
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly uniform distribution of Pt nanoparticles on N-doped hollow carbon spheres with enhanced durability for oxygen reduction reaction
Qiurong Shi a,
Chengzhou Zhua,
Mark H. Engelhardb,
Dan Dua and
Yuehe Lin*a
a,
Chengzhou Zhua,
Mark H. Engelhardb,
Dan Dua and
Yuehe Lin*a
aSchool of Mechanical and Material Engineering, Washington State University, Pullman, WA 99164, USA. E-mail: yuehe.lin@wsu.edu
bEnvironmental Molecular Sciences Laboratory, Pacific Northwest National Laboratory, Richland, WA 99352, USA
First published on 19th January 2017
Abstract
Carbon-supported Pt nanostructures currently exhibit great potential in polymer electrolyte membrane fuel cells. Nitrogen-doped hollow carbon spheres (NHCSs) with extra low density and high specific surface area are a promising carbon support for loading Pt NPs. The doped heteroatom of nitrogen not only contributes to the active activity for the oxygen reduction reaction (ORR), but also shows a strong interaction with Pt NPs for entrapping them to prevent dissolution/migration. This synergetic effect/interaction resulted in the uniform dispersion and strong combination of the Pt NPs on the carbon support and thus plays a significant role in hindering the degradation of the catalytic activities of Pt NPs. As expected, the as-obtained Pt/NHCSs displayed improved catalytic activity and superior durability toward the ORR.
Introduction
Currently, intensive efforts have been devoted to developing polymer electrolyte membrane fuel cell (PEMFC)-based energy conversion devices for their high power density, facile maintenance, environmental benignity and energy sustainable merits and so on. Therefore, PEMFCs are expected to be widely commercialized for use in transportation, stationary machines and portable electronic devices in the future.1–4 However, the cathode of PEMFCs employing Pt-based nanoparticles (NPs) dispersed on carbon black (CB) limits the fast development and mass production of PEMFCs-based devices for several critical issues. One problem is that a large amount of Pt loading is required to overcome the sluggish reaction kinetic of oxygen reduction reaction (ORR) to obtain a desirable mass activity (MA).5–10 Insufficient long-term operation durability is another serious problem that resulted from the serious dissolution, aggregation or Ostwald ripening of Pt-based NPs.9 One way to tackle the above-mentioned issues is to modify the shapes, morphologies, compositions of Pt-based electrocatalysts to reduce the total cost and enhance the overall catalytic performances.11–16 Another effective way is by fabrication of efficient carbon support to take advantage of the interaction and synergetic effect between catalysts and support.17–20Because of the large versatility of carbon structures, carbon nanotubes (CNTs), graphene sheet, aerogel or foam, carbon nanofibers, yolk–shell or hollow carbon spheres (HCSs), etc. have been chosen as substitutes of carbon black to gain an extended lifetime of PEMFCs.21–24 HCSs as one kind of novel carbon materials have long been appealing in various energy storage and conversion devices, biomedicine and environmental treatment due to their unique structures with low density, high surface to volume ratio and high mechanical strength.25–28 HCSs could effectively improve the dispersion of Pt-based electrolytes and stabilize Pt NPs from dissolution and aggregation via steric effect generated from pore confinement or encapsulation.29,30 Besides, introducing heteroatoms like nitrogen into the carbon materials is now very popular in enhancing the electrocatalytic activities, such as nitrogen-doped graphene,31 CNTs,32 polyaniline33 etc. They all exhibited enhanced electrochemical performances not only because of the synergetic effect between electrocatalysts and carbon support, but also due to their contribution to electron donor tendency and generation of carbon active sites for arching Pt seed.34–36
Here, we reported the in situ growth of Pt NPs on N-doped HCSs (NHCSs) with highly uniform distribution and sizes of about 3–5 nm at room temperature, employing formic acid as reducing agent. This reaction happens without any stirring or ultrasonic vibration, indicating a very simple and energy-saving method for mass production of the electrocatalysts. What's important is that the as-obtained Pt NPs are totally surfactant free, which could avoid the tedious work of washing process and is more favorable for direct use as cathode materials for ORR. The as-synthesized Pt NPs decorated NHCSs (Pt/NHCSs) exhibited excellent electrocatalytic performance toward ORR with MA 3.7 times higher than commercial Pt/C at 0.90 V, and half-potential shift of only 9.9 mV after accelerated durability test (ADT), which is much smaller than that of commercial Pt/C. This work demonstrates that Pt/NHCSs are truly promising cathode materials for PEMFCs and the facile synthesis strategy is applicable for a large-scale industrial production.
