
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRoom temperature synthesis of reduced TiO2 and its application as a support for catalytic hydrogenation†
Miao Zhang‡
ab,
Qijun Pei‡ab,
Weidong Chenab,
Lin Liu*a,
Teng He *a and
Ping Chenacd
*a and
Ping Chenacd
aDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: liulin@dicp.ac.cn; heteng@dicp.ac.cn
bUniversity of the Chinese Academy of Sciences, Beijing 100049, China
cState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
dCollaborative Innovation Center of Chemistry for Energy Materials, Dalian 116023, China
First published on 16th January 2017
Abstract
Reduced TiO2 (TiO2−x) materials have attracted increasing attention due to their large solar absorption and high photo-activity. However, their synthesis procedures usually involve harsh conditions, such as high temperature and/or high pressure. Herein, a facile solid ball-milling method for the synthesis of TiO2−x under ambient conditions was developed. By using finely dispersed Na/NaCl powders as the reducing agent and TiO2 (P25, Degussa) as the precursor, a series of TiO2−x of 20–30 nm with a controllable reduction degree can be successfully synthesized through adjusting the reaction conditions. The surface area of TiO2−x is much larger than that of pristine TiO2, showing its great potential as a catalyst support in chemical reactions. Our experimental results show that uniform Ru particles with particle size less than 1 nm can be well dispersed on the surface of the TiO2−x due to the enhanced surface area and plenty of oxygen vacancies in TiO2−x. As a result, Ru/TiO2−x exhibited superior activity upon catalytic hydrogenation of N-methylpyrrole in comparison with Ru/TiO2.
Introduction
Since Honda and Fujishima discovered hydrogen generation from water by using titanium dioxide (TiO2) as a photoelectrode,1 TiO2 has drawn much attention for its applications in pigments,2 sunscreens3 and photocatalysis.4,5 Apart from the applications in photo-catalysis, TiO2 is also a unique material widely employed as a catalyst support for selective catalytic hydrogenation,6–8 oxidation9,10 and electrochemical11,12 reactions.In 2011, Mao et al. found that the reduction of TiO2 nanoparticles through hydrogenation was an effective strategy to improve visible light absorption and photo-activity of TiO2.13 Since then, many studies have been devoted to the synthesis of reduced TiO2 (denoted as TiO2−x) that has abundant oxygen vacancies and Ti3+ species. Annealing TiO2 precursors in a reducing gas atmosphere (H2 or H2 plasma) under high temperature and/or high pressure is a common synthetic route for the preparation of TiO2−x.13,14 Other promising chemical methods including Al vapor,11 CaH2 (ref. 15) and NaBH4 (ref. 16) reduction etc., can also produce TiO2−x, where high reaction temperatures were required. Furthermore, electrons, Ar+ or other high energy particle bombardments have also been employed to produce TiO2−x materials.17 Despite those significant advances in the synthesis of TiO2−x, it is still highly desirable to develop facile and effective synthetic strategies for the scalable synthesis of TiO2−x under mild conditions.
As a TiO2 derived material with unique electronic properties, TiO2−x may show promises as a new kind of catalyst support, however, only limited investigations have been published so far. A model study of Au on a reduced titanium oxide ordered film has demonstrated that the strength of the interaction between over-layer Au and the support comprised of strong bonding between Au and Ti, yielding an electron-rich Au and exhibiting an exceptional high activity for CO oxidation.18 Very recently, the hydrogen treated TiO2 nanotube arrays with more oxygen vacancies and hydroxyl groups was synthesized which served as highly ordered nanostructured electrode supports and were able to significantly improve the electrochemical performance and durability of fuel cells.11 The unique properties of the TiO2−x supported catalysts may be derived from (1) the encapsulation of metal particles in TiO2−x, presumably because of the so-called strong metal support interaction (SMSI)19 between metal and TiO2−x, (2) the strong bonding between the metal atoms at the interface with surface defects (reduced Ti site) or (3) the electrons transfer between metal particles and TiO2−x.20 It is, therefore, very interesting to investigate the performance of TiO2−x supported catalysts in related chemical reactions.
In this paper, a solid ball-milling reduction process for the synthesis of nanosized TiO2−x from crystalline TiO2 (P25, Degussa) at room temperature was developed. The reductant is Na that has been well dispersed in NaCl powders. A series of TiO2−x samples with color changing from white to dark blue manifesting the increase in the reduction degree can be facilely prepared. The TiO2−x possesses much higher surface area and visible light absorption than those of the pristine TiO2. Highly dispersed Ru particles supported on TiO2−x were prepared and tested as catalyst for the hydrogenation of N-methylpyrrole, which obviously outperformed that of Ru particles supported on pristine TiO2, evidencing stronger promoting effect of TiO2−x on Ru.
