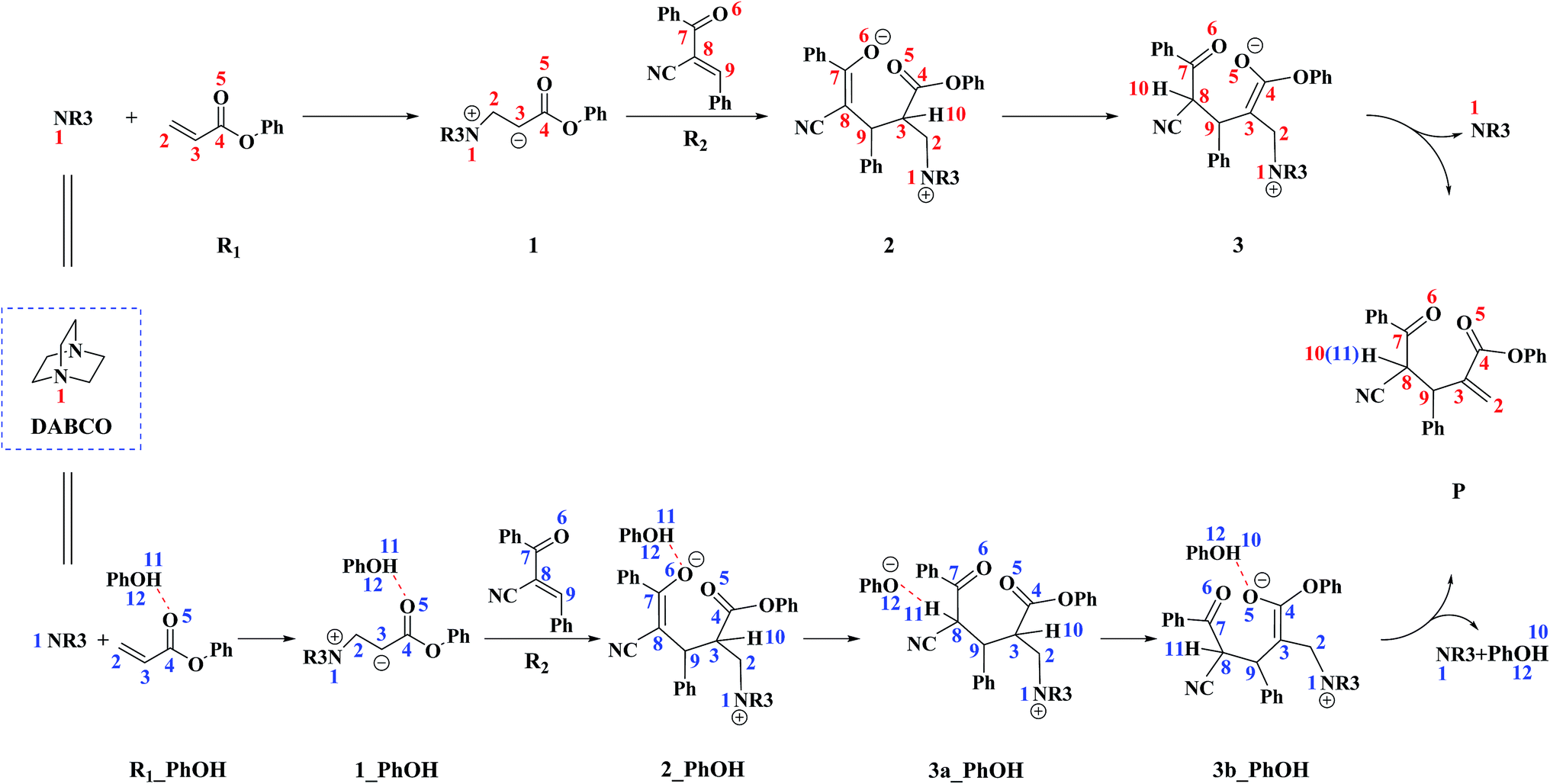 | ||
| Scheme 3 The reaction mechanism in the absence and in the presence of PhOH. | ||
 | ||
| Fig. 1 Free energy profile of the reaction mechanism in the absence of PhOH. The solvation-corrected relative free energies at SMD(CH3CN)/M06-2X/6-311++G(d,p) level are given in kcal mol−1. | ||
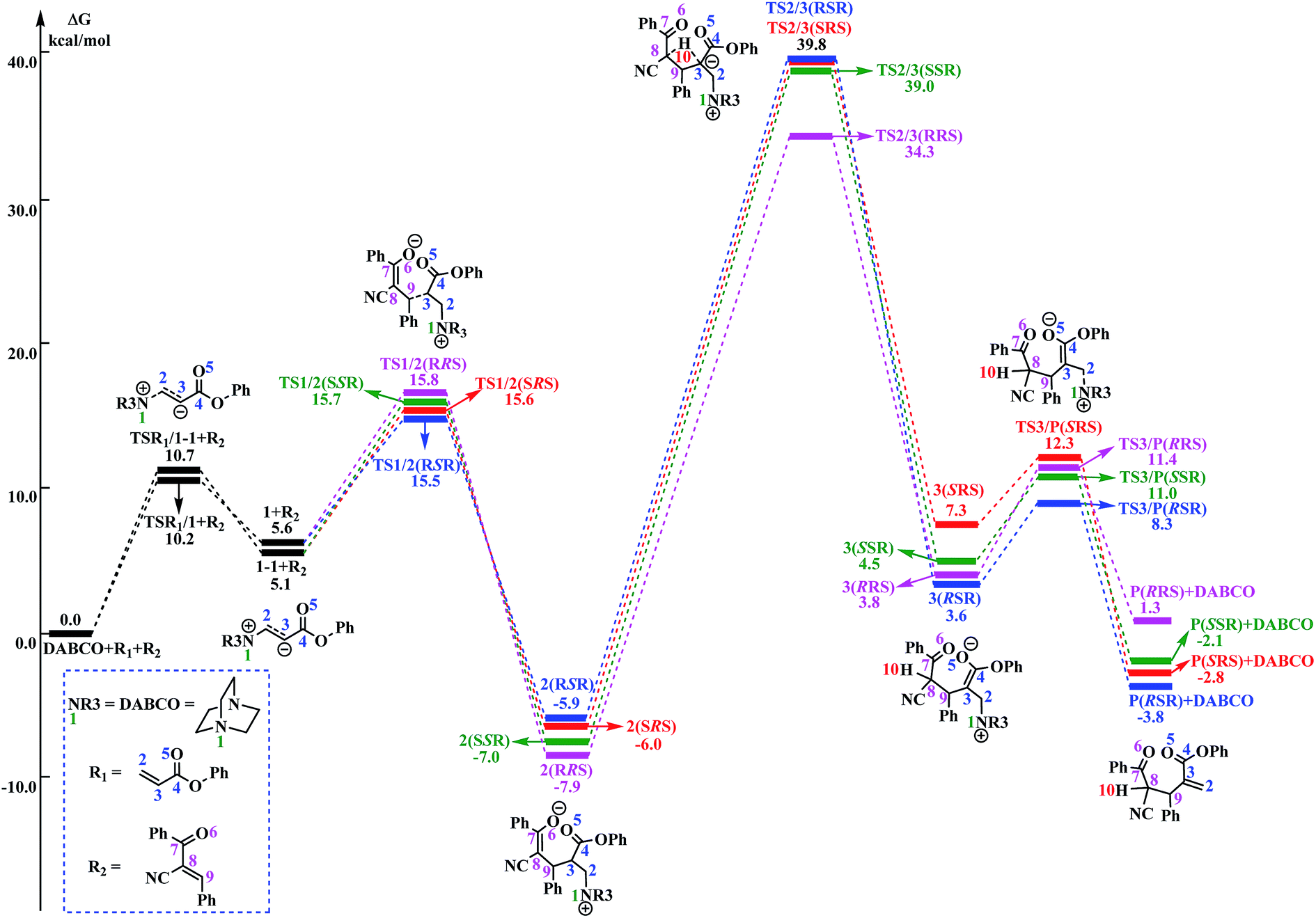
As shown in Fig. 1, the free energy barrier for the third step (2 → 3) is much higher than those of the other steps (R1 + DABCO → 1, 1 → 2, and 3 → P). Therefore, the third step is the rate-determining step. On the basis of our calculations, the energy barrier for the third step is inaccessibly high. Considering that the experiments were conducted under room temperature, we can easily deduce that the current RC reaction cannot take place via the direct H-shift mechanism.