Experimental
Chemical and materials
Chloroplatinic acid hexahydrate (H2PtCl6·6H2O), dopamine, tetraethyl orthosilicate (TEOS), ammonia hydroxide (NH3·H2O), formic acid, Tris base were all purchased from Sigma-Aldrich Co. Ltd. Ethanol were purchased from Decon Labs, Inc. All magnetic stirring bars and glasswares were cleaned using chloroazotic acid (HCl (37%) : HNO3 (65%) with 3 : 1 v/v) and then rinsed with deionized (DI) water for at least three times. (Caution: chloroazotic acid is very dangerous and shoule be used carefully.) The DI water used in experiments was all acquired from Millipore Milli-Q plus 185 purification system and the resistivity should be higher than 18.2 MΩ cm.Synthesis of hollow carbon spheres
Silica spheres were firstly synthesized as hard templates by Stöber method.37 Then, 80 mg of silica spheres were mixed with 80 mg of dopamine in 25 mL of Tris-buffer (0.01 M, pH = 8.5) for 24 h. The polymer/silica nanocomposite was collected via centrifugation. Then, carbonization was processed under N2 atmosphere firstly at 400 °C for 2 h with a heating rate of 1 °C min−1, followed by a second treatment at 800 °C for 3 h with a heating rate of 5 °C min−1. Hollow carbon spheres were collected after washing in sodium hydroxide at 70 °C for 12 h.Synthesis of Pt/NHCSs
Pt/NHCSs were obtained via mixing with H2PtCl6·6H2O, hollow carbon spheres and formic acid via ultrasonic for 15 min, followed by reaction for 48 h without stirring at room temperature, and washed three times with water.38Preparation and electrochemical measurements of electrode
The homogeneous Pt/NHCSs and commercial Pt/C catalyst ink (2 mg mL−1) were prepared via dispersing the sample powders into an aqueous solution containing 2 mL 2-propanol, 0.05 mL Nafion (5%) and 8 mL deionized water, respectively, followed by ultrasonic agitation.The electrochemical experiments were conducted via a standard three-electrode electrochemical workstation (CHI 630E). A Pt wire was used as the counter electrode and a Hg/HgCl2 electrode filled with saturated potassium chloride aqueous solution were employed as the reference electrode. The working electrode was prepared with a loading of 25.5 μg cm−2 of Pt on a rotating disk electrode (RDE) (0.196 cm2 in geometric area) and then dried at 60 °C under vacuum. 120 mL 0.1 M HClO4 solution was used as the electrolyte for ORR test.
Cyclic voltammetry (CV) and linear sweep voltammetry (LSV) measurements were performed on RDE connected with an electrode rotator (purchased from Princeton Applied Research). CV tests for ORR were conducted after purging nitrogen or oxygen into 0.1 M HClO4 solution for 30 min. LSV measurements were conducted in O2-saturated electrolyte.
Characterization
Transmission electron microscopy (TEM) images were acquired by Field Emission Instruments (FEI) (Philips CM200 UT, USA). Energy-dispersive X-ray (EDX) analysis was obtained using FEI sirion field emission scanning electron microscope (FESEM). The tube was operated at 15 mA current and 40 kV accelerating voltage. X-ray Diffraction (XRD) spectrum was obtained from Rigaku Miniflex 600. X-ray photoelectron spectroscopy (XPS) tests were conducted utilizing a Physical Electronics Quantera Scanning X-ray Microprobe. Excitation and a spherical section analyzer were obtained using a focused monochromatic Al Kα X-ray (1486.7 eV) source.Results and discussion
Polydopamine (PDA)@silica spheres were firstly obtained via polymerization of dopamine both as carbon and nitrogen source utilizing silica nanosphere as hard template. After carbonization, NHCSs with diameter of about 200 nm were obtained after removing silica nanospheres by sodium hydroxide. Decoration of uniform Pt NPs was achieved via adding H2PtCl6 and HCOOH aqueous solution at room temperature and stored for 48 h. Typically, the chemical reaction between H2PtCl6 and HCOOH is as follows:| H2PtCl6 + 2HCOOH → Pt + 6Cl− + 6H+ + CO2 |
Fig. 1A–F showed the as-synthesized Pt/NHCSs with different loading ratios from 66.1% (marked as Pt/NHCSs-1), 79.6% (Pt/NHCSs-2) to 88.6% (Pt/NHCSs-3), respectively. The Pt particle size with Pt loading of 66.1% is about 2.8 ± 0.5 nm, while those of 79.6% and 88.1% are about 3.4 nm ± 0.4 nm and 4.8 nm ± 0.6 nm, respectively. From TEM images of these three loading ratios, Pt NPs of Pt/NHCSs-1 appears to be a little bit sparse, while Pt NPs of Pt/NHCSs-3 seems to be too intensive, which may result in a larger particle size caused by the aggregation of NPs. While NHCSs with 79.6% loading shows a moderate size and a more homogeneous distribution on HCS.