Experimental section
Chemicals and materials
TiO2 were purchased from Degussa. Na, NaBH4, NaCl, and NaOH were purchased and used directly without further purification. Tetrahydrofuran (THF) was purchased from Merck, and dried by molecular sieve before usage.Preparation of Na/NaCl fine powders
Due to the soft and ductile nature of Na metal, NaCl powders were used to disperse Na metal by ball milling.21 In a typical experiment, Na metal and NaCl powders with a weight ratio of 1/10 were mechanically milled under argon atmosphere using a Retsch PM400 planetary ball milling. All the manipulations were conducted inside a glove box filled with purified argon. The ball milling was carried out at a milling rate of 150 rpm for 4 h at room temperature, and then black Na/NaCl fine powders can be obtained.Synthesis of TiO2−x
The Na/NaCl fine powder, composed of small Na particles dispersed by NaCl, is expected to be an effective reducing agent for the reduction of crystalline TiO2 to TiO2−x (Scheme 1). In a typical experiment, crystalline TiO2 was milled with Na/NaCl fine powders with a weight ratio of 1/n (n = 1–4) under argon atmosphere using a Retsch PM400 planetary ball milling, which was carried out at a series of milling rates, such as 80, 120, 150 and 180 rpm at room temperature. The samples milled for 0.25 to 4 hours were collected and washed with deionized water for several times to remove the Na and NaCl. Finally, the obtained TiO2−x products were dispersed in a small amount of deionized water and then vacuum-dried at room temperature to get TiO2−x powders. The synthesized TiO2−x samples are marked as TiO-n-v-t, where n, t and v stand for weight ratio between Na/NaCl and TiO2, ball milling rate and reaction time, respectively. For example, TiO-4-80-1 means that the obtained TiO2−x was synthesized at a milling rate of 80 rpm for 1 h, and the weight ratio of Na/NaCl fine powders to P25 is 4.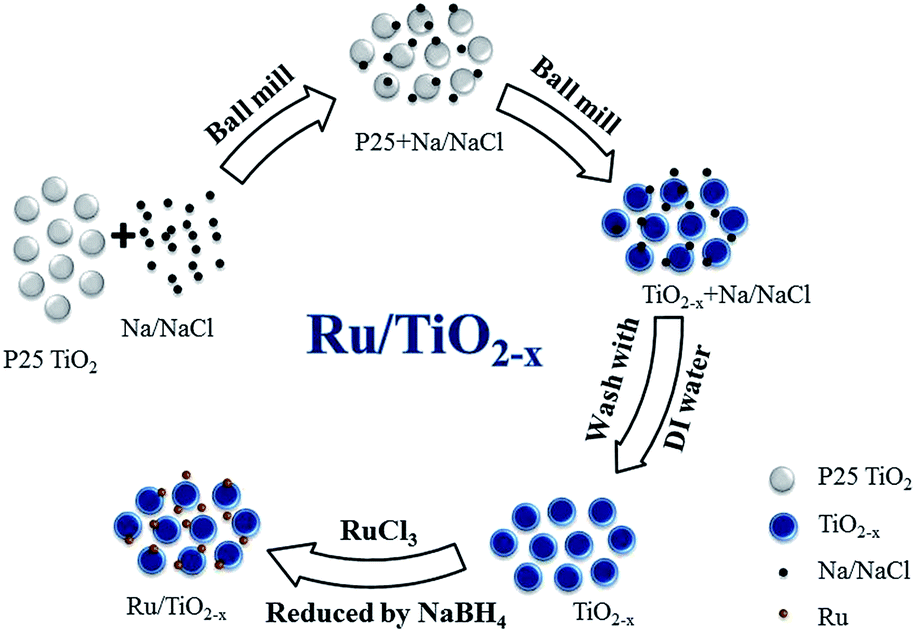 | ||
| Scheme 1 The route for preparation of Ru/TiO2−x. | ||
Preparations of catalysts and characterizations
5% Ru/TiO2 and 5% Ru/TiO2−x catalysts were both prepared by a deposition method using NaBH4 as the reducing agent (Scheme 1). The support TiO2 or TiO2−x was added to a certain concentration of RuCl3 aqueous solutions and stirred for 6 h. Then a NaBH4 solution was added slowly to reduce the Ru3+ cations with intensive stirring. Finally the powders were filtered, washed with deionized water and dried under vacuum overnight.Powder X-ray diffraction (XRD) patterns were recorded on an X'Pert Pro (PANalytical) diffractometer with Cu Kα radiation at 40 kV and 40 mA. Raman spectra were recorded with a Renishaw Raman spectrometer equipped with a He/Ne laser with a wavelength of 514 nm. Transmission electron microscopy (TEM) images were obtained on a JEOL 2000EX electronic microscope operating at 120 kV. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images were collected on a JEM-2100F instrument equipped with STEM dark-field (DF) detector operating at 200 kV. The specific surface area was measured on Autosorb-1 system (Quantachrome, USA) by N2 adsorption isotherm through BET method. The X-ray photoelectron spectroscopy (XPS) measurements were performed using an Escalab 250 Xi X-ray photoelectron spectrometer (Thermo Scientific) with non monochromatic Al Kα radiation (photon energy, 1486.6 eV). Due to the overlapped signal of C 1s and Ru 3d, all the samples were mixed with a certain amount of silicon that is used for calibration (Si 2p at 98.4 eV).22 The UV-Vis absorption spectra were measured on a Shimadzu UV 2600 UV/Vis spectrophotometer. The Ru loadings of the catalysts were determined by inductively coupled plasma spectrometry (ICP-OES, optima 7300DV, Perkin-Elmer, USA).
