
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermoplastic microfluidic devices for targeted chemical and biological applications†
Dan Voicua,
Gabriella Lestarib,
Yihe Wanga,
Michael DeBonoa,
Minseok Seoa,
Sangho Cho a and
Eugenia Kumachevaac
a and
Eugenia Kumachevaac
aDepartment of Chemistry, University of Toronto, 80 Saint George Street, Toronto, Ontario M5S 3H6, Canada. E-mail: ekumache@chem.utoronto.ca
bDepartment of Chemical Engineering and Applied Chemistry, University of Toronto, 200 College Street, Toronto, Ontario M5S 3E5, Canada
cInstitute of Biomaterials and Biomedical Engineering, University of Toronto, 164 College Street, Toronto, Ontario M5S 3G9, Canada
First published on 12th January 2017
Abstract
Combining photolithography and hot embossing offers the capability of cost-efficient and high-fidelity fabrication of polymer microfluidic devices, however, poor chemical resistance in nonpolar organic solvents and high gas permeability of the currently used polymers narrow the range of applications of the microfluidic devices. With the aim of specific chemical or biological applications, we report the fabrication of microfluidic devices in a broader range of thermoplastic polymers. For chemical reactions to be conducted in aromatic and hydrocarbon solvents, microfluidic reactors fabricated in high-density polyethylene (HDPE) showed excellent compatibility with a range of organic solvents. Microfluidic devices fabricated in polyvinylchloride (PVC) exhibited drastically reduced gas permeability, in comparison with devices fabricated in polydimethylsiloxane (PDMS). To address the needs of biorelated research, we fabricated polystyrene (PS) microfluidic devices containing high-density, two-dimensional arrays of aqueous droplets.
Introduction
Applications of microfluidics (MFs) in chemistry and biology offer advantages of low reagent consumption, enhanced control over heat and mass transfer, the ability to carry out multi-step reactions without exposure of intermediate products to the ambient environment, and the capability of gradual change in reagent concentrations in a high-throughput manner.Over the past two decades, materials used for the fabrication of MF devices have undergone a transition from silicone and glass to elastomeric and thermoplastic polymers. A growing interest in polymeric MF devices stems from their time-, labour-, and cost-efficient fabrication,1 however, their applications in chemistry and biology impose specific requirements for the polymer properties. Table 1 summarizes some of the key properties of polymers used for the fabrication of MF devices. In addition to the highly desirable low cost and ease of fabrication, many polymer characteristics are determined by the specific application of the MF device. For example, polycarbonate, cyclic olefin polymer, and poly(methyl methacrylate) are chemically stable in water and other polar liquids, however, MF reactors fabricated in these materials are not suitable for studies of chemical reactions conducted in aromatic and hydrocarbon solvents such as bromobenzene,2 tetrahydrofuran, or dichloromethane.3
| Characteristics | Requirements | Comments |
|---|---|---|
| Fabrication | High-fidelity, low cost, rapid prototyping | |
| The capability to scale up | ||
| Optical properties | Transparency at a desired wavelength | Important in optical characterization |
| No absorption or auto-fluorescence in the wavelength range used in MF experiments | Important for photoinitiated reactions, spectroscopy-based sensing, and fluorescence-based analysis | |
| Thermostability | High glass transition or melting temperature | Required for studies of high-temperature reactions or physical processes |
| Chemical stability | Resistance to swelling in polar or non-polar solvents | Minimal swelling can be tolerated in some applications |
| Gas permeability | High gas permeability | Useful for cell culture experiments and some polymerization reactions |
| Low gas permeability processes | Useful in studies of gas–liquid reactions and physical processes |
Gas permeability is another important characteristic of the polymer to be considered. For example, polydimethylsiloxane, polycarbonate, and cyclic olefin polymer have relatively high gas permeability.4,5 Gas permeation in MF devices fabricated in polydimethylsiloxane, has benefited cell culture experiments,6 evaporation-based protein crystallization,7 polymerase chain reaction,8 and supercritical fluid extraction.9 On the other hand, the requirement for low gas permeability becomes important when a gas resides in a MF device for a relatively long period of time or when gas diffusion in and out of the device has to be controlled.2
The method used for the fabrication of MF devices is another important factor to consider. Laser-ablation has a limitation in fabrication of smooth microchannel walls.10 Micromilling is limited by channel dimensions, which should not be smaller than ∼25 μm for standard bench-top mills, while the inherent vibrations caused by the milling lead to surface roughness of up to 1.5 μm.11 Soft lithography generally utilizes polydimethylsiloxane12 and polyurethane,13 which are not stable in aromatic and hydrocarbon organic solvents. While injection molding is a high-throughput method for producing polymeric MF reactors, prototyping of devices with complex designs may be limited by the cost of mold inserts. Similarly in hot embossing, the fabrication of a complex prototype embossing stamp may be prohibitively expensive.