3.2 Reaction mechanism in the presence of PhOH
The reaction mechanism in the presence of PhOH is also depicted in Scheme 3. The free energy profile for the complete process is presented in Fig. 2. It should be pointed out that, in order to have unbiased free energies, a molecule of PhOH is included in the reaction system throughout. As shown in Fig. 2, reactants N-phenylmaleimide and 2-benzoyl acrylate were complexed to a molecule of PhOH, which were labeled as R1_PhOH and R2_PhOH, respectively. Then the free energy of R1_PhOH + R2_PhOH + DABCO is set to 0.0 kcal mol−1 as reference in the potential energy profile.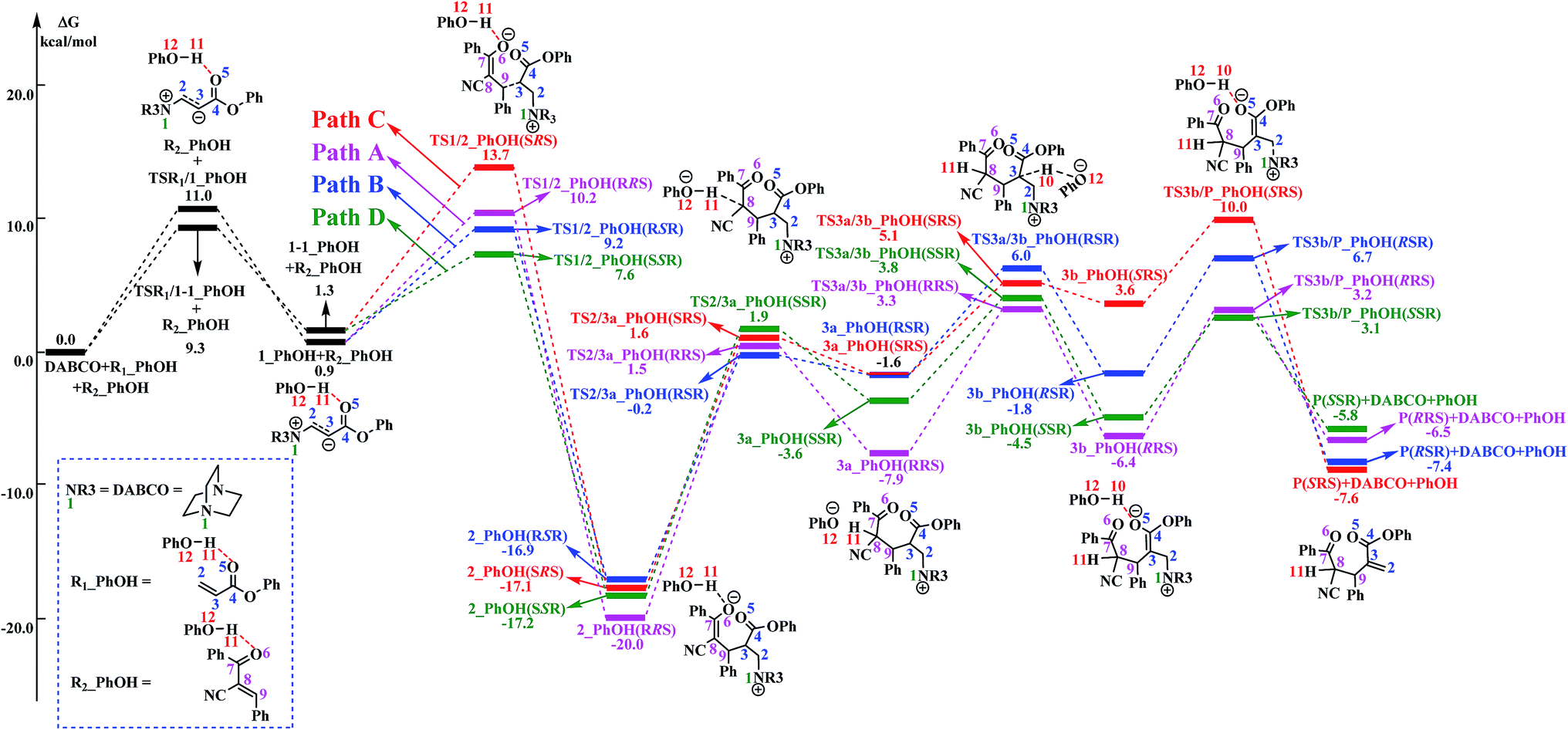 | ||
| Fig. 2 Free energy profile of the reaction mechanism in the presence of PhOH. The solvation-corrected relative free energies at SMD(CH3CN)/M06-2X/6-311++G(d,p) level are given in kcal mol−1. | ||
By the above, four pathways (paths A, B, C and D, see Fig. 2) for the DABCO-catalyzed RC reaction of phenyl acrylates with α,β-unsaturated ketone in the presence of PhOH have been discussed in detail. The four pathways start from the same reactants, go through different pathways leading to four isomers of product. As shown in Fig. 2, the overall energy barrier for paths A, B, C and D should be the energy difference of 2(RRS) with TS3a/3b(RRS), 2(RSR) with TS3b/P(RSR), 2(SRS) with TS3b/P(SRS), and 2(SSR) with TS3a/3b(SSR), which are calculated to be 23.3, 23.6, 27.1 and 21.0 kcal mol−1, respectively. Consequently, the energy barrier order of the four pathways increases as follows: path D → path A → path B → path C. Since the energy barrier determines the reaction rate of a given pathway, we can easily deduce that path D is the most energy favorable.
4. Comparison with experiments
It would be of interest to compare our calculation results with previous experimental findings. In the experimental report,36 the current RC reaction is initiated by conjugate addition of DABCO to aryl acrylate to form the zwitterionic intermediate A, which subsequently attacks the α,β-unsaturated ketone to give B, followed by the 1,3-hydrogen shift leads to C. Finally, catalyst DABCO releases from C affording the final product P. On the basis of our calculation results, the most energetically