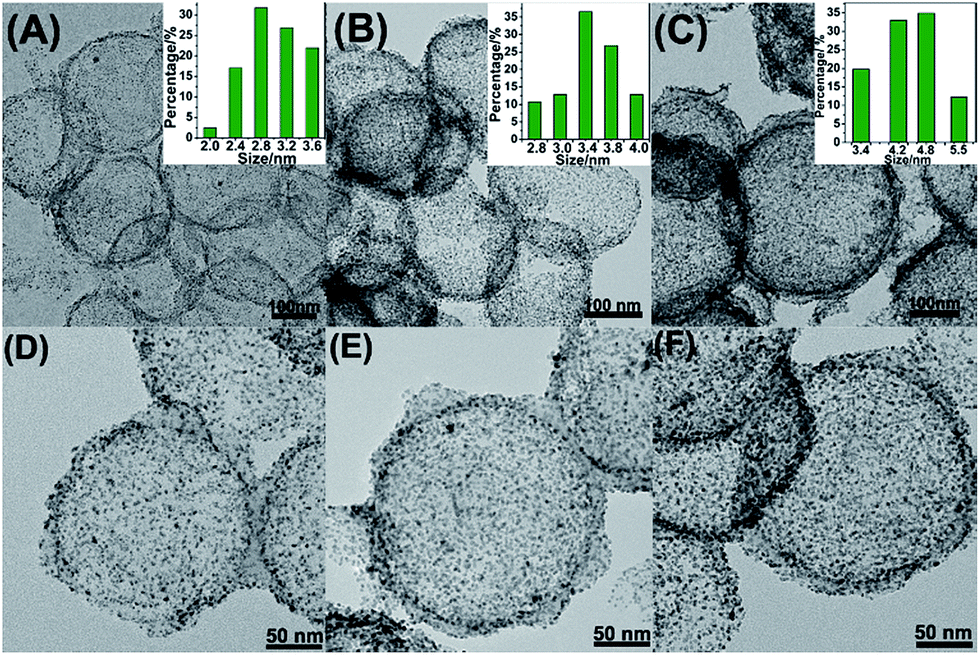 | ||
| Fig. 1 TEM images of as-synthesized Pt/NHCSs with Pt loading amount of 66.1% (A and D), 79.6% (B and E) and 88.6% (C and F), respectively. The inset pictures in (A–C) are their related size distribution. | ||
The composition of as-fabricated Pt/NHCSs-2 was further confirmed via X-ray diffraction (XRD) shown in Fig. 2A. The diffraction peaks were typical face-centered cubic Pt lattice, shown as (111), (200), (211), (311) and (222). The EDX result shown in Fig. 2B also verified the main composition of Pt/NHCSs was Pt. The Pt loading amount of Pt/NHCSs-2 were confirmed via thermogravimetric analysis (TGA), shown in Fig. 2C. A total weight loss of about 22.3% were obviously observed after the temperature reached about 800 °C, which was ascribed to the oxidation of NHCSs at 800 °C. The weight loss is almost in agreement with the desired Pt loading of about 79.6% in Pt/NHCSs-2, indicating a complete yield of Pt via formic acid reduction method.
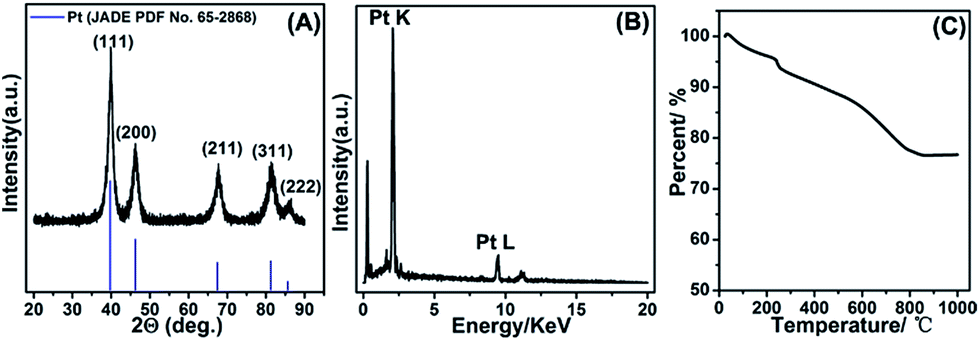 | ||
| Fig. 2 XRD pattern (A), EDX (B) and TGA curve (C) of as-fabricated Pt/NHCSs-2. | ||
We further confirmed the composition of as-synthesized Pt/NHCSs-2 via employing XPS. The peak appeared at 400.2 eV and 398.1 eV in Fig. 3A are corresponding to N 1s, which could be further deconvoluted into four peaks including pyridinic N (398.4 eV), pyrrolic N (400.3 eV), graphitic N (401.2 eV) and pyridine-N oxide (403.9 eV), respectively. The content of these four types of nitrogen is about 44.7%, 31.1%, 11.6% and 12.6%, respectively. The peak of N-HCs in Fig. 3C could be further deconvoluted into four peaks including pyridinic N (398.1 eV), pyrrolic N (399.8 eV), graphitic N (400.8 eV) and pyridine-N oxide (402.9 eV), respectively. The origin content of these four types of nitrogen is about 24.7%, 20.0%, 37.8% and 17.5%, respectively. The N content in N-doped hollow carbon sphere is about 3.44% and that in Pt/NHCSs with Pt loading amount of 79.6% is about 3.84%. It could be seen that the content of pyridinic N and pyrrolic N have been increased and also the binding energy shifted due to the interaction with metal NPs. A first principles study reported that pyrrolic and pyridinic N offered the defect site for the in situ nucleation and growth of Pt NPs due to the induced partial charges on neighboring carbon atoms.32,39 An improved performances toward ORR of mesoporous N-doped carbons containing mainly quaternary N-groups was highlighted as a result of the chemical interaction between Pt NPs and N-defects.40 Besides, pyridinic and graphitic N acted as active sites for contributing to the ORR performances.35,41–43 Therefore, it is believed that they could display a catalytic behaviour toward ORR. The N-doped carbon sphere is beneficial in modifying the electronic structure of Pt, leading to the reduced binding energy and adsorption of intermediate on Pt surface.31,43,45 Similarly, the peaks in Fig. 3B appeared at 74.6 eV and 71.3 eV are corresponding to the Pt 4f5/2 and Pt 4f7/2, respectively. They also can be deconvoluted into four peaks at 74.6 eV, 71.0 eV and 76.6 eV, 72.4 eV, which are assigned to be metallic state (Pt0) and oxide state (Pt2+) of Pt, respectively. It is obvious Pt is mainly consists of Pt0. Besides, the Pt 4f peaks showed a positive shift compared to commercial Pt/C (70.3 eV and 74.0 eV, respectively).44 The binding energy shift of the N 1s and Pt 4f both indicated a strong interaction between the doped nitrogen atoms and Pt NPs, which would be critical and beneficial in maintaining the long-term catalytic abilities for its unique capability of “entrapping” NPs from aggregation, migration or dissolution.