Recently, we reported the use of photo-lithographically fabricated photoresist-on-copper stamps for the fabrication of MF devices in polycarbonate, cyclic olefin polymer, and poly(methyl methacrylate).14 The drawback of these polymers is poor chemical resistance in aromatic and hydrocarbon solvents, which limits their applications as chemical reactors. In addition, these polymers have high gas permeability and are thus not suitable for long-term studies of gas–liquid reactions.
In the present work, we provide guidance for the fabrication of thermoplastic MF devices for specific chemical and biological applications. High-density polyethylene was selected as the material of choice to fabricate MF reactors compatible with nonpolar, polar aprotic, and polar protic solvents in the temperature range from 25 to 80 °C. To address the requirement of low gas permeability, we utilized polyvinyl chloride. In addition, we report the fabrication of polystyrene MF reactors for applications in biology and biomedical research.
Experimental
High-density polyethylene (HDPE) and polyvinylchloride (PVC, Type 1) sheets were purchased from McMaster Carr (USA). Polystyrene (PS) was purchased from Home Depot (0.05′′ × 11′′ × 14′′, Plaskolite Inc. A-81, OH, USA). One mm thick, single-side polished copper sheets were purchased from McMaster Carr (USA). SU-8 photoresist was purchased from Microchem (SU-8 3050, Microchem Corp, USA).Fabrication of microfluidic devices
Microfluidic devices were fabricated using SU-8-on-copper stamps using thermo-embossing in a Carver press (Model M, Carver Inc., USA), as described elsewhere.14 All embossing and bonding steps were conducted in a custom built vacuum chamber that was integrated with the hot press. The de-embossing step was easier performed for lower aspect ratio features. Following the bonding of an embossed sheet with a planar sheet of the same polymer, the MF devices were cooled under applied pressure to room temperature and removed from the press.To reduce the thickness of the supplied HDPE sheets, they were sandwiched between two polished silicon wafers on the hot press, heated for 10 min at 190 °C (above the softening temperature of the polymer) and subjected to a pressure of 20.3 atm. Metal spacers with the thicknesses 300, 500, 800, and 1500 μm were used to control the ultimate film thickness. Polydimethylsiloxane devices were fabricated by the soft-lithography method using SU-8 on silicon masters.12
Characterization methods
An optical microscope (Olympus BX41, Olympus, Japan) coupled with a CCD camera (Evolution VF, Media Cybernetics, USA) was used to image MF devices in the course of experiments. Image analysis was conducted using Image Pro Plus software (v. 5.0, Media Cybernetics, USA). The variation in transmission of 2 × 2 cm HDPE films with different thicknesses was examined in the spectral range from 300 to 800 nm with a resolution of 2 nm using a UV-visible spectrometer (Cary 5000, Agilent Technologies, USA).Contact angles of deionized water on HDPE and PS surfaces were measured using a drop shape analyzer (DSA100S, KRUSS Gmbh, Germany). To change the surface properties of HDPE sheets, they were exposed to gas plasma in a plasma cleaner (PDC-32G, Harrick Plasma, USA). Ambient air, oxygen, or nitrogen (both Linde, USA) were supplied to the chamber under 600 mTorr vacuum for the time interval varying from 45 to 180 s.
The compatibility of MF devices with organic solvents was tested by purging each solvent in a MF device for 5 h (typical for MF experiments), and inspecting the change in dimensions of the microchannels. Experiments at elevated temperature were conducted by immersing the MF reactor in an oil bath and, subsequently, purging the specific solvent through the channels.2
Gas permeability of MF reactors fabricated in PVC was tested by examining the change in the length of nitrogen gaseous plugs separated with aqueous segments, all introduced and maintained in the microchannel at a pressure in the range from 1.34 to 2 atm. The results of these experiments were compared with those conducted in MF devices fabricated in PDMS.