favorable pathway is path D (as depicted in Fig. 2). The intermediates A, B, C, and P correspond to 1-1_PhOH, 2_PhOH(SSR), 3b_PhOH(SSR) and P_PhOH(SSR), respectively in our result. Furthermore, Zhao and co-workers suggested that phenol PhOH can stabilize A, and accelerate the reaction between A and R2. Our calculation results indicate that complexation of 1-1 by one molecule of PhOH induces a decrease of reaction energy of 3.8 kcal mol−1 (see Fig. 1 and 2). Moreover, the energy barrier for 1-1 + R2 → 2 in the presence of PhOH is lower than that in the absence of PhOH (6.3 vs. 10.6 kcal mol−1, associated with SSR-configuration), and 2_PhOH is much more stable than 2 (−17.2 vs. −7.0 kcal mol−1, associated with SSR configuration). Therefore, based on both kinetical and thermodynamical considerations, the reaction of 1 + R2 in the presence of PhOH is much more favorable than the corresponding reaction in the absence of PhOH. As for these aspects, our calculation results are in good agreement with the experimental results reported by Zhao and co-workers.36 However, there exist discrepancies. Zhao and co-workers suggested that the addition of A (corresponds to 1-1_PhOH in our result) to R2 is the rate-determining step. According to our calculations, the overall energy barrier for the hydrogen-shift process is 21.0 kcal mol−1 (corresponds to the energy difference between 2_PhOH and TS3a/3b_PhOH, see path D in Fig. 2), which is much higher than that for the addition of 1-1_PhOH to R2 (6.3 kcal mol−1 relative to 1-1_PhOH and 7.6 kcal mol−1 with respect to reactants). In other words, we predicted the first step of hydrogen-shift process is the rate-determining step. In view of this discrepancy, future reinvestigation of the current RC reaction is strongly desired.5. Conclusions
The mechanisms and stereoselectivities of DABCO-catalyzed RC reaction of phenyl acrylate R1 with (E)-2-benzoyl-3-phenyl-acrylonitrile R2 both in the absence and presence of phenol (PhOH) have been investigated at the M06-2X/6-311++G(d,p)//M06-2X/6-31G(d) level in CH3CN solvent using the SMD solvation model. Our calculation results can be summarized as follows:In the absence of PhOH, the reaction proceeds via four steps, which includes activation of R1 by DABCO, addition to R2, direct 1,3-hydrogen shift, catalyst DABCO liberation. The calculated results show that the 1,3-hydrogen shift is rate-limiting and the activation energy barrier for this step is inaccessibly high (42.2–46.0 kcal mol−1), indicating this reaction process is kinetically unfavorable.