Hydrogenation of N-methylpyrrole
5% Ru/TiO2 and 5% Ru/TiO2−x catalysts were employed for catalytic hydrogenation of N-methylpyrrole (C5H7N), which is a commercial chemical with large annual global production. The reactions were carried out in the autoclave reactor (PARR®5500 series compact reactor). 100 mg catalyst (Ru/TiO2 or Ru/TiO2−x), 425 μL N-methylpyrrole and 30 mL THF (as the solvent) were put into the autoclave filled with Ar. The temperature programmer began to heat the reactor with stirring speed of 500 rpm. When the temperature of the reactor was stable at the value we set, 30 atm hydrogen was filled into the reactor. At this time, the reaction started and the pressure of the reactor was recorded to monitor the hydrogenation progress. The final products were analysed by the gas chromatography (Agilent 7890-B).Results and discussion
A highly efficient and economic viable reducing agent is needed to synthesize scalable TiO2−x under mild condition. Previous study reported that TiO2 could be reduced by Al vapor at high temperatures,23 showing the strong reducing potential of metals. To perform the reduction of TiO2 at room temperature, a more powerful reductant and a better contact between TiO2 and reductant are needed. Recently, Na metal dispersed in inert medium (for instance NaCl) has received attention for its use as a very strong reducing agent in the synthesis of NaB3H8.21 The well dispersed Na metal in Na/NaCl may function as a promising reductant for the reduction of TiO2. Thus, we chose a commercially available crystalline TiO2 (P25, Degussa), consisting of mixed phases of anatase and rutile, as precursor for the synthesis of TiO2−x. Ball milling of P25 with Na/NaCl fine powders at room temperature and different mechano-chemical conditions led to reduction of P25 into TiO2−x of different oxygen vacancies content as evidenced by the visible color changes after reduction reaction (Fig. 1). The degree of reduction can be facilely controlled by varying the reaction conditions, i.e., weight ratio of P25 nanocrystals to Na/NaCl fine powders, milling rate and reaction time. The synthesized TiO2−x samples are marked as TiO-n-v-t, where n, t and v stand for weight ratio between Na/NaCl and TiO2, ball milling rate and reaction time, respectively. The color of post-reduced P25 samples ranges from white to light blue and finally to dark blue with the increase of ball milling speed, reaction time and weight ratio of Na/NaCl and P25 (Scheme 1). The corresponding UV-Vis diffuse reflectance spectra clearly show the intensity of the absorption in the visible light region (400–800 nm) gradually increases with the increases of reduction degree, which is consistent with the color changes of the TiO2−x samples (Fig. S1†). | ||
| Fig. 1 Photographs of P25 nanocrystals and TiO2−x. (a) P25 nanocrystals; (b) TiO-1-80-0.5; (c) TiO-1-80-1; (d) TiO-1-120-4; (e) TiO-1-150-4; (f) TiO-1-180-4; (g) TiO-2-180-4; (h) TiO-3-180-4 and (i) TiO-4-180-4. | ||
TEM images (Fig. 2) were collected to show the morphology and particle size of the P25 and TiO-4-180-4. The particle size of TiO-4-180-4 sample is similar to that of P25 nanocrystals (20–30 nm), which indicates that such a solid reduction treatment has little influence on the particle size of the reduced TiO2 samples. Different from that of the pristine P25, disordered layer can be observed on the surface of TiO-4-180-4 particles, which is probably resulted from ball-milling treatment and/or surface reaction between TiO2 and Na. As a consequence, a remarkable increase of BET surface area was obtained, i.e., the TiO-4-180-4 (113 m2 g−1) has a BET surface area that is ca. 2.5 times of the pristine P25 (45 m2 g−1).
 | ||
| Fig. 2 TEM images of (a) P25 nanocrystals and (b) TiO-4-180-4. | ||
The crystal structure of TiO2−x is characterized by XRD and compared with that of P25 nanocrystals. As shown in Fig. S2,† a mild reduction treatment with ball milling speed of 80 rpm has little influence on the crystallinity of TiO2−x. However, obvious changes occurred under harsh solid ball milling conditions (longer ball milling time with high Na/TiO2 ratio or at a high ball milling speed, Fig. 3). Compared with that of the pristine TiO2, there is no obvious change in the intensities of diffraction peaks corresponding to the rutile phase in the TiO2−x samples treated with different ball milling speeds. However, the intensities of peaks corresponding to the anatase phase significantly weaken with the increase of ball milling speed. The maintenance of rutile phase in the synthesized TiO2−x may be due to its chemical stability compared with that of anatase.
