
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel total synthesis of aculeatin A via a stepwise approach†
Zhibin Zhanga,
Xiao Lengb,
Shenkun Yanga,
Cheng Lianga,
Shuangping Huang*a and
Xiaoji Wang *c
*c
aSchool of Pharmacy, Jiangxi Science and Technology Normal University, Fenglin Road 605, Nanchang 330013, China. E-mail: hsping02@@gmail.com
bSchool of Chemistry & Chemical Engineering, Jiangxi Science and Technology Normal University, Fenglin Road 605, Nanchang 330013, China
cSchool of Life Science, Jiangxi Science and Technology Normal University, Fenglin Road 605, Nanchang 330013, China. E-mail: 2012207455@tju.edu.cn
First published on 2nd February 2017
Abstract
Aculeatin A, isolated from the plant Amomum aculeatum, is a dispirocyclic compound having antimalarial activity. In this paper, we describe a novel synthetic approach to aculeatin A from 1-tetradecanal in nine steps via a stepwise strategy. The key features of this approach include the 1,3-diketone preparation from Claisen condensation of ketone and acyl chloride, cyclodehydration, and intramolecular oxa-Michael addition to 2,3-dihydro-4H-pyran-4-one.
Introduction
Traditional medicines, which usually contain rich bioactive natural products with fascinating architectures, play a significant role in the cure of diseases such as malaria, and are therefore important sources of lead compounds. Malaria is the most important parasitic disease, causing serious effects to infected humans and killing more than 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 every year.1 Artemisinin and chloroquine are commonly used clinically to reduce morbidity and mortality. With the widespread use of these agents, however, resistance to them has emerged in many regions.2 Therefore, identifying new and effective antimalarial agents, especially with novel mechanisms of action and neoteric chemical structures, is an important problem to be addressed by scientists. In Papua New Guinea, the plant Amomum aculeatum has long been used in folk medicine for the treatment of fever and malaria. In 2000 and 2001, Heilmann and coworkers reported four novel dispirocyclic natural products from petroleum extracts of its rhizomes, namely, aculeatins A–D (Fig. 1).3
000 every year.1 Artemisinin and chloroquine are commonly used clinically to reduce morbidity and mortality. With the widespread use of these agents, however, resistance to them has emerged in many regions.2 Therefore, identifying new and effective antimalarial agents, especially with novel mechanisms of action and neoteric chemical structures, is an important problem to be addressed by scientists. In Papua New Guinea, the plant Amomum aculeatum has long been used in folk medicine for the treatment of fever and malaria. In 2000 and 2001, Heilmann and coworkers reported four novel dispirocyclic natural products from petroleum extracts of its rhizomes, namely, aculeatins A–D (Fig. 1).3
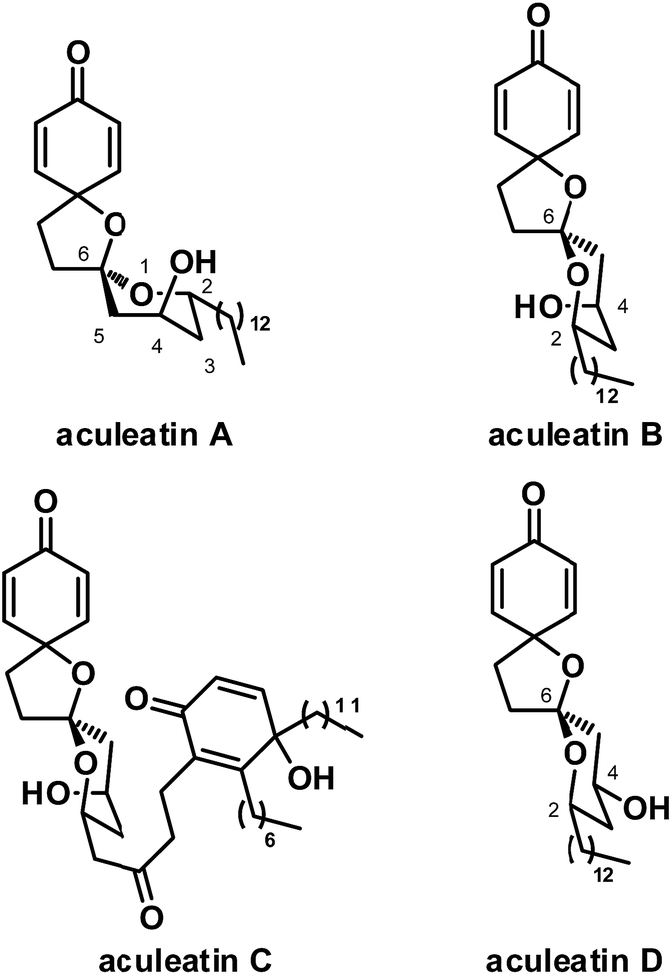 | ||
| Fig. 1 Structures of aculeatins A–D. | ||
Aculeatins A–D have attracted tremendous interest from scientists since their isolation. This new novel class of natural products exhibit interesting antimalarial activities at submicromolar level, as shown by in vitro experiments. For example, they have an IC50 of 0.18–3.0 μM against Plasmodium falciparum, Trypanosoma brucei rhodesiense and Trypanosoma cruzi. Aculeatins, however, contain a novel 1,7-dioxdispiro[5.1.5.2]pentadecane skeleton, which poses a significant challenge in synthesis. Their potency as antimalarial agents and their complex structure have thus made them popular synthetic targets in recent years.4
Over the past 15 years, more than 20 total syntheses of aculeatins have been developed using various strategies such as asymmetric allylation,4c–f,q kinetic resolution,4l and chiral pool approaches.4g–j,p,m In 2015, we described a versatile approach to the syntheses of aculeatins A/B/D/6-epi-D via an asymmetric Mukaiyama aldol reaction,4t ending with the protocol developed by Prof. Wong which characterized a [bis(trifluoroacetoxy)iodo]benzene (PIFA)-mediated oxidative spirocyclization (Scheme 1). However, what we obtained are the mixture of aculeatins A and B or D and 6-epi-D because of the absence of significant diaselectivity of the spirocyclization. Recently, Tong's group developed a novel double intramolecular oxa-Michael addition (DIOMA) of secondary/tertiary alcohols and subsequent selective reduction to synthesize aculeatin A, as illustrated in Scheme 1.4s However, details of the sequence of DIOMA are not given in their paper, although he hypothesized in his newest review paper that 6-endo-dig double oxa-Michael addition might occur and precede a second 5-exo-trig cyclization.5 This synthesis implies that we can obtain the single spiroisomer of aculeatin if we construct the spiro structure stepwise. Herein, we report a novel and straightforward approach to the total synthesis of aculeatin A via a stepwise strategy.