Results and discussion
Fabrication of thermoplastic microfluidic devices
For each polymer, the pressure, temperature, and total microfabrication time, including heating, embossing, and de-embossing times, were optimized to ensure high-fidelity replication of the features from the stamp to the polymer MF device, strong bonding between the embossed and planar polymer sheets, and minimal damage of the stamp during the embossing process. Bonding of the embossed polymer sheet and a planar sheet was achieved at 140 °C for HDPE, that is, 6 °C below Tm, and at 100 °C for PVC and PS, which was 10 and 5 °C above Tg of these polymers, respectively. The bonding temperatures were selected to allow sufficient molecular mobility at the interface between the embossed and planar sheets and to avoid deformation of the embossed features during the bonding process. Importantly, cooling the polymer and stamp to room temperature before de-embossing resulted in the stamp features tearing off the stamp substrate. Table 2 summarizes the optimized fabrication conditions for the fabrication of features of MF devices in HDPE, PVC, and PS.| High-density polyethylene | Polyvinylchloride | Polystyrene | |
|---|---|---|---|
| a Information provided by the manufacturer. | |||
| Polymer state | Semi-crystalline15 | Semi-crystalline16 | Amorphous17 |
| Glass transition temperature, Tg (°C) | −123 (ref. 18) | 90 (ref. 19) | 95a |
| Melting temperature, Tm (°C) | 146 (ref. 20) | 210a | — |
| Embossing temperature (°C) | 160 | 125 | 125 |
| Embossing time (min) | 1 | 3 | 2 |
| Embossing pressure (atm) | 22.1 | 20.4 | 15.3 |
| De-embossing temperature (°C) | 130 | 65 | 105 |
| Bonding temperature (°C) | 140 | 100 | 100 |
| Bonding time (min) | 6 | 6 | 5 |
| Bonding pressure (atm) | 22.1 | 13.6 | 12 |
Fabrication of microfluidic devices operating in aromatic and hydrocarbon solvents
We selected HDPE as the polymer for the fabrication of MF reactors operating in aromatic and hydrocarbon solvents. As specified in Table 1, optical transmission was an important factor to consider when chemical reactions occurring in MF reactors are characterized by microscopy. Since HDPE is a translucent polymer, due to the scattering of light by its crystalline domains,15 we increased light transmission by reducing the thickness of walls of the MF device. To optimize wall thickness, light transmission was examined in the spectral range from 300 to 800 nm for a HDPE sheet varying in thickness from 300 to 1500 μm (Fig. 1a). For the wavelength of 500 nm, light transmission increased from 12 to 64%, when the sheet thickness reduced from ∼1500 to 300 μm, respectively. Notably, with reducing thickness, the flexibility of the HDPE sheets increased which, for thinner films, led to the sagging and warping of the sheet in the bonding stage and the resulting distortion of the channel dimensions. At the thickness of the HDPE sheet larger than ∼1500 μm (for 150 μm-tall channels), segmented gas–liquid flow in the fabricated MF reactor could not be discerned by optical microscopy.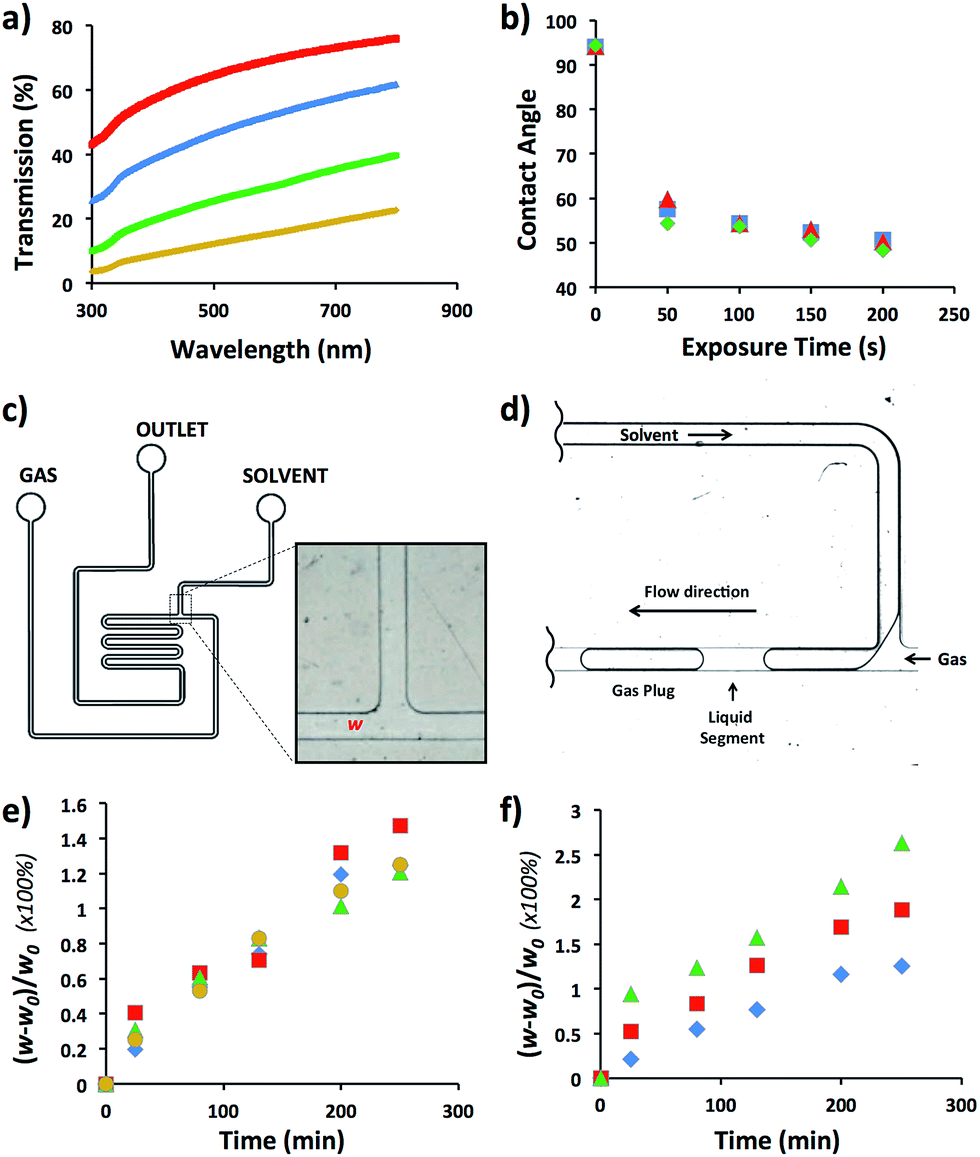 | ||