In the presence of PhOH, four reaction pathways (paths A, B, C and D) associated with RRS, RSR, SRS and SSR configuration products have been investigated and compared. The most favorable pathway (path D) include four stages: the reaction is initiated with the conjugate addition of DABCO to R1 to form the zwitterionic intermediate 1-1_PhOH (stage I), which subsequently attacks R2 to produce 2_PhOH(SSR) (stage II). 1,3-H-shift of 2_PhOH(SSR) is demonstrated to proceed via a stepwise mechanism: the C8 atom of 2_PhOH(SSR) abstracts H11 from O12 to form intermediate 3a_PhOH(SSR), and then the C3 can donate H10 to O12 to complete the H shift process (from C8 to C3) and produce intermediate 3b_PhOH(SSR) (stage III). Finally, the catalyst DABCO releases from 3b_PhOH(SSR) to generate the final product P(SSR) (stage IV). The calculated results show that the first step of H-shift process (2 → 3a) is the rate-determining step. The addition of 1-1_PhOH to R2 is calculated to be the stereoselectivity-determining step.
The calculated results reveal that the presence of PhOH does stabilize the intermediates and transition states. Moreover, the overall energy barrier for the 1,3-H shift step in the absence and presence of PhOH is calculated to be 46.0 and 21.0 kcal mol−1 (associated with SSR configuration), respectively, which is consistent with previous experimental findings (rate acceleration in the presence of PhOH). The present paper may provide a useful guide for understanding other analogous reactions.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 21403024).Notes and references
- M. M. Rauhut and H. Currier, U.S. Pat., 3074999, American Cyanamide, 1963Chem. Abstr., 1963, 58, 11224a.
- J. L. Methot and W. R. Roush, Adv. Synth. Catal., 2004, 346, 1035 CrossRef CAS.
- C. E. Aroyan, A. Dermenci and S. J. Miller, Tetrahedron, 2009, 65, 4069 CrossRef CAS.
- P. Z. Xie and Y. Huang, Eur. J. Org. Chem., 2013, 6213 CrossRef CAS.