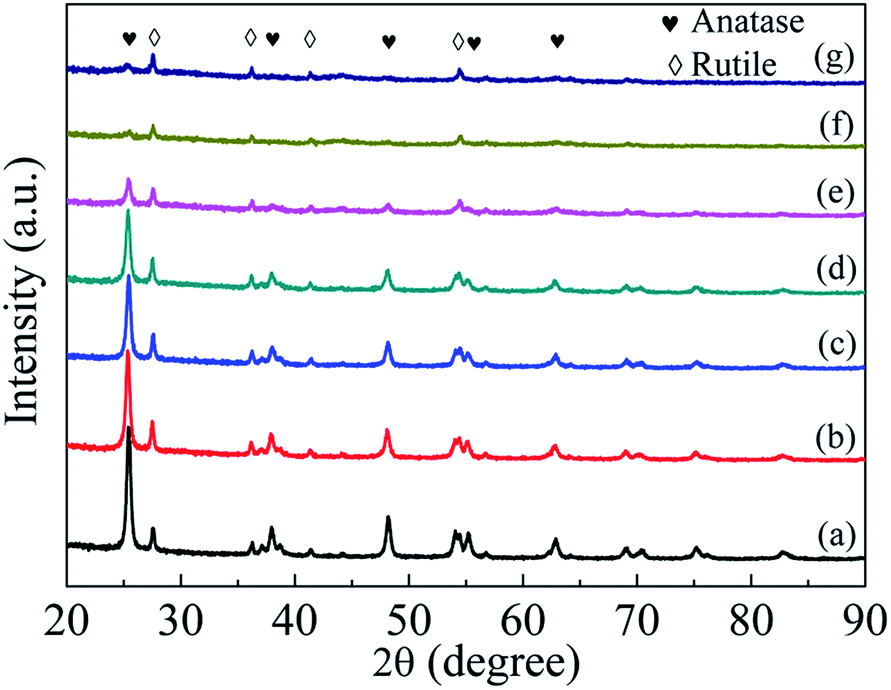 | ||
| Fig. 3 XRD patterns of P25 nanocrystals and TiO2−x (a) P25 nanocrystals; (b) TiO-1-120-4; (c) TiO-1-150-4; (d) TiO-1-180-4; (e) TiO-2-180-4; (f) TiO-3-180-4 and (g) TiO-4-180-4. | ||
As shown in Fig. 4, P25 nanocrystals display the typical anatase Raman active modes with frequencies at 144, 197, 399, 515, 519 (superimposed with the 515 cm−1 band), and 639 cm−1 together with modes at 447 and 612 (should peak) cm−1 corresponding to the rutile phase.24 The relatively low intensity of rutile mode may originate from the low Raman response and low content of rutile in the P25.25 For TiO2−x samples, intensities of peaks corresponding to the rutile phase did not show any obvious change. However, intensities of the peaks corresponding to the anatase phase decreased obviously (Fig. 4e). More importantly, a blue shift of the strongest Eg mode at 144 cm−1 for anatase can be detected. As reported in previous research, this peak shift was mainly caused by the stoichiometry defects in TiO2.26 Therefore, the shift in the present study suggests the formation of oxygen vacancies in TiO2−x during solid reduction treatment. Since the anatase was detected by Raman technique, the disappearance of anatase phase in XRD should be attributed to its amorphous state. Noting that the reduction of TiO2 could mainly occur on its surface or near surface, the synthesized TiO2−x here may have a core–shell structure with reduced shell and unchanged core, which is similar to the results reported in literature.13
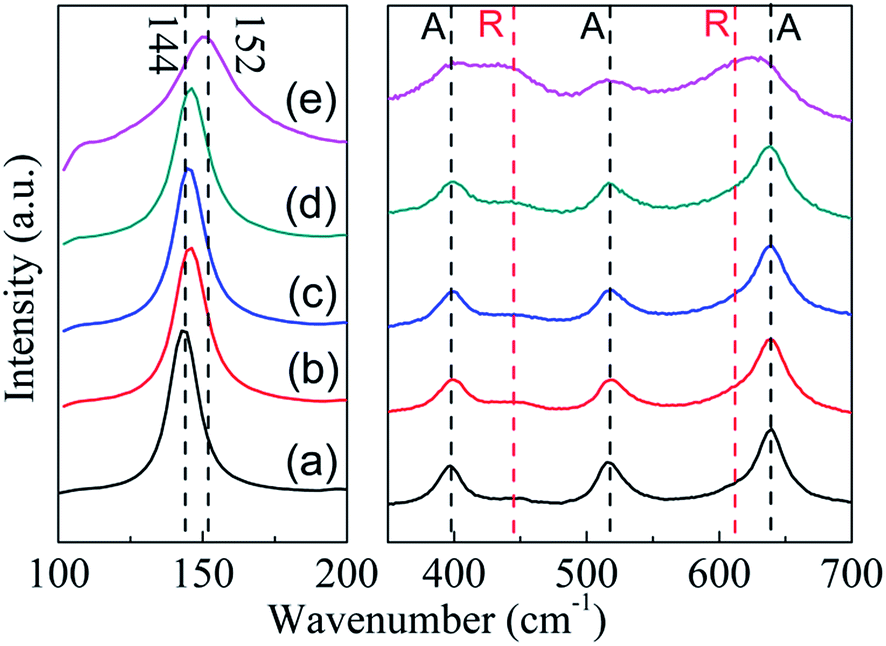 | ||
| Fig. 4 Raman spectra of P25 nanocrystals and TiO2−x (a) P25 nanocrystals; (b) TiO-1-120-4; (c) TiO-1-150-4; (d) TiO-1-180-4; (e) TiO-4-180-4. A: anatase, R: rutile. | ||