Fig. 1 (a) Variation in transmission of HDPE sheets with thickness of 300 ( ), 500 ( ), 500 ( ), 800 ( ), 800 ( ), and 1500 μm ( ), and 1500 μm ( ) in the spectral range 300 to 800 nm. (b) Contact angles of water on HDPE sheets after their exposure to oxygen ( ) in the spectral range 300 to 800 nm. (b) Contact angles of water on HDPE sheets after their exposure to oxygen ( ), nitrogen ( ), nitrogen ( ), or air plasma ( ), or air plasma ( ). (c) Schematic of the MF T-junction bubble generator with the width of channel at T-junction, w, measured in the course of experiments. The channel width is 300 μm and channel height is 50 μm. (d) Schematic of the T-junction region at the point of segmented flow generation. (e) Relative change in the channel width at T-junction, plotted as a function of time of MF bubble generation in THF ( ). (c) Schematic of the MF T-junction bubble generator with the width of channel at T-junction, w, measured in the course of experiments. The channel width is 300 μm and channel height is 50 μm. (d) Schematic of the T-junction region at the point of segmented flow generation. (e) Relative change in the channel width at T-junction, plotted as a function of time of MF bubble generation in THF ( ), DCM ( ), DCM ( ), hexanes ( ), hexanes ( ), and BB ( ), and BB ( ). (f) Relative change in the channel width at the T-junction, plotted as a function of time of MF bubble generation in DCM at 25 ( ). (f) Relative change in the channel width at the T-junction, plotted as a function of time of MF bubble generation in DCM at 25 ( ), 60 ( ), 60 ( ), and 80 °C ( ), and 80 °C ( ). ). | ||
In the next step, we examined the ability to vary the surface energy of HDPE by subjecting it to plasma treatment and measuring the contact angle, θ, of deionized water on the sheet as a function of plasma exposure time. Fig. 1b shows that after 50 s exposure of HDPE to oxygen plasma, the value of θ reduced from 92 to 53°; and it did not further change at longer plasma treatment time. Full hydrophobic recovery did not occur for at least, 7 days upon storage of HDPE sheets under ambient conditions. Exposure of HDPE sheets to nitrogen and air plasma yielded similar θ values (Fig. 1b).
The compatibility of HDPE with hydrocarbon and aromatic solvents was examined by fabricating a T-junction MF device (Fig. 1c), generating nitrogen gas plugs dispersed in dichloromethane (DCM), tetrahydrofurane (THF), bromobenzene (BB), or hexanes, and examining the change in the original width, w0, of the T-junction. Fig. 1d shows the schematic of gaseous plug generation. The value of T-junction width, w, was analyzed every 30 min for 4.5 h during exposure to a selected solvent, that is, the duration of a typical MF experiment. The degree of swelling of the microchannel walls was characterized as [(w0 − w)/w0] × 100%. Fig. 1e shows that in the course of the generation of gaseous plugs the degree of swelling of the channel wall was in the range of 1.2–1.5%. The maximum swelling occurred in DCM after 4.5 h. No de-bonding, excessive channel distortion, or compromised reactor structural integrity was observed. For comparison, the swelling ratio of PDMS in organic solvents such as THF and hexanes was 138 and 135%, respectively.21
The performance of the MF device at elevated temperatures was examined in DCM in the temperature range from 25 to 80 °C. Fig. 1f shows the variation in the degree of swelling after 4.5 h device exposure to DCM at 25, 60, and 80 °C. The degree of swelling was 1.2, 1.9 and 2.7%, respectively (Fig. 1e). No de-bonding or change in structural integrity of the HDPE reactor was observed.