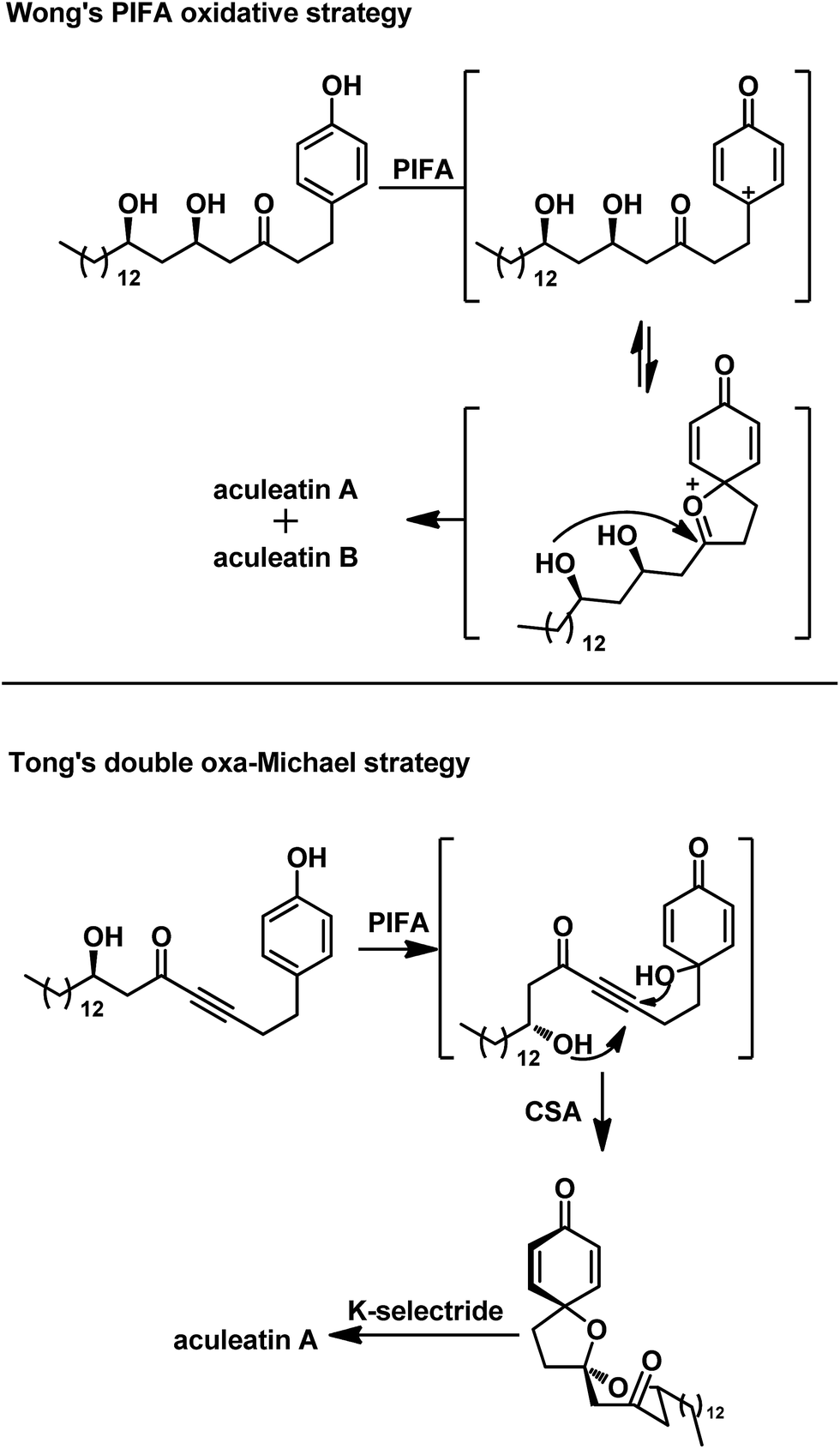 | ||
| Scheme 1 Previous syntheses of aculeatin A by Wong and Tong. | ||
Our retrosynthetic analysis is depicted in Scheme 2. Inspired by Tong's work, we envisioned that aculeatin A could be prepared by chemo- and diaselective reduction of the precursor 1. For intermediate 1, we assumed that we could carry out PIFA oxidation of the phenol and acid catalyzed intramolecular oxa-Michael addition to provide it. Therefore, we disconnected 1 from 2. We envisioned that the hydroxyl in 2 would attack from the less hindered direction of the 2,3-dihydro-4H-pyran-4-one ring, resulting in the desired spiro structure during cyclization. Compound 2 could be prepared from 3 under standard conditions for PIFA oxidation. The desired 2,3-dihydro-4H-pyran-4-one 3 was hypothesized to be derived from the diketone 4.