XPS was also applied to characterize the chemical state of surface elements in TiO-4-180-4. The full XPS survey (Fig. S3†) reveals that no elements other than C, O, Ti can be detected on the surface of P25 and TiO-4-180-4. The C 1s at approximate 284.5 eV may arise from adventitious carbon on the surface of samples.27 For P25, the Ti 2p3/2 and Ti 2p1/2 peaks center at binding energies of 458.6 and 464.4 eV, respectively, which are characteristics of the Ti4+–O bonds in TiO2. However, these two peaks shift to lower binding energies of 458.2 and 464.0 eV for TiO-4-180-4 (Fig. 5a), which can be an indication of the existence of Ti3+ in the TiO-4-180-4. The binding energy of O 1s of TiO-4-180-4 shows a slight decrease compared with that of P25 (Fig. 5b). It has been reported that the shift of binding energies to lower region can be observed in the hydrogen reduced TiO2 nanotube,11 which is similar to our result. Consistent with Raman result, the XPS data also evidence that the formation of oxygen vacancies in TiO-4-180-4.
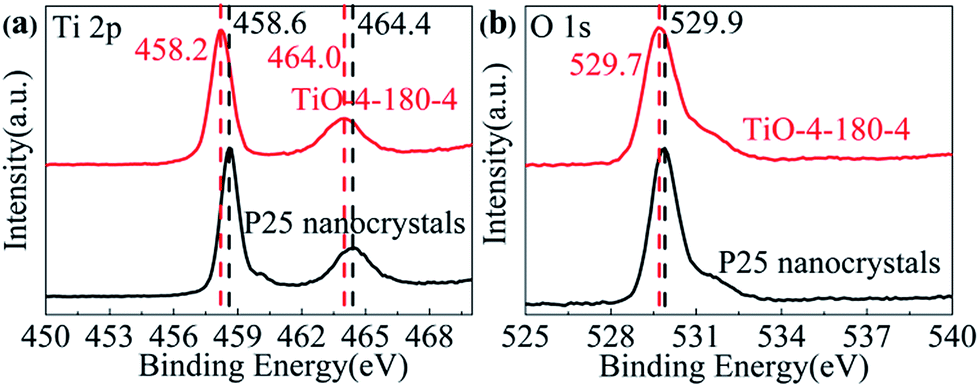 | ||
| Fig. 5 (a) Ti 2p and (b) O 1s XPS of P25 nanocrystals and TiO-4-180-4. | ||
TiO2 has been widely used as a support for various metal catalysts for selective catalytic hydrogenation,6–8 oxidation9,10 and electrochemical11,12 reactions. There is a SMSI between TiO2 and metal particles that induces unique catalytic performance. However, the SMSI was usually observed at high temperatures under reducing atmosphere, where TiO2 was highly likely to be reduced.28 Moreover, the specific surface area of TiO2−x (TiO-4-180-4) is about 2.5 times as large as pristine P25. Therefore, it is very interesting to investigate the prepared TiO2−x as catalyst support. There are growing research activities in employing hydrocarbons and N-heterocycles as liquid organic hydrogen carriers (LOHCs) in recent years because of their compatibleness with existing gasoline infrastructure facilitating the switch to hydrogen energy system.29–31 Catalyst development for the hydrogenation and dehydrogenation of those LOHCs are of practical importance. In the context, the TiO2−x supported Ru catalyst is prepared and tested for the hydrogenation of N-methylpyrrole, a commercially available LOHC with large annual global production.