Microfluidic reactors with low gas permeability
Gas permeation of polymer substrates depends on the type of polymer and gas, the temperature, and the pressure gradient across the substrate. Gas permeability increases with the flexibility of polymer chains, that is, with the increase in polymer free volume.22 We selected PVC for the fabrication of MF devices with low gas permeability owing to the high-density packing and weak segmental motion of the polymer molecules.23 Since PDMS has a high gas permeability and is extensively used for the fabrication of MF devices, we used this polymer as a reference material. The design of the MF devices fabricated in PDMS and PVC was similar to the design shown in Fig. 1c. A 6.35 mm-thick PVC sheet embossed with features of the MF device was bonded to a 100 μm-thick planar PVC sheet. An identical MF device was fabricated in PDMS. The side walls of the microchannels had the thickness of at least, 6.35 mm to support the assumption that the majority of gas diffused through the 100 μm thick ceiling of the microchannel during the time span of the experiments.Alternating segments of gaseous nitrogen and water were generated in the MF device using pressure-driven flow. Subsequently, the inlets and the outlet of the MF device were both pressurized at 1.73 or 2.44 atm, thereby stopping flow and making the gas plugs stationary. The pressure difference between 1.73 or 2.44 atm (inside the plugs) and 1 atm (outside the device) was the driving force for gas mass transfer across the 100 μm-thick PVC or PDMS channel ceiling.
The length of the experiment was 5 h. To ensure that the shrinkage of gaseous plugs was not caused by nitrogen dissolution in water, the plugs were generated 15 cm upstream of the characterization zone (corresponding to 25 s residence time) in the device prior to the beginning of monitoring the change in plug dimensions. Since physical dissolution of gas bubbles in MF experiments occurs in ≤10 s,2 any subsequent plug shrinkage was attributed to gas mass transfer through the 100 μm-thick ceiling of the MF device.
The relative change in the plug volume was characterized as Vt/V0, where V0 is the plug volume when it reached the characterization zone and remained stationary (ESI†), and Vt is the volume of the stationary plug at time t. For the nitrogen plugs in the MF device fabricated in PDMS, we observed a strong decay in plug dimensions with time (Fig. 2). After 5 h, the plug volumes at 1.73 and 2.44 atm were 45 and 21% of the initial values, respectively (Fig. 2). In contrast, under similar conditions for nitrogen plugs confined to the microchannel in the PVC device at the same applied pressures, the change in plug dimensions was not significant (Fig. 2). Notably, the pressure of 2.44 atm used in these experiments exceeded the pressures used in studies of gas–liquid CO2 reactions.2,3
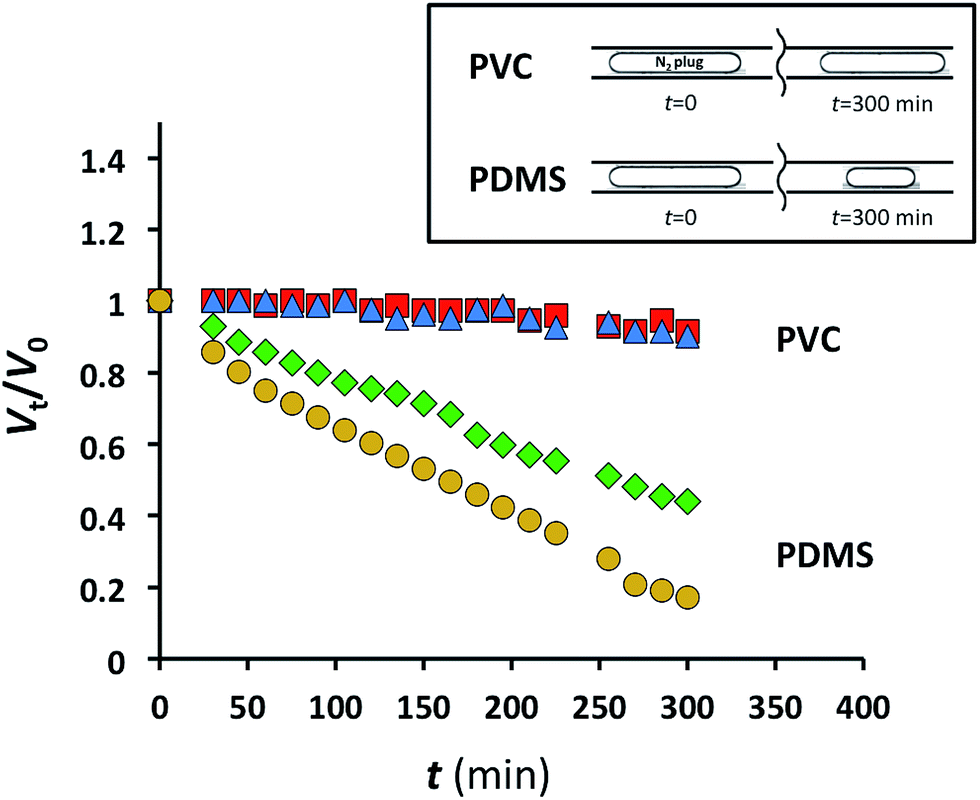 | ||
Fig. 2 Normalized gaseous nitrogen plug volumes plotted as a function of time for PDMS MF devices at 1.73 atm ( ) and 2.44 atm N2 ( ) and 2.44 atm N2 ( ), and for PVC MF devices at 1.73 atm ( ), and for PVC MF devices at 1.73 atm ( ) and 2.44 atm N2 ( ) and 2.44 atm N2 ( ). ). | ||
These experiments highlight the applicability of MF devices fabricated in PVC for long-term MF studies of gas–liquid reactions and physical processes.