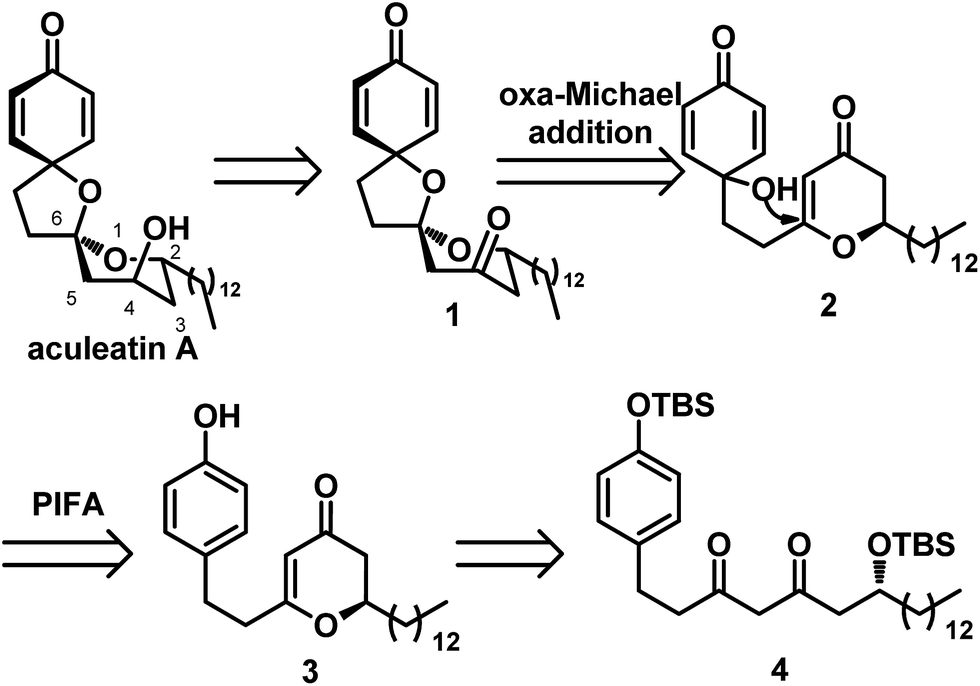 | ||
| Scheme 2 Retrosynthetic analysis of aculeatin A. | ||
We first prepared the intermediate 4 (Scheme 3). Its route commences with the commercially available 1-tetradecanal 6. We introduced the chiral center via a Crimmins modification of the Evans aldol reaction; in the presence of TiCl4, DIPEA, and NMP, the aldol reaction between 6 and 5 thus affords the aldol adduct 7 at 65% overall yield with a diastereoselectivity of 93:7.6 In a preliminary study, we attempted an addition between an enolate anion with a Weinreb amide to obtain 4. The alcohol 7 was treated with MeONHMe·HCl to afford 8.7 Subsequently, the hydroxyl in 8 was protected with TBSCl in the presence of imidazole to provide Weinreb amide 9, which was then subjected to addition with ketone 10 prepared from raspberry ketone through a known procedure.8 However, we found that the enolate anion thus formed from 10 using LDA hardly reacted with 9. Harris once obtained a similar result, in which the lithium salt of acetophenone with N-methoxy-N-methylacetamide failed to give anything other than the starting materials.9
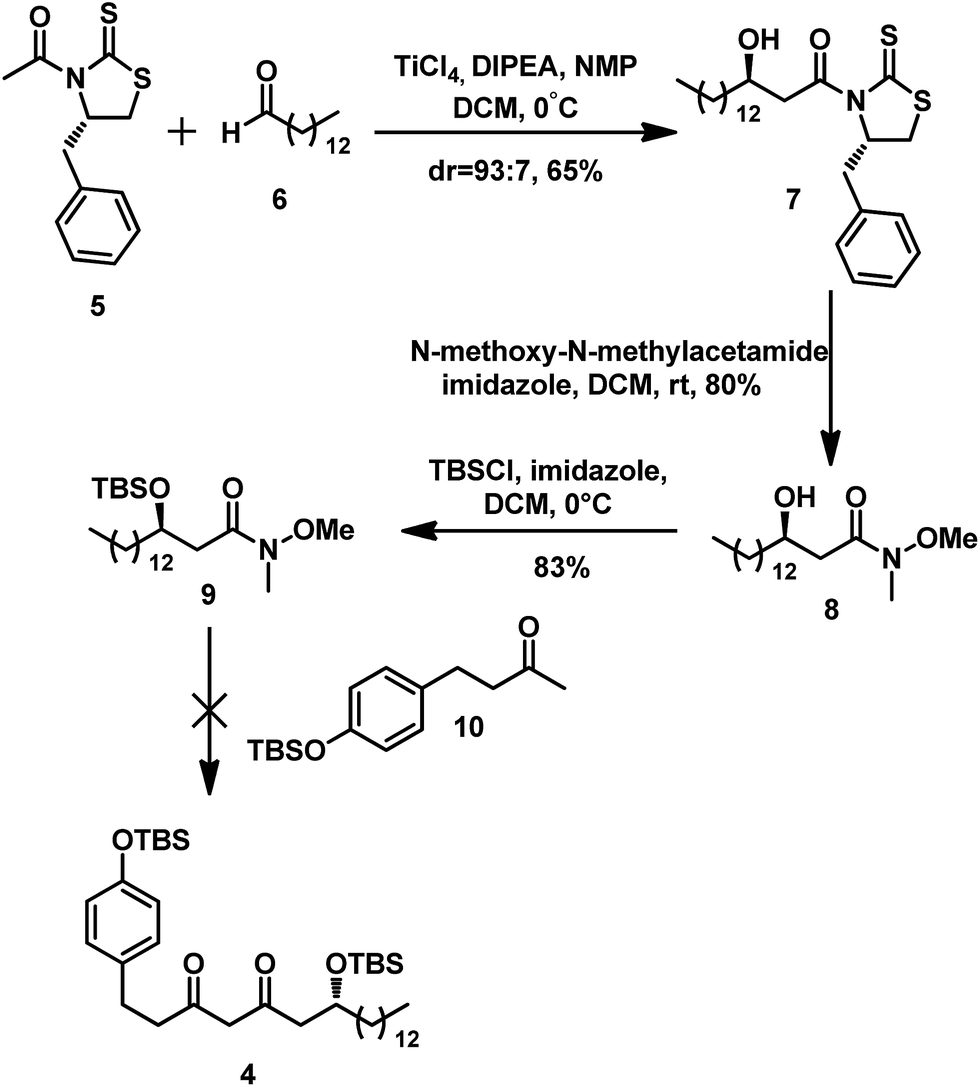 | ||
| Scheme 3 Initial approach to the synthesis of the intermediate 4 via Weinreb ketone synthesis. | ||
Thus, we adopted a Claisen condensation strategy in order to produce 4 in our retrosynthesis. We hypothesized that the condensation between the enolate anion of 10 with lactone 12 could afford 4 (Scheme 4). Therefore, removal of the auxiliary by hydrolysis with LiOH and H2O2 afforded the β-hydroxy acid 11.10 However, we hardly obtained the desired β-lactone 12. To simplify the preparation of 12, we turned to the approach shown in Scheme 5; another synthetic route using acyl chloride was used to produce the intermediate 4.