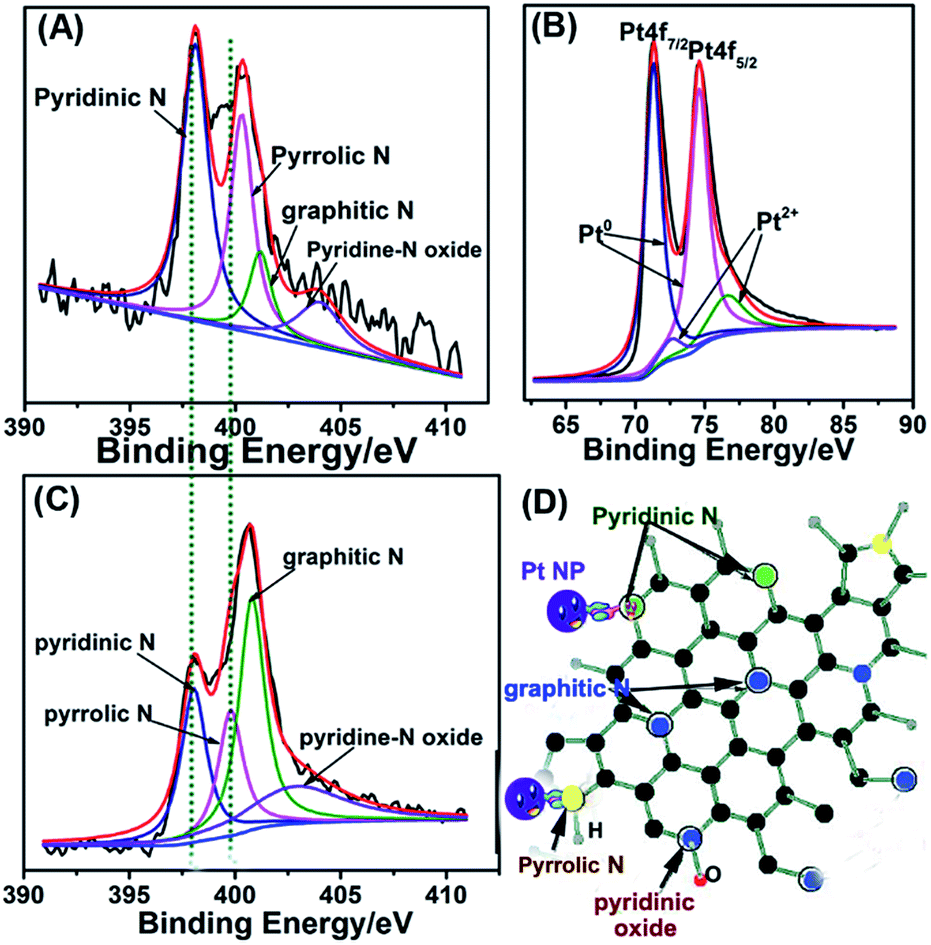 | ||
| Fig. 3 X-ray photoelectron spectroscopy (XPS) of the N 1s (A), Pt 4f (B) of as-synthesized Pt/NHCSs-2 and N 1s (C) of the as-obtained NHCSs, respectively. (D) Schematic illustration of the four types of nitrogen and their strong interaction with Pt NPs. | ||
According to the XPS result, the interaction/synergetic effect between Pt NPs and nitrogen-doped carbon support may play an important role in boosting the electrocatalytic activities and long-term operation stabilities.46 The electrochemical active surface areas (ECSAs) of the as-fabricated Pt/NHCSs-1, Pt/NHCSs-2, Pt/NHCSs-3 and commercial Pt/C were calculated from the hydrogen adsorption/desorption peak (0.08–0.26 V) in CV curves in N2-saturated 0.1 M HClO4 aqueous solution, shown in Fig. 4A. The ECSA results of as-obtained Pt/NHCSs were listed in Table 1, which were comparable to commercial Pt/C. From the inset in Fig. 4A, we can see that Pt/NHCSs-2 shows a more positive Pt oxide reduction peak (0.833 V) than commercial Pt/C (0.810 V), indicating a weaker bond connection between Pt surfaces and oxygen containing species, which is favorable for ORR catalytic performances.47
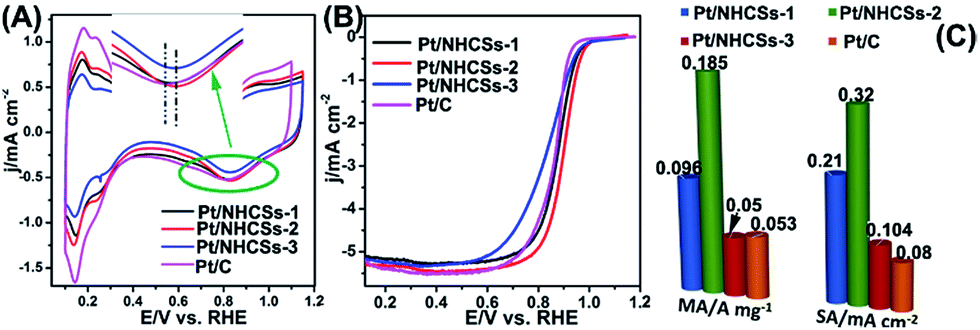 | ||
| Fig. 4 (A) CV curves of as-obtained Pt/NHCSs-1, Pt/NHCSs-2, Pt/NHCSs-3 and commercial Pt/C in N2-saturated 0.1 M HClO4 aqueous solution. (B) LSV curves of the four electrocatalysts at a rotation rate of 1600 rpm and a scan rate of 10 mV s−1. (C) MA and SA of the four electrocatalysts at 0.9 V. | ||
| ECSA (m2 g−1) | Onset potential/V | E1/2 (V) | MA (A mg−1) | SA (mA cm−2) | |
|---|---|---|---|---|---|
| Pt/NHCSs-1 | 45.9 | 0.951 | 0.875 | 0.096 | 0.209 |
| Pt/NHCSs-2 | 57.56 | 0.963 | 0.892 | 0.185 | 0.320 |
| Pt/NHCSs-3 | 48.11 | 0.953 | 0.870 | 0.050 | 0.104 |
| Pt/C | 66.70 | 0.921 | 0.876 | 0.053 | 0.080 |
Fig. 4B exhibited the LSV curves for ORR utilizing the four kinds of electrocatalysts on a rotating disk electrode (RDE) in O2-saturated 0.1 M HClO4 solution at a rotating rate of 1600 rpm and a scan rate of 10 mV s−1. The onset potential of the four electrocatalysts was listed in Table 1. It is found that Pt/NHCSs-2 exhibited a more positive onset potential than the other three samples, implying a more active behavior toward ORR. Half-potential (E1/2) is another key indicator for evaluating the activity of the electrocatalysts. From Table 1, Pt/NHCSs-2 displayed a 17.02 mV, 22.0 mV and 16.0 mV positive shift compared with Pt/NHCSs-1, Pt/NHCSs-3 and commercial Pt/C, respectively, which also confirmed it a more active electrocatalysts.