Microfluidic devices for biological and biomedical applications
Microfluidic systems offer a micrometer-to-millimeter length scale range for spatially and temporally-controlled environments in biochemistry, cell biology and biomedical research. For cell biology experiments, PS would be a material of choice for the fabrication of MF devices, since biologists have a long history of using PS as the material of e.g., Petri dishes and well plates.24 The transition to other materials is partly counteracted by the stigma of using unfamiliar substrates. Unfortunately, microfabrication in PS is more challenging than in PDMS, which limits the utilization of PS-based MF devices for cell biology research. In addition, PDMS absorbs some of the reagents, e.g., amines, which may lead to uncertainty in chemical characterization.For a showcase, we used the fabrication of PS devices for the generation of high-density two-dimensional (2D) aqueous droplet arrays. The hot embossed PS devices were manufactured using an identical process and with similar throughput as the HDPE and PVC devices, thereby allowing for customized MF devices to be fabricated for bio-related applications.
We used the design of the MF device and the method of self-digitization of a liquid stream into droplets that have been developed by Chiu et al.25 This method has been used in studies of polymorph crystallization,25 DNA amplification,26 and recently, extended to the generation of 2D arrays of cell-laden hydrogel modules.27
The devices were fabricated by thermal bonding of two PS sheets, one of which contained features of a 2D array of wells connected with a common supply microchannel26 and the other one, a planar PS sheet. Prior to bonding, the PS sheets were exposed for 60 min to oxygen plasma at a vacuum of 1000 mTorr. After bonding, the MF device was filled for 10 min at 65 °C with (heptadecafluoro-1,1,2,2-tetrahydrodecyl) trimethoxysilane, which resulted in the increase of the wetting angle of water on the modified PS surface from 76 to 110°. A 2D high-density array of droplets of the mixture of water and glycerol was generated in the PS device, as shown in Fig. 3a. First, the entire device was filled with fluorinated oil mixed with 1 wt% triblock copolymer perfluoropolyether-b-poly(ethylene oxide-co-polypropylene oxide)-b-perfluoropolyether (top row, Fig. 3a). Then, the fluorinated oil was replaced with a mixture of water and glycerol containing 50 vol% glycerol mixed with a green food dye, as shown in the second row in Fig. 3a. Next, the fluorinated oil was introduced in the MF device, again, to replace the aqueous solution in the supplying microchannel but retain it in the wells.
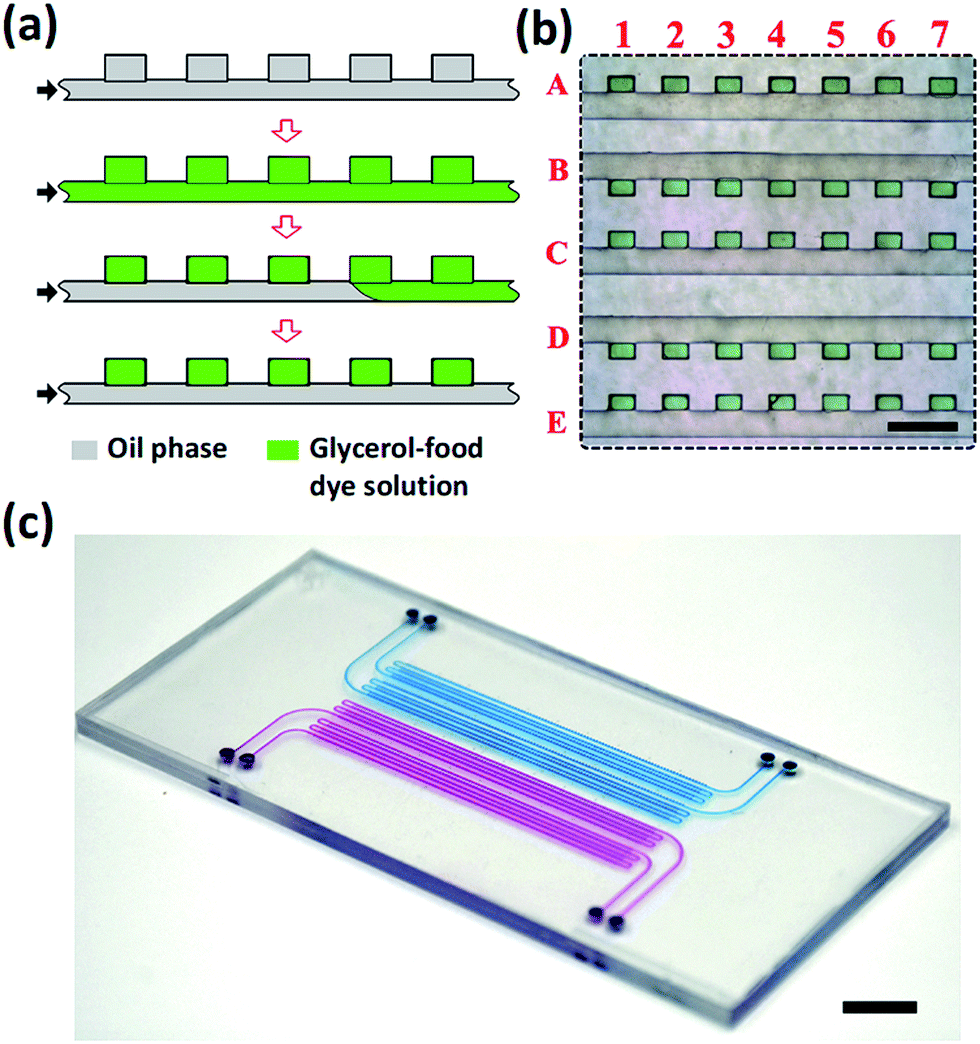 | ||
| Fig. 3 Polystyrene MF well plate device hosting a dense 2D array of droplets. (a) Schematic of aqueous droplet generation. (b) Optical microscopy image of the fragment of indexed 2D droplet array (the droplets are labeled with a green food dye). Scale bar is 500 μm. (c) Photograph of a PS device containing 2000 wells filled with a blue and red food dyes, with each row of 500 wells connected to an individual supplying channel. Scale bar is 1 cm. | ||
As a result, water–glycerol droplets resided in the wells, while the supplying channel was filled with the oil phase (Fig. 3a, bottom row). Following this procedure, we generated 5 parallel rows, each containing 500 dye-labeled aqueous droplets and connected to a common supplying channel.
Fig. 3b shows a fragment of the droplet rows residing in the wells. The green dye-labeled droplets were indexed by the numbers and letters, corresponding to the columns and rows, respectively. The size of each well was 130 × 200 × 100 μm. The volume of droplets was ∼2.6 nL, with a coefficient of variance of droplets not exceeding 15%.
Fig. 3c shows a photograph of the MF device fabricated in PS, filled with an aqueous solution of a blue food dye (two rows of wells) or a red food dye (two rows of wells). The MF device occupied the area 40 × 50 mm2 and contained 4 inlets, each supplying a liquid to 500 wells.
Conclusion
We have developed a cost- and time-efficient method for fabrication of thermoplastic polymer MF devices for targeted chemical and biological applications. For each particular requirement to the material of MF devices, we identified a specific thermoplastic polymer to be used for microfabrication. In particular, MF devices fabricated in high-density polyethylene showed excellent compatibility with aromatic and hydrocarbon solvents and thus can be used for studies of chemical reactions involving these solvents. Polyvinylchloride was used to showcase low gas permeability of MF devices fabricated in this polymer in comparison with the device fabricated in PDMS. The fabrication of MF well plates in polystyrene extends the range of potential applications of MFs in bio-analytical chemistry, cell biology, and bioengineering.Acknowledgements
EK appreciates funding from NSERC Canada (Discovery grant and Canada Research Chair Program). DV and YW thank support by the Microfluidic Applications and Training in Cardiovascular Health (MATCH) and NSERC ENGAGE programs. EK and YW thank the Medicine-by-Design program (University of Toronto) for financial support. Consultations with FlowJEM, Ltd. are highly appreciated.Notes and references
- H. Becker and L. E. Locascio, Talanta, 2002, 56, 267 CrossRef CAS PubMed.
- D. Voicu, M. Abolhasani, R. Choueiri, G. Lestari, C. Seiler, G. Menard, J. Greener, A. Guenther, D. W. Stephan and E. Kumacheva, J. Am. Chem. Soc., 2014, 136, 3875 CrossRef CAS PubMed.
- G. Lestari, M. Abolhasani, D. Bennett, P. Chase, A. Gunther and E. Kumacheva, J. Am. Chem. Soc., 2014, 136, 11972 CrossRef CAS PubMed.
- T. C. Merkel, V. I. Bondar, K. Nagai, B. D. Freeman and I. Pinnau, J. Polym. Sci., Part B: Polym. Phys., 2000, 38, 415 CrossRef CAS; A. Singh, B. D. Freeman and I. Pinnau, J. Polym. Sci., Part B: Polym. Phys., 1998, 36, 289 CrossRef.
- L. W. McKeen, The Effects of Sterilization on Plastics and Elastomers, Elsevier, Oxford, UK, 2012, p. 354 Search PubMed.
- M. Mehling and S. Tay, Curr. Opin. Biotechnol., 2014, 25, 95 CrossRef CAS PubMed.
- Y. Yu, X. Wang, D. Oberthur, A. Meyer, M. Perbandt, L. Duan and Q. J. Kang, J. Appl. Crystallogr., 2012, 45, 53 CrossRef CAS.
- J. Chen, D. Chen, Y. Xie, X. Chen, K. Wang, D. Cui, H. Du and Z. J. Wang, Appl. Phys. Lett., 2015, 106, 247 Search PubMed.
- N. Assmann, S. Kaiser and P. R. von Rohr, J. Supercrit. Fluids, 2012, 67, 149 CrossRef CAS.
- R. Suriano, A. Kuznetsov, S. M. Eaton, R. Kiyan, G. Cerullo, R. Osellame, B. N. Chichkov, M. Levi and S. Turri, Appl. Surf. Sci., 2011, 257, 6243 CrossRef CAS.
- D. J. Guckenberger, T. E. de Groot, A. M. D. Wan, D. J. Beebe and E. W. K. Young, Lab Chip, 2015, 15, 2364 RSC.
- Y. N. Xia and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 550 CrossRef CAS.
- M. Seo, Z. Nie, S. Xu, M. Mok, P. C. Lewis, R. Graham and E. Kumacheva, Langmuir, 2005, 21, 11614 CrossRef CAS PubMed; D. Qin, Y. Xia and G. M. Whitesides, Nat. Protoc., 2010, 5, 491 CrossRef PubMed.
- J. Greener, W. Li, J. Ren, D. Voicu, V. Pakharenko, T. Tang and E. Kumacheva, Lab Chip, 2010, 10, 522 RSC.
- E. J. Mark, Polymer Data Handbook, Oxford University Press, UK, 2nd edn, 2009, p. 500 Search PubMed.
- G. M. Michler, Electron Microscopy of Polymers, Springer Laboratory, USA, 2008, p. 277 Search PubMed.
- J. R. Wunsch, Polystyrene: Synthesis, Production, and Applications, Rapra Publishing, 2000, p. 15 Search PubMed.
- M. L. Dannis, J. Appl. Polym. Sci., 1959, 1, 121 CrossRef CAS; F. C. Stehling and L. Mandelkern, Macromolecules, 1970, 3, 242 CrossRef; H. Flocke, Kolloid Z. Z. Polym., 1962, 180, 118 Search PubMed.
- G. Ceccorulli, M. Pizzoli and G. Pezzin, J. Macromol. Sci., Part B: Phys., 1977, 4, 499 CrossRef.
- A. M. Rijke and L. Mandelkern, J. Polym. Sci., Part A-1: Polym. Chem., 1970, 8, 225 CrossRef CAS; M. Gopalan and L. Mandelkern, J. Phys. Chem., 1967, 71, 3833 CrossRef; R. A. Chivers, P. J. Barham, I. Matinez-Salazar and A. Keller, J. Polym. Sci., Polym. Phys. Ed., 1982, 20, 1717 CrossRef.
- J. Ng Lee, C. Park and G. M. Whitesides, Anal. Chem., 2003, 75, 6544–6554 CrossRef PubMed.
- T. C. Merkel, V. I. Bondar, K. Nagai, B. D. Freeman and I. Pinnau, J. Appl. Polym. Sci., 2000, 38, 415 CrossRef CAS.
- C. Pritchard, Polym. Int., 1995, 36, 102 CrossRef.
- A. Paguirigan and D. J. Beebe, BioEssays, 2008, 30, 811 CrossRef CAS PubMed.
- D. E. Cohen, T. Schneider, M. Wang and D. T. Chiu, Anal. Chem., 2010, 82, 5707 CrossRef CAS PubMed.
- A. Gansen, A. M. Herrick, I. K. Dimov, L. P. Lee and D. T. Chiu, Lab Chip, 2012, 12, 2247 RSC.
- Y. Wang, Y. Li, H. Therien-Aubin, J. Ma, P. W. Zandstra and E. Kumacheva, Biomicrofluidics, 2016, 10, 014110 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra27592c |
| This journal is © The Royal Society of Chemistry 2017 |