- J. Peng, X. S. Takizawa, T. M.-N. Nguyen, A. Grossmann, D. Enders and H. Sasai, Angew. Chem., Int. Ed., 2012, 51, 5423 (Angew. Chem., 2012, 124, 5519) CrossRef PubMed.
- F. Zheng and Y. C. Chen, Org. Lett., 2013, 15, 5534 CrossRef PubMed.
- M. M. Baizer and J. D. Anderson, J. Org. Chem., 1965, 30, 1357 CrossRef CAS.
- J. D. McClure, J. Org. Chem., 1970, 35, 3045 CrossRef CAS.
- L. Yu, J. Wang, X. Zhang, H. E. Cao, G. L. Wang, K. H. Ding, Q. Xu and M. Lautens, RSC Adv., 2014, 4, 19122 RSC.
- D. Basavaiah, V. V. L. Gowriswari and T. K. Bharathi, Tetrahedron Lett., 1987, 28, 4591 CrossRef CAS.
- H. Amri, M. Rambsud and J. Villieras, Tetrahedron Lett., 1989, 30, 7381 CrossRef CAS.
- P. Shanbhag, P. R. Nareddy, M. Dadwal, S. M. Mobin and I. N. N. Namboothiri, Org. Biomol. Chem., 2010, 8, 4867 CAS.
- X. L. Dong, L. Liang, E. Q. Li and Y. Huang, Angew. Chem., Int. Ed., 2015, 54, 1621 CrossRef CAS PubMed.
- W. J. Yao, Y. H. Wu, G. Wang, Y. P. Zhang and C. Ma, Angew. Chem., 2009, 121, 9893 CrossRef.
- P. Shanbhag, P. R. Nareddy, M. Dadwal, S. M. Mobin and I. N. N. Namboothiri, Org. Biomol. Chem., 2010, 8, 4867 CAS.
- P. Z. Xie, Y. Huang, W. Q. Lai, X. T. Meng and R. Y. Chen, Org. Biomol. Chem., 2011, 9, 6707 CAS.
- R. Kumar, T. Kumar, S. M. Mobin and I. N. N. Namboothiri, J. Org. Chem., 2013, 78, 5073 CrossRef CAS PubMed.
- W. Liu and G. Zhao, Org. Biomol. Chem., 2014, 12, 832 CAS.
- W. Zhou, X. Su, M. N. Tao, C. E. Zhu, Q. J. Zhao and J. L. Zhang, Angew. Chem., 2015, 127, 15066 CrossRef.
- Q. Zhao, C. Pei, X. Guan and M. Shi, Adv. Synth. Catal., 2011, 353, 1973 CrossRef CAS.
- S. Takizawa, T. M. N. Nguyen, A. Grossmann, D. Enders and H. Sasai, Tetrahedron, 2013, 69, 1202 CrossRef CAS.
- X. Dong, L. Liang, E. Li and Y. Huang, Angew. Chem., Int. Ed., 2015, 54, 1621 CrossRef CAS PubMed.
- J. H. Robert, O. G. Scanes, G. André and R. S. David, Org. Lett., 2015, 17, 2462 CrossRef PubMed.
- X. Zhao, J. J. Gong, K. Yuan, F. Sha and X. Y. Wu, Tetrahedron Lett., 2015, 56, 2526 CrossRef CAS.
- W. Zhou, X. Su, M. Tao, C. Zhu, Q. Zhao and J. Zhang, Angew. Chem., Int. Ed., 2015, 54, 14853 CrossRef CAS PubMed.
- X. Su, W. Zhou, Y. Li and J. Zhang, Angew. Chem. Int. Ed., 2015, 54, 6874 (Angew. Chem., 2015, 127, 6978) CrossRef CAS PubMed.
- W. Zhou, P. Chen, M. Tao, X. Su, Q. Zhao and J. Zhang, Chem. Commun., 2016, 52, 7612 RSC.