MA and SA are usually employed to precisely evaluate the activity of the Pt-based electrocatalysts toward ORR. MA is obtained from kinetic current density (jk) via the Levich–Koutecky equation shown as follows:
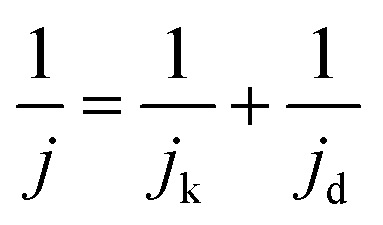
The ADT test was conducted in O2-saturated 0.1 M HClO4 solution at a scan rate of 100 mV s−1 with the potential between 0.6 V and 1.1 V for 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles. Fig. 5A–D displayed the CV curves and LSV curves of the Pt/NHCSs-2 and commercial Pt/C before and after ADT test, respectively. The ECSA degradation of Pt/NHCSs-2 and commercial Pt/C were about 4.8% and 24.7%, respectively. The E1/2 of Pt/NHCSs-2 showed a relatively smaller negative shift of about 9.9 mV compared to that of commercial Pt/C (30.0 mV), indicating an excellent long-term operation durability. Fig. 6A and B exhibited the TEM images of commercial Pt/C and Pt/NHCSs-2 after 10000 cycle test, respectively. It can be observed that Pt NPs of commercial Pt/C suffered from serious aggregation after ADT, resulting in a poor stability toward ORR, while the morphologies of Pt/NHCSs-2 could be well maintained with the size only changed about 0.6 nm after 10000 cycle test. It is believed that the strong interaction between the nitrogen sites and Pt NPs plays a significant role in confining Pt NPs from dissolution and aggregation for avoiding the loss of activity.40,42,47
000 cycles. Fig. 5A–D displayed the CV curves and LSV curves of the Pt/NHCSs-2 and commercial Pt/C before and after ADT test, respectively. The ECSA degradation of Pt/NHCSs-2 and commercial Pt/C were about 4.8% and 24.7%, respectively. The E1/2 of Pt/NHCSs-2 showed a relatively smaller negative shift of about 9.9 mV compared to that of commercial Pt/C (30.0 mV), indicating an excellent long-term operation durability. Fig. 6A and B exhibited the TEM images of commercial Pt/C and Pt/NHCSs-2 after 10000 cycle test, respectively. It can be observed that Pt NPs of commercial Pt/C suffered from