As shown in Fig. 6a, uniformly distributed Ru on the pristine TiO2 has a mean particle size of about 1.1 nm (Fig. S4†). The Ru supported on TiO2−x, on the other hand, is hardly distinguishable by TEM (Fig. S5†), indicating even smaller particle size of Ru on TiO2−x. Fortunately, the HAADF-STEM image evidences the presence of Ru particles (particle size < 1 nm, Fig. 6b), but the specific particle size is difficult to be calculated because of the ambiguous boundary of those particles. Considering the larger surface area and “rough” surface morphology of TiO2−x, Ru may have better dispersion on TiO2−x. Furthermore, oxygen vacancies or Ti3+ in TiO2−x may also be helpful for anchoring metal particles and, thus, leading to better dispersion than that on pristine P25.18
 | ||
| Fig. 6 (a) TEM image of 5 wt% Ru/TiO2. (b) HAADF-STEM image of 5 wt% Ru/TiO2−x. | ||
Recent reports showed that the electron-rich transition metals could facilitate the hydrogenation of N-heterocycles.32 The reduced state of TiO2−x may also be favorable in electron transfer between support and Ru. As we expected, the XPS of Ru 3d5/2 signal shows that the binding energy of Ru supported on TiO2−x down shifts 0.5 eV as compared with that on TiO2 (Fig. S6†), indicating an electron rich state of Ru on TiO2−x, which also suggests a stronger interaction between support and metal particles.33 Although Ru is known to catalyze the hydrogenation of N-methylpyrrole well,34 the effect of catalyst support has not been well investigated. As shown in Fig. 7a, about 80% and 88% N-methylpyrrole can be hydrogenated to N-methylpyrrolidine in 60 min at 90 °C and 100 °C by using Ru/TiO2 catalyst, respectively. Ru/TiO2−x, on the other hand, shows superior catalytic performance compared to that of Ru/TiO2, i.e., about 91% and 95% N-methylpyrrole can be hydrogenated under the same condition. Furthermore, the initial hydrogenation rate on Ru/TiO2−x at 100 °C is about twofold as that with Ru/TiO2 (Table S1†), showing encouraging promotion effect of TiO2−x as support. Calculated from the Arrhenius plots (Fig. 7b), the activation energies for the hydrogenation reaction are 50.9 kJ mol−1 and 50.0 kJ mol−1 for Ru/TiO2 and Ru/TiO2−x, respectively, suggesting similar hydrogenation mechanism for both catalysts. Therefore, the improved catalytic activity of Ru/TiO2−x can be probably attributed to the better dispersion of Ru on the support, which is confirmed by the larger pre-exponential factor (A) as shown in Table S1.† We suggest that in addition to the larger specific surface area of TiO2−x (113 m2 g−1), oxygen vacancies or Ti3+ in TiO2−x may intensify the interaction between Ru particles and TiO2−x, and therefore, enhancing the better dispersion of Ru particles by inhibiting Ru migration and agglomeration.11 However, the strong interaction between TiO2−x and metal particles is an interesting subject that needs to be further investigated and elucidated over reactions that are sensitive to the electronic state of transition metals.