- A. Dermenci, P. S. Selig, R. A. Domaoal, K. A. Spasov, K. S. Anderson and S. J. Miller, Chem. Sci., 2011, 2, 1568 RSC.
- Y. Y. Zhang, R. Gurubrahamam and K. Chen, Adv. Synth. Catal., 2015, 357, 2457 CrossRef CAS.
- V. Mane, T. Kumar, S. Pradhan, S. Katiyar and I. N. N. Namboothiri, RSC Adv, 2015, 5, 69990 RSC.
- G. Wang, R. Rexiti, F. Sha and X.-Y. Wu, Tetrahedron, 2015, 71, 4255 CrossRef CAS.
- A. Albrecht, A. Skrzynska, A. Przydacz and L. Albrecht, Synlett, 2015, 26, 2679 CrossRef CAS.
- A. Bhunia, S. R. Yetra, R. G. Gonnadec and A. T. Biju, Org. Biomol. Chem., 2016, 14, 5612 CAS.
- X. F. Wang, L. Peng, J. An, C. Li, Q. Q. Yang, L. Q. Lu, F. L. Gu and W. J. Xiao, Chem.–Eur. J., 2011, 17, 6484 CrossRef CAS PubMed.
- S. Osuna, A. Dermenci, S. J. Miller and K. N. Houk, Chem.–Eur. J., 2013, 19, 14245 CrossRef CAS PubMed.
- W. Liu and G. Zhao, Org. Biomol. Chem., 2014, 12, 832 CAS.
- J. Staunton, in Comprehensive Organic Chemistry, ed. P. G. Sammes, Pergamon Press, Oxford, England, 1979, vol. 4, p. 629 Search PubMed.
- T. Tombray, A. Blanc, J.-M. Weibel and P. Pale, Org. Lett., 2010, 12, 5362 CrossRef PubMed.
- D. Liu, X.-M. Li, L. Meng, C.-S. Li, S.-S. Gao, Z. Shang, P. Proksch, C.-G. Huang and B.-G. Wang, J. Nat. Prod., 2011, 7, 7487 Search PubMed.
- G. A. Suárez-Ortiz, C. M. Cerda-García-Rojas, A. Hernández-Rojas and R. Pereda-Miranda, J. Nat. Prod., 2013, 76, 72 CrossRef PubMed.
- C. D. Donner, Tetrahedron, 2013, 69, 377 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- W. L. Hehre, L. Radom, P. R. Schleyer and J. A. Pople, Ab initio molecular orbital theory, Wiley, New York, 1986, p. 332 Search PubMed.
- E. H. Krenske, E. C. Davison, I. T. Forbes, J. A. Warner, A. L. Smith, A. B. Holmes and K. N. Houk, J. Am. Chem. Soc., 2012, 134, 2434 CrossRef CAS PubMed.
- H. Yang and M. W. Wong, J. Am. Chem. Soc., 2013, 135, 5808 CrossRef CAS PubMed.
- H. Yang and M. W. Wong, J. Org. Chem., 2011, 76, 7399 CrossRef CAS PubMed.
- M. W. Wong and A. M. E. Ng, Aust. J. Chem., 2014, 67, 1100 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157 CrossRef CAS PubMed.
- H. Xue, D. Jiang, H. Jiang, C. W. Kee, H. Hirao, T. Nishimura, M. W. Wong and C. H. Tan, J. Org. Chem., 2015, 80, 5745 CrossRef CAS PubMed.
- C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed.
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO Version 3.1, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 1996 Search PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899 CrossRef CAS.
- J. P. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102, 7211 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, R. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, J. Cossi, N. Rega, J. M. Millam, M. Klene, M. Knox, J. B. Cross, V. Bakken, V. Adamo, J. Jaramillo, J. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, R. L. Morokuma, V. G. Zakrzewski, G. A. Voth, G. A. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 (revision B. 01), Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25311c |
| This journal is © The Royal Society of Chemistry 2017 |