serious aggregation after ADT, resulting in a poor stability toward ORR, while the morphologies of Pt/NHCSs-2 could be well maintained with the size only changed about 0.6 nm after 10000 cycle test. It is believed that the strong interaction between the nitrogen sites and Pt NPs plays a significant role in confining Pt NPs from dissolution and aggregation for avoiding the loss of activity.40,42,47
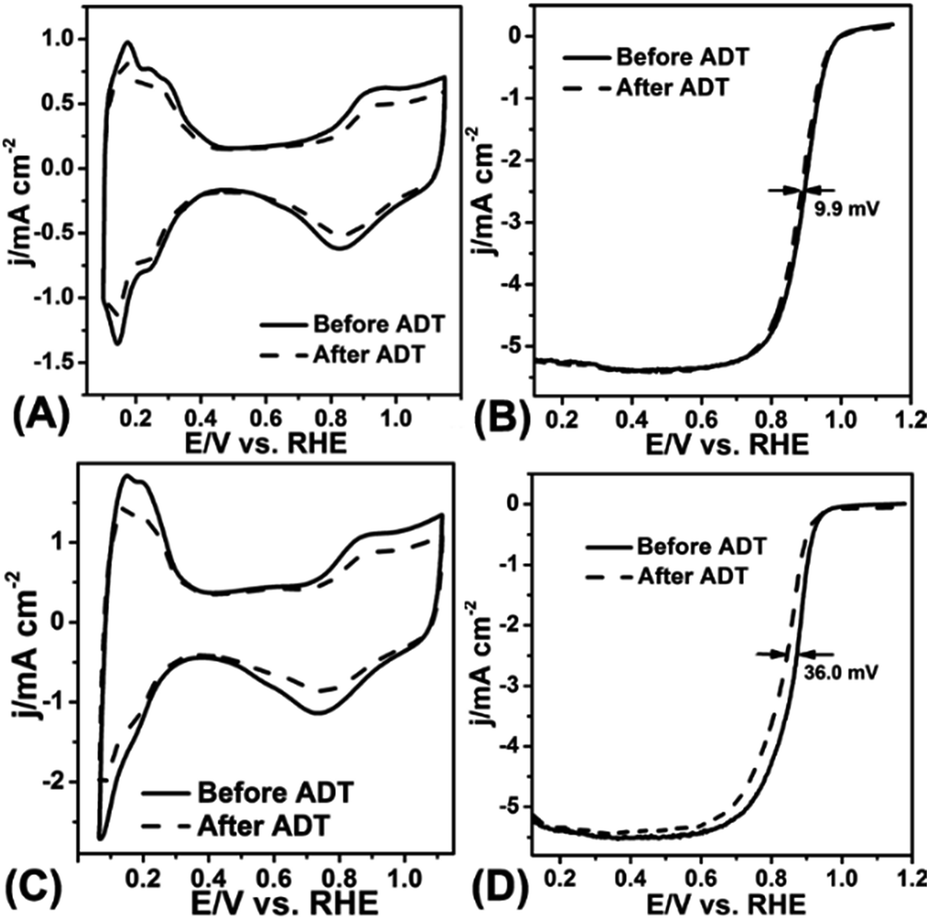 | ||
| Fig. 5 CV curves of Pt/NHCSs-2 (A) and commercial Pt/C (C) on RDE before and after 10000 cycles test in N2-saturated 0.1 M HClO4 solution with a scan rate of 50 mV s−1. LSV curves of Pt/NHCSs-2 (B) and commercial Pt/C (D) on RDE before and after ADT test at a rotation rate of 1600 rpm at a scan rate of 10 mV s−1. | ||
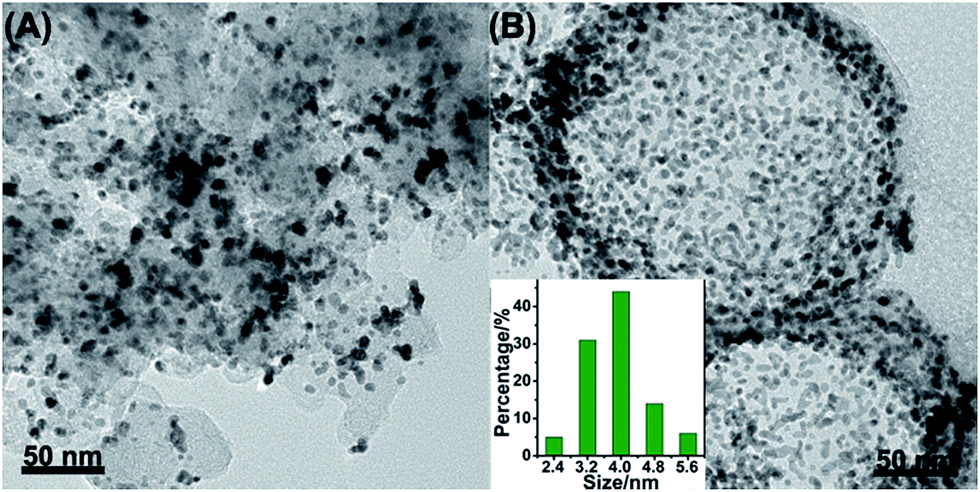 | ||
| Fig. 6 TEM images of commercial Pt/C (A) and Pt/NHCSs-2 (B) after ADT. The inset in (B) is the size distribution of Pt NPs after ADT. | ||
Conclusions
In summary, we have synthesized NHCSs with Pt NPs loading via a facile and green method applied for ORR. The HCSs as carbon support have the advantage of low density and high specific density. What's more important is the heteroatom doping hold the key for improving the overall catalytic performance toward ORR: (I) nitrogen doping could help the in situ deposition of Pt ions for growth without decorating any other functional groups; (II) the synergetic effect between nitrogen sites and Pt NPs, and pyridinic and graphitic N acting as active sites are critical in enhancing the catalytic activities; (III) the strong interaction between nitrogen sites and Pt catalysts is of great importance in obtaining the long-term operation durability for ORR via preventing the Pt NPs from dissolution or aggregation. Overall, the optimized Pt/NHCSs is promising for mass production in application in PEMFCs.Acknowledgements
This work was sponsored by Washington State University start-up fund. We thank Franceschi Microscopy & Image Center at Washington State University for TEM and SEM measurement. We also thank EMSL, a national scientific user facility for XPS measurement, which is sponsored by the Department of Energy's Office of Biological and Environmental Research located at Pacific Northwest National Laboratory. Q. S. thanks the China Scholarship Council for the financial support.Notes and references