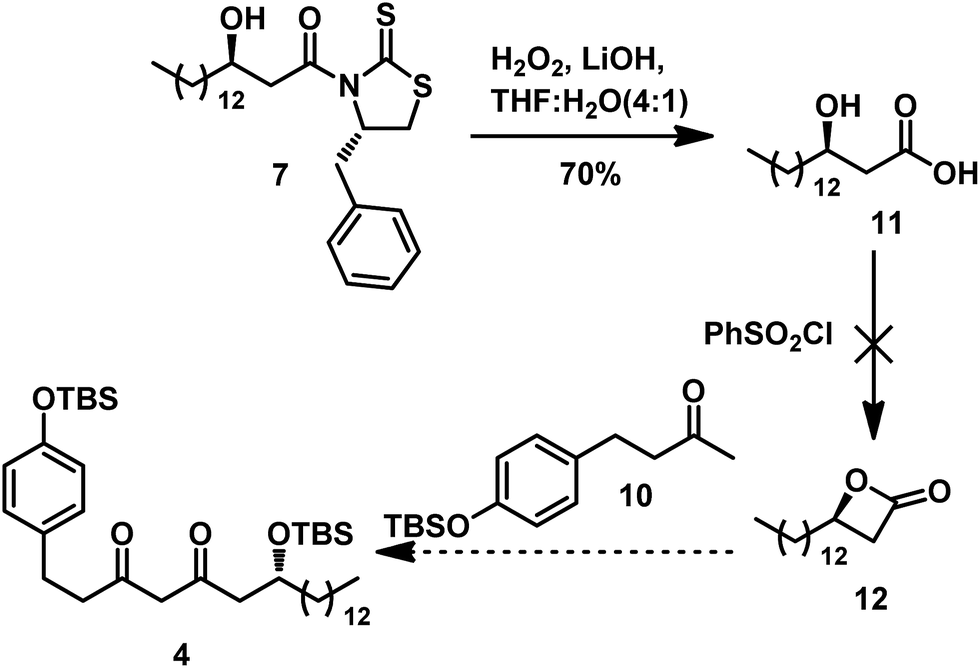 | ||
| Scheme 4 Initial synthetic approach to intermediate 4 via Claisen condensation strategy. | ||
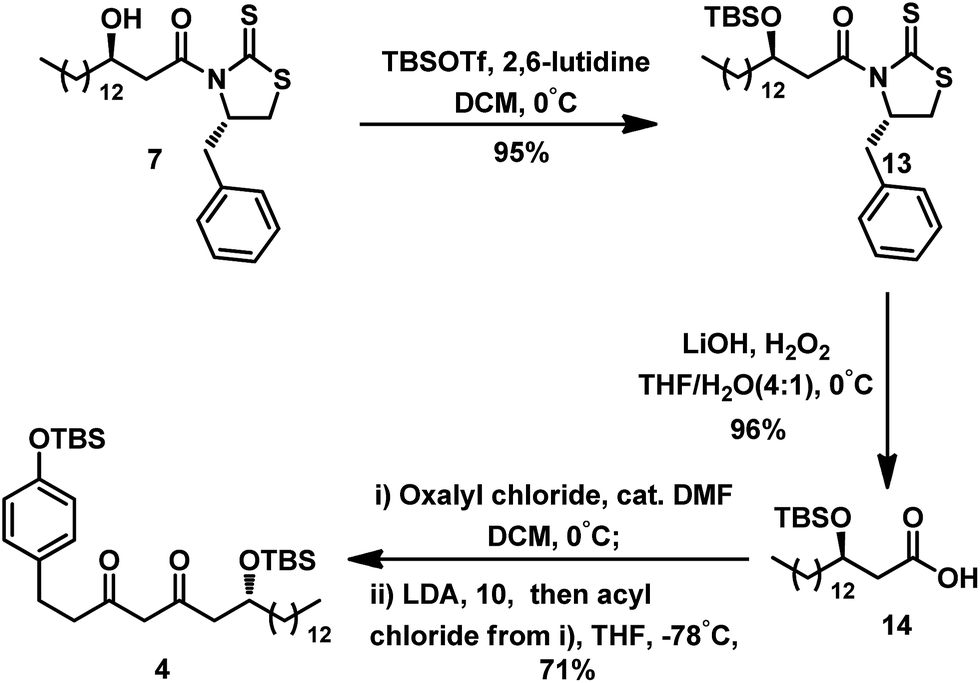 | ||
| Scheme 5 Synthesis of intermediate 4 via acyl chloride strategy. | ||
As illustrated in Scheme 5, protection of the hydroxyl group of compound 7 with TBSOTf gave TBS-ether 13 in 95% yield. Similarly, removal of the auxiliary in compound 13 by hydrolysis with LiOH and H2O2 afforded acid 14. To avoid deprotection of the TBS group, mild condition was used to prepare the acyl chloride of acid 14. Treatment using the catalyst DMF at 0 °C in the presence of oxalyl chloride resulted in smooth conversion of 14 to its acyl chloride,11 which was immediately subjected to Claisen condensation with the enolate anion derived from 10.12 This step generated the desired diketone intermediate 4 at 71% yield.
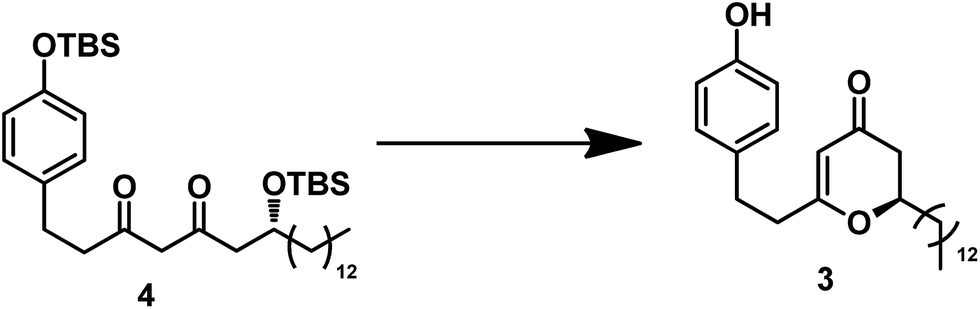
With diketone 4 in hand, we proceeded to construct the 2,3-dihydro-4H-pyran-4-one unit (Table 1) under typical conditions13 including acid and TBAF. In the presence of TFA in DCM, the desired product was not obtained even after an extended period. One equivalent of TsOH·H2O in the solvent DCM could promote the cyclization, leading to a yield of 53%. Increasing the amount of TsOH·H2O to 5 equiv. resulted in a higher yield (80%) and shorter reaction time (10 h). Because of the poor solubility of excess TsOH·H2O in DCM, we replaced the solution with acetone. A similar yield of 3 was thus obtained in much shorter time (5 h). Using TBAF as the deprotection reagent, we obtained 3 at relatively low yield (62%). We also conducted the reaction using the Lewis acid of BF3·OEt2;14 however, it gave a very low yield (30%) after 20 h.
Preparation of the precursor 3 enabled us to study the construction of the spiro unit of aculeatin A. Here, PIFA is an effective oxidizing agent; it is a common reagent for the oxidation of phenol to a cyclohexadienone ring with a tertiary hydroxyl. This hydroxyl can undergo intramolecular oxa-Michael addition to the 2,3-dihydro-4H-pyran-4-one unit, as described by Tong. We thus attempted the oxidation of 3 using PIFA, following the protocol of Tong.4s We first treated 3 with PIFA in a solvent mixture (acetone/H2O at 9:1 ratio) and then removed the solvent without further purification. The product was immediately subjected to CSA because of the possible instability of intermediate 2. Fortunately, we successfully obtained a single isomer of the desired polyspirocyclic compound 1 in 58% yield from compound 3. The latter was easily and diaselectively reduced to aculeatin A as a single isomer through a lithium tri-sec-butylborohydride (L-selectride)/MnO2 strategy at 42% overall yield (Scheme 6).
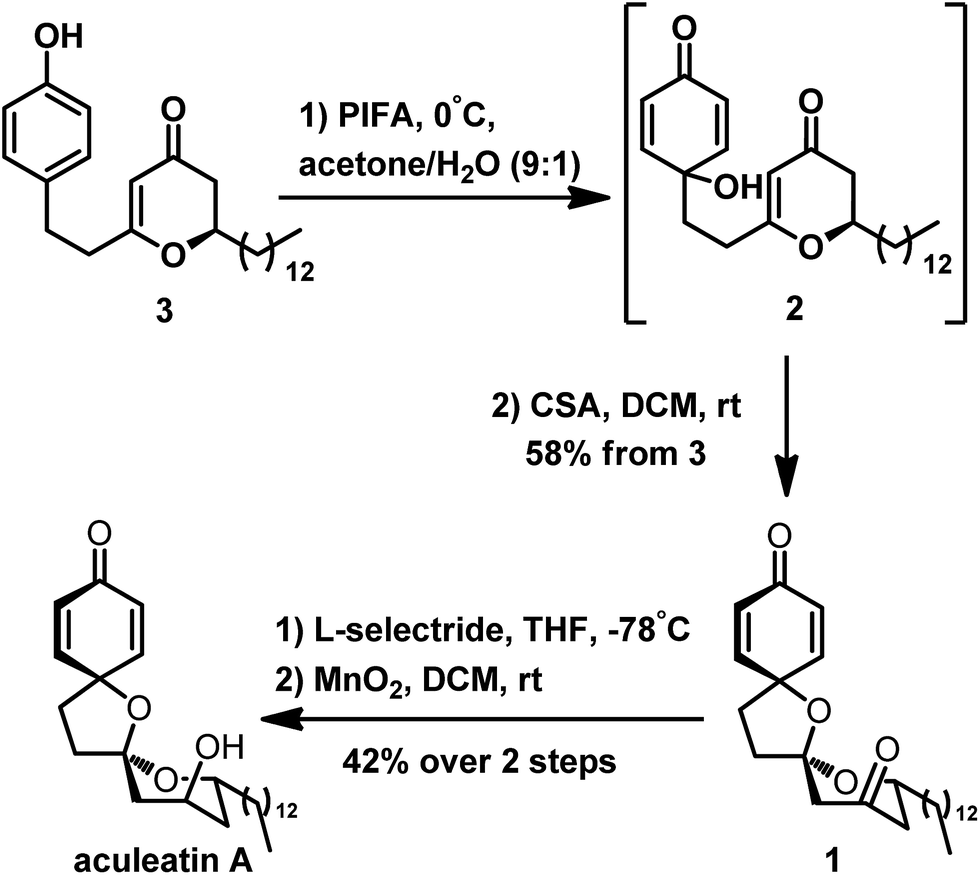 | ||
| Scheme 6 Total synthesis of aculeatin A. | ||
In our approach, preparation of aculeatin A depended critically on the highly diastereoselective intramolecular oxa-Michael addition of 2. We assumed that the spirocyclization proceeded via the transition state T-2 (Fig. 2), in which the hydrogen occupied an axial position. Under the condition of acid, hydroxyl at C-8 attacked the enone from the less-hindered Re-face side, that is, the side of hydrogen at C-2. This step produced diastereoselectively the desired stereocenter of aculeatin A without aculeatin B. The physical properties, as well as 1H and 13C NMR spectra of the product are consistent with those reported in the literature.4s
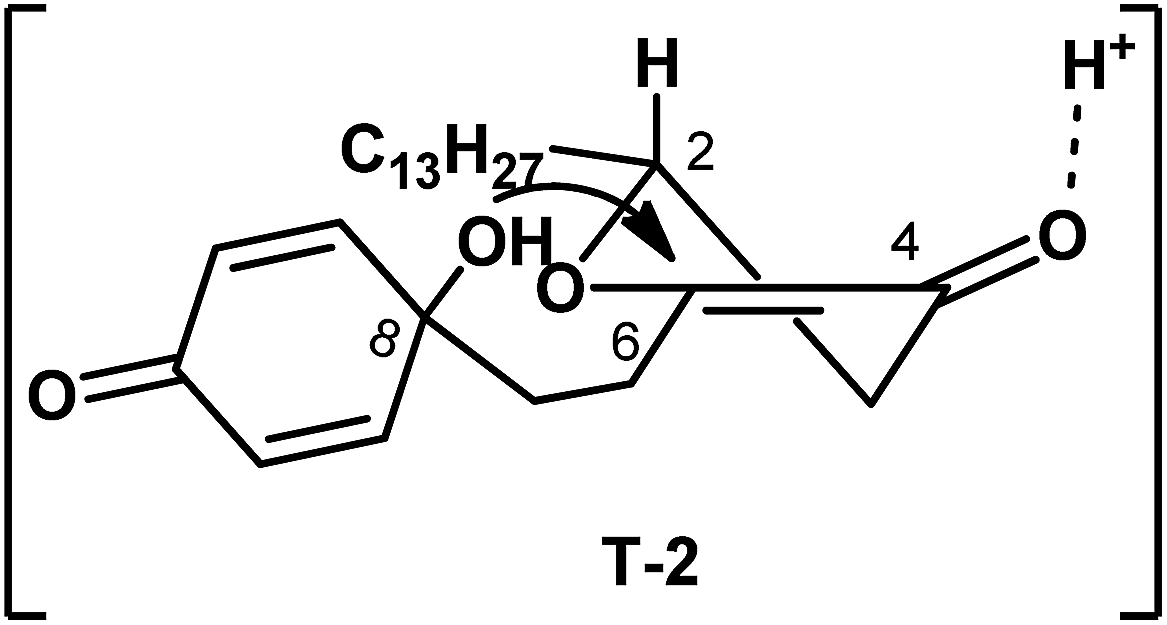 | ||
| Fig. 2 Transition state in the diastereoselective oxa-Michael addition. | ||
Conclusions
In summary, we have developed a novel and straightforward approach to the total synthesis of aculeatin A via a stepwise strategy, using commercially available 1-tetradecanal. This approach is the second example of a process that leads to aculeatin A as a single spiroisomer and is also the second example that does not end in PIFA-mediated cascade reaction involving oxidative cyclization (oxidation/spiroketalization). The total synthesis features 1,3-diketone preparation from Claisen condensation of ketone and acyl chloride, deprotection–cyclodehydration in a one-pot reaction, and intramolecular oxa-Michael addition of 2,3-dihydro-4H-pyran-4-one. The highly diastereoselective spirocyclization can occur during the intramolecular 5-exo-dig oxa-Michael addition in which the hydroxyl attacks from the less-hindered side. This stepwise strategy was demonstrated to be an effective protocol for constructing the spiro structure of aculeatin A, which can be useful in the syntheses of analogues of aculeatins and other natural products having a similar structure.Experimental
General
All reactions were carried out under N2 atmosphere with dry solvents unless otherwise noted and monitored by thin-layer chromatography (TLC) carried out on 0.25 mm silica gel plates (60F-254). Silica gel (200–300 mesh) supplied by Qingdao Marine chemical factory in China was used for flash column chromatography. Anhydrous THF was distilled from sodium-benzophenone. DCM (CH2Cl2) and DMF were distilled from CaH2. Other solvents or reagents weren't purified. Yield refers to chromatographically and spectroscopically (1H, 13C NMR), unless otherwise stated. NMR spectra were recorded on either a 400 MHz spectrometer (1H: 400 MHz, 13C: 100 MHz) or 500 MHz (1H: 500 MHz, 13C: 125 MHz). High-resolution mass spectra were obtained from a MALDI-TOF Mass Spectrometer. IR spectra were recorded on