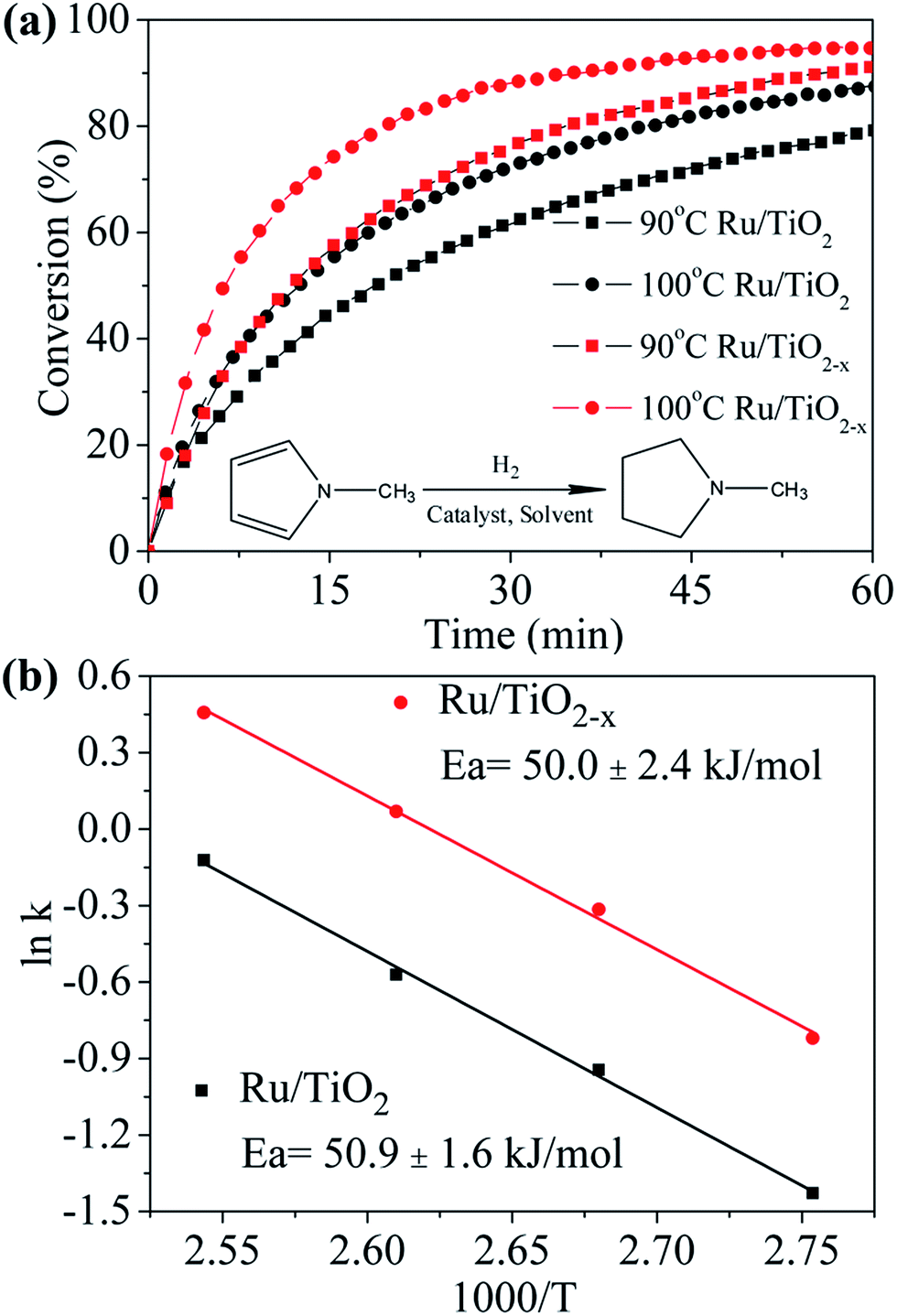 | ||
Fig. 7 (a) Hydrogenation of N-methylpyrrole with catalysts Ru/TiO2 or Ru/TiO2−x at 30 atm H2 pressure, 90 °C and 100 °C, catalyst/substrate molar ratio is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100. (b) The Arrhenius plots in the temperature range of 363–393 K. :100. (b) The Arrhenius plots in the temperature range of 363–393 K. | ||
Conclusions
In summary, we have developed a room temperature solid reduction approach for the synthesis of nanosized TiO2−x from TiO2 crystals. A series of TiO2−x with controllable reduction degree have been successfully synthesized by ball-milling of TiO2 crystal with finely dispersed Na/NaCl powders. The obtained TiO2−x with high surface area can be employed as an effective support for Ru particles and the Ru/TiO2−x catalyst exhibited superior activity in catalytic hydrogenation of N-methylpyrrole, a commercial available heterocycle with large annual global production. We believe that this highly efficient room temperature reduction approach for the production of TiO2−x offers a promising opportunity for the practical applications of TiO2−x in different areas.Acknowledgements
The authors would like to acknowledge financial support from the projects of National Natural Science Foundation of China (Grant No. U1232120, 21473181, 51671178 and 51472237), support from the Youth Innovation Promotion Association (CAS) and the CAS-Helmholtz Association Collaborative Funding.Notes and references
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- J. H. Braun, A. Baidins and R. E. Marganski, Prog. Org. Coat., 1992, 20, 105–138 CrossRef CAS.
- R. Zallen and M. Moret, Solid State Commun., 2006, 137, 154–157 CrossRef CAS.
- A. L. Linsebigler, G. Lu and J. T. Yates Jr, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- Z. Zhang and J. T. Yates Jr, Chem. Rev., 2012, 112, 5520–5551 CrossRef CAS PubMed.
- M. Vannice and R. Garten, J. Catal., 1980, 66, 242–247 CrossRef CAS.
- V. Kratky, M. Kralik, M. Mecarova, M. Stolcova, L. Zalibera and M. Hronec, Appl. Catal., A, 2002, 235, 225–231 CrossRef CAS.
- X. Han, R. Zhou, G. Lai and X. Zheng, Catal. Today, 2004, 93, 433–437 CrossRef.
- S. Bonanni, K. Ait-Mansour, H. Brune and W. Harbich, ACS Catal., 2011, 1, 385–389 CrossRef CAS.
- A. Yoshida, Y. Mori, T. Ikeda, K. Azemoto and S. Naito, Catal. Today, 2013, 203, 153–157 CrossRef CAS.
- C. Zhang, H. Yu, Y. Li, Y. Gao, Y. Zhao, W. Song, Z. Shao and B. Yi, ChemSusChem, 2013, 6, 659–666 CrossRef CAS PubMed.
- L. Zhao, Z. B. Wang, J. Liu, J. J. Zhang, X. L. Sui, L. M. Zhang and D. M. Gu, J. Power Sources, 2015, 279, 210–217 CrossRef CAS.
- X. B. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- Z. Wang, C. Yang, T. Lin, H. Yin, P. Chen, D. Wan, F. Xu, F. Huang, J. Lin, X. Xie and M. Jiang, Adv. Funct. Mater., 2013, 23, 5444–5450 CrossRef CAS.
- S. Tominaka, Inorg. Chem., 2012, 51, 10136–10140 CrossRef CAS PubMed.
- H. Tan, Z. Zhao, M. Niu, C. Mao, D. Cao, D. Cheng, P. Feng and Z. Sun, Nanoscale, 2014, 6, 10216–10223 RSC.
- X. Y. Pan, M. Q. Yang, X. Z. Fu, N. Zhang and Y. J. Xu, Nanoscale, 2013, 5, 3601–3614 RSC.
- M. Chen and D. Goodman, Science, 2004, 306, 252–255 CrossRef CAS PubMed.
- S. Tauster, S. Fung and R. Garten, J. Am. Chem. Soc., 1978, 100, 170–175 CrossRef CAS.
- D. Goodman, Catal. Lett., 2005, 99, 1–4 CrossRef CAS.
- W. Chen, G. Wu, T. He, Z. Li, Z. Guo, H. Liu, Z. Huang and P. Chen, Int. J. Hydrogen Energy, 2016, 41(34), 15371–15476 CrossRef.
- F. Sirotti, M. De Santis and G. Rossi, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 8299 CrossRef CAS.
- Z. Wang, C. Yang, T. Lin, H. Yin, P. Chen, D. Wan, F. Xu, F. Huang, J. Lin, X. Xie and M. Jiang, Energy Environ. Sci., 2013, 6, 3007–3014 CAS.
- J. Zhang, M. Li, Z. Feng, J. Chen and C. Li, J. Phys. Chem. B, 2006, 110, 927–935 CrossRef CAS PubMed.
- V. Likodimos, A. Chrysi, M. Calamiotou, C. Fernández-Rodríguez, J. Doña-Rodríguez, D. Dionysiou and P. Falaras, Appl. Catal., B, 2016, 192, 242–252 CrossRef CAS.
- A. Li Bassi, D. Cattaneo, V. Russo, C. E. Bottani, E. Barborini, T. Mazza, P. Piseri, P. Milani, F. O. Ernst, K. Wegner and S. E. Pratsinis, J. Appl. Phys., 2005, 98, 074305 CrossRef.
- C. Elmasides, D. I. Kondarides, W. Grunert and X. E. Verykios, J. Phys. Chem. B, 1999, 103, 5227–5239 CrossRef CAS.
- G. L. Haller and D. E. Resasco, Adv. Catal., 1989, 36, 173–235 CAS.
- A. C. Cooper, D. E. Fowler, A. R. Scott, A. H. Abdourazak, H. S. Cheng, L. D. Bagzis, F. C. Wilhelm, B. A. Toseland, K. M. Campbell and G. P. Pez, Abstr. Pap. Am. Chem. S., 2005, 229, U868 Search PubMed.
- R. H. Crabtree, Energy Environ. Sci., 2008, 1, 134–138 CAS.
- T. He, Q. Pei and P. Chen, J. Energy Chem., 2015, 24, 587–594 CrossRef.
- T. He, L. Liu, G. Wu and P. Chen, J. Mater. Chem. A, 2015, 3, 16235–16241 CAS.
- A. Lewera, L. Timperman, A. Roguska and N. Alonso-Vante, J. Mater. Chem. C, 2011, 115, 20153–20159 CAS.
- L. Hegedus, T. Mathe and A. Tungler, Appl. Catal., A, 1997, 161, 283 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: More figures of XRD, XPS, TEM, particle size distribution and a table of data. See DOI: 10.1039/c6ra26667c |
| ‡ These authors contributed equally to this study. |
| This journal is © The Royal Society of Chemistry 2017 |