- N. Tian, Z. Y. Zhou, S. G. Sun, Y. Ding and Z. L. Wang, Science, 2007, 316, 732–735 CrossRef CAS PubMed.
- B. Lim, M. J. Jiang, P. H. C. Camargo, E. C. Cho, J. Tao, X. M. Lu, Y. M. Zhu and Y. N. Xia, Science, 2009, 324, 1302–1305 CrossRef CAS PubMed.
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4269 CrossRef CAS PubMed.
- M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS PubMed.
- H. A. Gasteiger and N. M. Markovic, Science, 2009, 324, 48–49 CrossRef CAS PubMed.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS.
- C. Z. Zhu, H. Li, S. F. Fu, D. Du and Y. H. Lin, Chem. Soc. Rev., 2016, 45, 517–531 RSC.
- C. Z. Zhu, D. Du, A. Eychmuller and Y. H. Lin, Chem. Rev., 2015, 115, 8896–8943 CrossRef CAS PubMed.
- Y. Y. Shao, J. Liu, Y. Wang and Y. H. Lin, J. Mater. Chem., 2009, 19, 46–59 RSC.
- S. Zhang, Y. Y. Shao, G. P. Yin and Y. H. Lin, J. Mater. Chem. A, 2013, 1, 4631–4641 CAS.
- S. J. Guo, S. Zhang and S. H. Sun, Angew. Chem., Int. Ed., 2013, 52, 8526–8544 CrossRef CAS PubMed.
- B. Y. Xia, H. B. Wu, Y. Yan, X. W. Lou and X. Wang, J. Am. Chem. Soc., 2013, 135, 9480–9485 CrossRef CAS PubMed.
- X. H. Sun, K. Z. Jiang, N. Zhang, S. J. Guo and X. Q. Huang, ACS Nano, 2015, 9, 7634–7640 CrossRef CAS PubMed.
- L. Z. Bu, J. B. Ding, S. J. Guo, X. Zhang, D. Su, X. Zhu, J. L. Yao, J. Guo, G. Lu and X. Q. Huang, Adv. Mater., 2015, 27, 7204–7212 CrossRef CAS PubMed.
- C. Chen, Y. J. Kang, Z. Y. Huo, Z. W. Zhu, W. Y. Huang, H. L. L. Xin, J. D. Snyder, D. G. Li, J. A. Herron, M. Mavrikakis, M. F. Chi, K. L. More, Y. D. Li, N. M. Markovic, G. A. Somorjai, P. D. Yang and V. R. Stamenkovic, Science, 2014, 343, 1339–1343 CrossRef CAS PubMed.
- X. Q. Huang, Z. P. Zhao, L. Cao, Y. Chen, E. B. Zhu, Z. Y. Lin, M. F. Li, A. M. Yan, A. Zettl, Y. M. Wang, X. F. Duan, T. Mueller and Y. Huang, Science, 2015, 348, 1230–1234 CrossRef CAS PubMed.
- S. J. Guo and S. H. Sun, J. Am. Chem. Soc., 2012, 134, 2492–2495 CrossRef CAS PubMed.
- W. T. Yu, M. D. Porosoff and J. G. G. Chen, Chem. Rev., 2012, 112, 5780–5817 CrossRef CAS PubMed.
- X. Q. Huang, Z. P. Zhao, Y. Chen, E. B. Zhu, M. F. Li, X. F. Duan and Y. Huang, Energy Environ. Sci., 2014, 7, 2957–2962 CAS.
- S. H. Sun, F. Jaouen and J. P. Dodelet, Adv. Mater., 2008, 20, 3900–3904 CrossRef CAS.