a Shimadzu FT-IR spectrophotometer. Optical rotations were measured on a digital polarimeter in CHCl3 at 25 °C.:1) to give compound 13 (3.13 g, 95%). [α]25D = 174.6 (c 2.3, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.38–7.35 (m, 2H), 7.31–7.27 (m, 3H), 5.30–5.26 (m, 1H), 4.38–4.33 (m, 1H), 3.58 (dd, J = 16.7, 8.2 Hz, 1H), 3.35 (dd, J = 11.5, 7.3 Hz, 1H), 3.27 (dd, J = 13.2, 3.8 Hz, 1H), 3.17 (dd, J = 16.7, 3.8 Hz, 1H), 3.06 (dd, J = 13.1, 10.7 Hz, 1H), 2.89 (d, J = 11.6 Hz, 1H), 1.56–1.52 (m, 2H), 1.30–1.21 (m, 22H), 0.90 (t, J = 6.9 Hz, 3H), 0.88 (s, 9H), 0.11 (s, 3H), 0.07 (s, 3H); 13C NMR (125 MHz, CDCl3): 201.0, 172.5, 136.6, 129.4, 128.9, 127.2, 69.3, 68.7, 45.8, 37.8, 36.5, 32.2, 31.9, 29.7–29.6 (br, 6C), 29.3, 25.8, 24.9, 22.7, 18.0, 14.1, −4.4, −4.7; IR (film): 2925, 2853, 1699, 1464, 1362, 1341, 1257, 1165, 1042, 836, 775, 701 cm−1; HRMS (ESI): m/z calcd for C32H56NO2S2Si+ [M + H]+: 578.3516, found 578.3519.:1) at 0 °C. The mixture was then stirred at 0 °C for 1 h. When the TLC monitor indicated compound 13 was consumed, the reaction was quenched with NH4Cl and the pH was adjusted to 0 using 1 N HCl. The organic layer was separated and the aqueous layer was extracted with EtOAc (100 mL × 3). The combined organic layer was then washed with sat. NaCl solution, dried over sodium sulfate, filtered, and concentrated in reduced pressure. The residue was purified with flash chromatography (hexane/EtOAc = 15:1) to afford the desired acid 14 as a white solid (3.84 g, 96%). [α]25D = 0.49 (c 2.6, CHCl3). 1H NMR (500 MHz, CDCl3): δ 4.14–4.11 (m, 1H), 2.48 (d, J = 5.5 Hz, 2H), 1.59–1.50 (m, 2H), 1.30–1.21 (m, 22H), 0.90 (t, J = 6.5 Hz, 3H), 0.88 (s, 9H), 0.08 (s, 3H), 0.07 (s, 3H); 13C NMR (125 MHz, CDCl3): 177.4, 69.4, 42.3, 37.5, 31.9, 29.7–29.6 (br, 5C), 29.3, 25.8, 25.0, 22.7, 18.0, 14.1, 1.0, −4.5, −4.8; IR (film): 2926, 2855, 1715, 1466, 1258, 1219, 1098, 1082, 836, 773 cm−1; HRMS (ESI): m/z calcd for C22H47O3Si+ [M + H]+: 387.3289, found 387.3281.Under a N2 atmosphere, n-BuLi (1.14 mL, 2.86 mmol, 2.2 equiv.) was added dropwise to the stirred solution of DIPA (0.40 mL, 2.86 mmol, 2.2 equiv.) in anhydrous THF (10 mL) at −78 °C. After stirring for 20 min at −78 °C, the solution of 10 (0.72 g, 2.60 mmol, 2.0 equiv.) in THF (0.5 mL) was added slowly to the freshly prepared LDA. The reaction was then stirred at −78 °C for another 10 min followed by addition of the above acyl chloride. When the TLC monitor indicated compound 14 was consumed, the reaction was quenched with 1 N HCl. Subsequently, the resultant mixture was extracted with DCM (50 mL × 3) and the combined organic layer was washed with sat. NaCl, dried over anhydrous sodium sulfate, filtered through a funnel, and concentrated under reduced pressure. Purification by flash chromatography (hexane/EtOAc = 50:1) gave the desired compound 4 as a yellow oil (0.60 g, 71%). [α]25D = 61.6 (c 0.33, CHCl3). 1H NMR (500 MHz, CDCl3): δ 15.50 (br, 0.5H), 7.04 (d, J = 8.0 Hz, 1.6H), 7.02–6.99 (m, 0.4H), 6.78–6.77 (m, 0.4H), 6.76 (d, J = 8.0 Hz, 1.6H), 5.50 (s, 0.7H), 4.20–4.13 (m, 0.25H), 4.11–4.07 (m, 0.75H), 3.74–3.53 (m, 0.3H), 2.86 (t, J = 8.0 Hz, 1.6H), 2.82–2.77 (m, 0.4H), 2.65–2.62 (m, 0.4H), 2.56 (t, J = 8.0 Hz, 1.6H), 2.40–2.32 (m, 1.6H), 2.23–2.08 (m, 0.5H), 1.48–1.45 (m, 2H), 1.30–1.21 (m, 22H), 0.99 (s, 9H), 0.89 (t, J = 6.8 Hz, 3H), 0.87 (s, 9H), 0.19 (s, 6H), 0.08–0.03 (m, 6H); 13C NMR (125 MHz, CDCl3): 194.3, 191.0, 154.0, 133.4, 129.1, 120.0, 69.9, 46.2, 40.6, 37.8, 31.9, 30.7, 29.7–29.5 (br, 5C), 29.3, 25.8, 25.7, 25.0, 22.7, 18.2, 18.0, 14.1, 1.0, −4.4, −4.6, −4.9; IR (film): 3471, 2958, 2927, 2856, 1612, 1511, 1472, 1258, 1023, 917, 837, 773 cm−1; HRMS (ESI): m/z calcd for C38H71O4Si2+ [M + H]+: 647.4885, found 647.4894.
:1) offered the desired compound 3 in yield of 80% (20 mg). [α]25D = 64.0 (c 0.53, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.01 (d, J = 8.0 Hz, 2H), 6.77 (d, J = 8.0 Hz, 2H), 5.31 (s, 1H), 4.32–4.30 (m, 1H), 2.80 (t, J = 7.5 Hz, 2H), 2.57–2.48 (m, 2H), 2.46–2.35 (m, 2H), 1.81–1.76 (m, 1H), 1.67–1.61 (m, 1H), 1.49–1.42 (m, 1H), 1.40–1.23 (m, 21H), 0.88 (t, J = 6.7 Hz, 3H); 13C NMR (125 MHz, CDCl3): 193.9, 177.4, 154.6, 131.7, 129.3, 115.4, 104.3, 79.3, 40.8, 36.8, 34.4, 31.9, 31.7, 29.6–29.3 (br, 8C), 24.9, 22.6, 14.1; IR (film): 3270, 2925, 2854, 1654, 1595, 1517, 1466, 1400, 1340, 1221, 1023, 826, 773 cm−1; HRMS (ESI): m/z calcd for C26H41O3+ [M + H]+: 401.3050, found 401.3052.:1) at 0 °C. The reaction was then kept stirred in the dark for 2 h. When the TLC monitor indicated compound 3 was consumed, anhydrous Na2SO4 and anhydrous NaHCO3 were added to the reaction. The mixture was then filtered and concentrated under reduced pressure. Without further purification, the residence was dissolved in DCM (20 mL) and CSA (0.24 g, 1.05 mmol, 1.0 equiv.) was added to this solvent. The resultant mixture was then kept stirred overnight and quenched with sat. NaHCO3. The organic layer was separated and the aqueous layer was extracted with DCM (20 mL × 3). Subsequently, the combined organic layer was washed with sat. NaCl, dried over sodium sulfate, filtered and concentrated in vacuum. The residence was purified by flash chromatography (hexane/EA = 3:1) to provide compound 1 (0.25 g, 58% from compound 3).[α]25D = −7.8 (c 1.0, CHCl3). 1H NMR (400 MHz, CDCl3): δ 6.77 (ddd, J = 20.8, 9.6, 2.8 Hz, 2H), 6.16–6.10 (m, 2H), 4.17–4.11 (m, 1H), 2.77 (d, J = 14.4 Hz, 1H), 2.56 (d, J = 14.4 Hz, 1H), 2.44–2.40 (m, 3H), 2.27–2.24 (m, 1H), 2.10–2.09 (m, 2H), 1.67–1.65 (m, 1H), 1.65–1.53 (m, 1H), 1.30–1.22 (m, 22H), 0.88 (t, J = 6.4 Hz, 3H); 13C NMR (125 MHz, CDCl3): 204.3, 185.1, 150.2, 148.4, 127.6, 127.2, 109.6, 79.7, 69.7, 49.8, 46.7, 38.7, 36.1, 34.9, 31.9, 29.6–29.3 (br, 8C), 25.4, 22.6, 14.1; IR (film): 3490, 2924, 2854, 1727, 1674, 1467, 1021, 883, 857, 773 cm−1; HRMS (ESI): m/z calcd for C26H41O4+ [M + H]+: 417.3006, found 417.3000.
:1) to provide aculeatin A (21 mg, 42% from compound 1). [α]25D = −13.5 (c 0.2, CHCl3). 1H NMR (300 MHz, CDCl3): δ 6.85 (dd, J = 10.0, 2.8 Hz, 1H), 6.75 (dd, J = 10.0, 2.8 Hz, 1H), 6.16–6.09 (m, 2H), 4.13–4.07 (m, 2H), 2.39–2.36 (m, 1H), 2.26–2.20 (m, 1H), 2.05–2.00 (m, 3H), 1.92 (br d, J = 14.0 Hz, 1H), 1.79 (br d, J = 12.0, 1H), 1.52–1.40 (m, 5H), 1.38–1.20 (m, 20H), 0.88 (t, J = 6.8 Hz, 3H); 13C NMR (75 MHz, CDCl3): 185.3, 150.9, 148.7, 127.3, 127.0, 109.0, 79.7, 65.3, 64.8, 39.1, 39.0, 37.9, 35.8, 34.1, 31.9, 29.6–29.3 (br, 8C), 25.6, 22.6, 14.1; IR (film): 3552, 2925, 2854, 1674, 1632, 1465, 1392, 1220, 1099, 1046, 1000, 853, 772 cm−1; HRMS (ESI): m/z calcd for C26H43O4+ [M + H]+: 419.3156, found 419.3156.Acknowledgements
We thank the National Science Foundation of China (21362012, 21062088, 21562020), the Science and Technology Research Projects of the Education Department of Jiangxi Province (No. GJJ14579), the Natural Science Foundation of Jiangxi Province (No. 20151BAB203007), and the Science and Technology Plan Project of Jiangxi Province (No. 20142BBE50006, 20151BBE50004, 20151BBG70028) for the funding support.Notes and references
- World Health Organization, World Malaria Report 2015, World Health Organization, Geneva, Switzerland, 2015, http://www.who.int/malaria/publications/world_malaria_report_2014/en/.
- A. M. Dondorp, F. Nosten, P. Yi, D. Das, A. P. Phyo, J. Tarning, K. M. Lwin, F. Ariey, W. Hanpithakpong, S. J. Lee, P. Ringwald, K. Silamut, M. Imwong, K. Chotivanich, P. Lim, T. Herdman, S. S. An, S. Yeung, P. Singhasivanon, N. P. Day, N. Lindegardh, D. Socheat and N. J. White, N. Engl. J. Med., 2009, 361, 455 CrossRef CAS PubMed.
- (a) J. Heilmann, S. Mayr, R. Brun, T. Rali and O. Sticher, Helv. Chim. Acta, 2000, 83, 2939 CrossRef CAS; (b) J. Heilmann, R. Brun, S. Mayr, T. Rali and O. Sticher, Phytochemistry, 2001, 57, 1281 CrossRef CAS PubMed.
- (a) Y.-S. Wong, Chem. Commun., 2002, 686 RSC; (b) J. E. Baldwin, R. M. Adlington, V. W.-W. Sham, R. Márquez and P. G. Bulger, Tetrahedron, 2005, 61, 2353 CrossRef CAS; (c) E. Falomir, P. Álvarez-Bercedo, M. Carda and J. A. Marco, Tetrahedron Lett., 2005, 46, 8407 CrossRef CAS; (d) P. Álvarez-Bercedo, E. Falomir, M. Carda and J. A. Marco, Tetrahedron, 2006, 62, 9641 CrossRef; (e) M. Peuchmaur and Y.-S. Wong, J. Org. Chem., 2007, 72, 5374 CrossRef CAS PubMed; (f) S. Chandrasekhar, Ch. Rambabu and T. Shyamsunder, Tetrahedron Lett., 2007, 48, 4683 CrossRef CAS; (g) C. V. Ramana and B. Srinivas, J. Org. Chem., 2008, 73, 3915 CrossRef CAS PubMed; (h) V. Suresh, J. J. P. Selvam, K. Rajesh and Y. Venkateswarlu, Tetrahedron: Asymmetry, 2008, 19, 1509 CrossRef CAS; (i) Z.-B. Zhen, J. Gao and Y. Wu, J. Org. Chem., 2008, 73, 7310 CrossRef CAS PubMed; (j) A. Kamal, P. V. Reddy, S. Prabhakar and M. Balakrishna, Tetrahedron: Asymmetry, 2009, 20, 2861 CrossRef CAS; (k) V. Malathong and S. D. Rychnovsky, Org. Lett., 2009, 11, 4220 CrossRef CAS PubMed; (l) A. Harbindu and P. Kumar, Synthesis, 2010, 1479 CAS; (m) C. V. Ramana and S. K. Pandey, Tetrahedron, 2010, 66, 390 CrossRef CAS; (n) J. S. Yadav, K. V. R. Rao, K. Ravindar and B. V. S. Reddy, Synlett, 2010, 51 CrossRef CAS; (o) J. S. Yadav, N. Thrimurtulu, M. Venkatesh and A. R. Prasad, Synthesis, 2010, 431 CrossRef CAS; (p) J. S. Yadav, Y. G. Rao, D. Chandrakanth, K. Ravindar and B. V. S. Reddy, Helv. Chim. Acta, 2010, 93, 2426 CrossRef CAS; (q) B. Das, M. Krishnaiah and C. Sudhakar, Bioorg. Med. Chem. Lett., 2010, 20, 2303 CrossRef CAS PubMed; (r) B. Das, M. Krishnaiah, S. Nagendra and C. R. Reddy, Lett. Org. Chem., 2011, 8, 244 CrossRef CAS; (s) H. B. Yao, L. Y. Song and R. B. Tong, J. Org. Chem., 2014, 73, 1498 CrossRef PubMed; (t) S. P. Huang, S. P. Chen, G. P. Wang, J. T. Zhang, L. J. Tang, G. Y. Du and X. J. Wang, Synthesis, 2015, 47, 1303 CrossRef CAS.
- L. Y. Song, H. L. Yao, Y. J. Dai, M. W. Wu and R. B. Tong, Tetrahedron Lett., 2016, 57, 4257 CrossRef CAS.
- (a) M. T. Crimmins and J. She, Synlett, 2004, 8, 1371 Search PubMed; (b) M. T. Crimmins, B. W. King, E. A. Tabet and K. Chaudhary, J. Org. Chem., 2001, 66, 894 CrossRef CAS PubMed.
- (a) M. T. Crimmins, B. W. King and E. A. Tabet, J. Am. Chem. Soc., 1997, 119, 7883 CrossRef CAS; (b) J. I. Levin, E. Turos and S. M. Weinreb, Synth. Commun., 1982, 12, 989 CrossRef CAS; (c) A. Basha, M. Lipton and S. M. Weinreb, Tetrahedron Lett., 1977, 18, 4171 CrossRef.
- D. J. Trader and E. E. Carlson, J. Org. Chem., 2013, 78, 7349 CrossRef CAS PubMed.
- T. A. Oster and T. M. Harris, Tetrahedron Lett., 1983, 24, 1851 CrossRef CAS.
- S. Kawamura, Y. Unno, A. Asai, M. Arisawa and S. Shuto, Org. Biomol. Chem., 2013, 11, 6615 CAS.
- K. Anderson, F. Calo, T. Pfaffeneder, A. J. P. White and A. G. M. Barrett, Org. Lett., 2011, 13, 5748 CrossRef CAS PubMed.
- P. A. Wender, A. V. W. Mayweg and C. L. VanDeusen, Org. Lett., 2003, 5, 277 CrossRef CAS PubMed.
- H. Wang, B. J. Shuhler and M. Xian, J. Org. Chem., 2007, 72, 4280 CrossRef CAS PubMed.
- D. R. Kelly and S. M. Roberts, Synth. Commun., 1979, 9, 295 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra27140e |
| This journal is © The Royal Society of Chemistry 2017 |