- R. Kou, Y. Y. Shao, D. H. Wang, M. H. Engelhard, J. H. Kwak, J. Wang, V. V. Viswanathan, C. M. Wang, Y. H. Lin, Y. Wang, I. A. Aksay and J. Liu, Electrochem. Commun., 2009, 11, 954–957 CrossRef CAS.
- H. J. Qiu, X. C. Dong, B. Sana, T. Peng, D. Paramelle, P. Chen and S. Lim, ACS Appl. Mater. Interfaces, 2013, 5, 782–787 CAS.
- J. L. Zhu, G. Q. He, Z. Q. Tian, L. Z. Liang and P. K. Shen, Electrochim. Acta, 2016, 194, 276–282 CrossRef CAS.
- S. Takenaka, H. Miyamoto, Y. Utsunomiya, H. Matsune and M. Kishida, J. Phys. Chem. C, 2014, 118, 774–783 CAS.
- F. Bottger-Hiller, P. Kempe, G. Cox, A. Panchenko, N. Janssen, A. Petzold, T. Thurn-Albrecht, L. Borchardt, M. Rose, S. Kaskel, C. Georgi, H. Lang and S. Spange, Angew. Chem., Int. Ed., 2013, 52, 6088–6091 CrossRef PubMed.
- Y. H. Ng, S. Ikeda, T. Harada, S. Higashida, T. Sakata, H. Mori and M. Matsumura, Adv. Mater., 2007, 19, 597–601 CrossRef CAS.
- Z. C. Yang, Y. Zhang, J. H. Kong, S. Y. Wong, X. Li and J. Wang, Chem. Mater., 2013, 25, 704–710 CrossRef CAS.
- J. A. Zhang, K. X. Wang, S. J. Guo, S. P. Wang, Z. Q. Liang, Z. M. Chen, J. W. Fu and Q. Xu, ACS Appl. Mater. Interfaces, 2014, 6, 2192–2198 CAS.
- C. Galeano, J. C. Meier, V. Peinecke, H. Bongard, I. Katsounaros, A. A. Topalov, A. H. Lu, K. J. J. Mayrhofer and F. Schuth, J. Am. Chem. Soc., 2012, 134, 20457–20465 CrossRef CAS PubMed.
- C. Yang, M. Zhou and Q. Xu, Nanoscale, 2014, 6, 11863–11870 RSC.
- J. B. Zhu, M. L. Xiao, X. Zhao, K. Li, C. P. Liu and W. Xing, Chem. Commun., 2014, 50, 12201–12203 RSC.
- D. C. Higgins, D. Meza and Z. W. Chen, J. Phys. Chem. C, 2010, 114, 21982–21988 CAS.
- J. Melke, B. Peter, A. Habereder, J. Ziegler, C. Fasel, A. Nefedov, H. Sezen, C. WÖll, H. Ehrenberg and C. Roth, ACS Appl. Mater. Interfaces, 2016, 8, 82–90 CAS.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC.
- K. P. Gong, F. Du, Z. H. Xia, M. Durstock and L. M. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- T. Ikeda, M. Boero, S. F. Huang, K. Terakura, M. Oshima and J. Ozaki, J. Phys. Chem. C, 2008, 112, 14706–14709 CAS.
- A. F. Wener Stöber, J. Colloid Interface Sci., 1968, 62–69 CrossRef.
- A. Postma, Y. Yan, Y. Wang, A. N. Zelikin, E. Tjipto and F. Caruso, Chem. Mater., 2009, 21, 3042–3044 CrossRef CAS.
- T. Holme, Y. Zhou, R. Pasquarelli and R. O'Hayre, Phys. Chem. Chem. Phys., 2010, 12, 9461–9468 RSC.
- L. Perini, C. Durante, M. Favaro, V. Perazzolo, S. Agnoli, O. Schneider, G. Granozzi and A. Gennaro, ACS Appl. Mater. Interfaces, 2015, 7, 1170–1179 CAS.
- L. T. Qu, Y. Liu, J. B. Baek and L. M. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS PubMed.
- S. B. Yang, X. L. Feng, X. C. Wang and K. Mullen, Angew. Chem., Int. Ed., 2011, 50, 5339–5343 CrossRef CAS PubMed.
- Y. J. Zhang, K. Fugane, T. Mori, L. Niu and J. H. Ye, J. Mater. Chem., 2012, 22, 6575–6580 RSC.
- X. Zhao, J. B. Zhu, L. Liang, J. H. Liao, C. P. Liu and W. Xing, J. Mater. Chem., 2012, 22, 19718–19725 RSC.
- N. Dimakis, N. E. Navarro, T. Mion and E. S. Smotkin, J. Phys. Chem. C, 2014, 118, 11711–11722 CAS.
- S. Zhang, Y. Y. Shao, G. P. Yin and Y. H. Lin, J. Mater. Chem., 2009, 19, 9289 CAS.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. F. Wang, P. N. Ross, C. A. Lucas and N. M. Markovic, Science, 2007, 315, 493–497 CrossRef CAS PubMed.
- M. G. Del Popolo, E. P. M. Leiva, M. Mariscal and W. Schmickler, Surf. Sci., 2005, 597, 133–155 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |